
Targeting the KRAS Oncogene for Patients with Metastatic Colorectal Cancer


Simple Summary
Abstract
1. Introduction
2. Molecular Biology of KRAS Mutations
3. Biological Implications of KRAS Mutations
3.1. Tumor Initiation and Progression
3.2. Impact on the TME
3.3. Role in Metastasis
3.4. Therapeutic Resistance
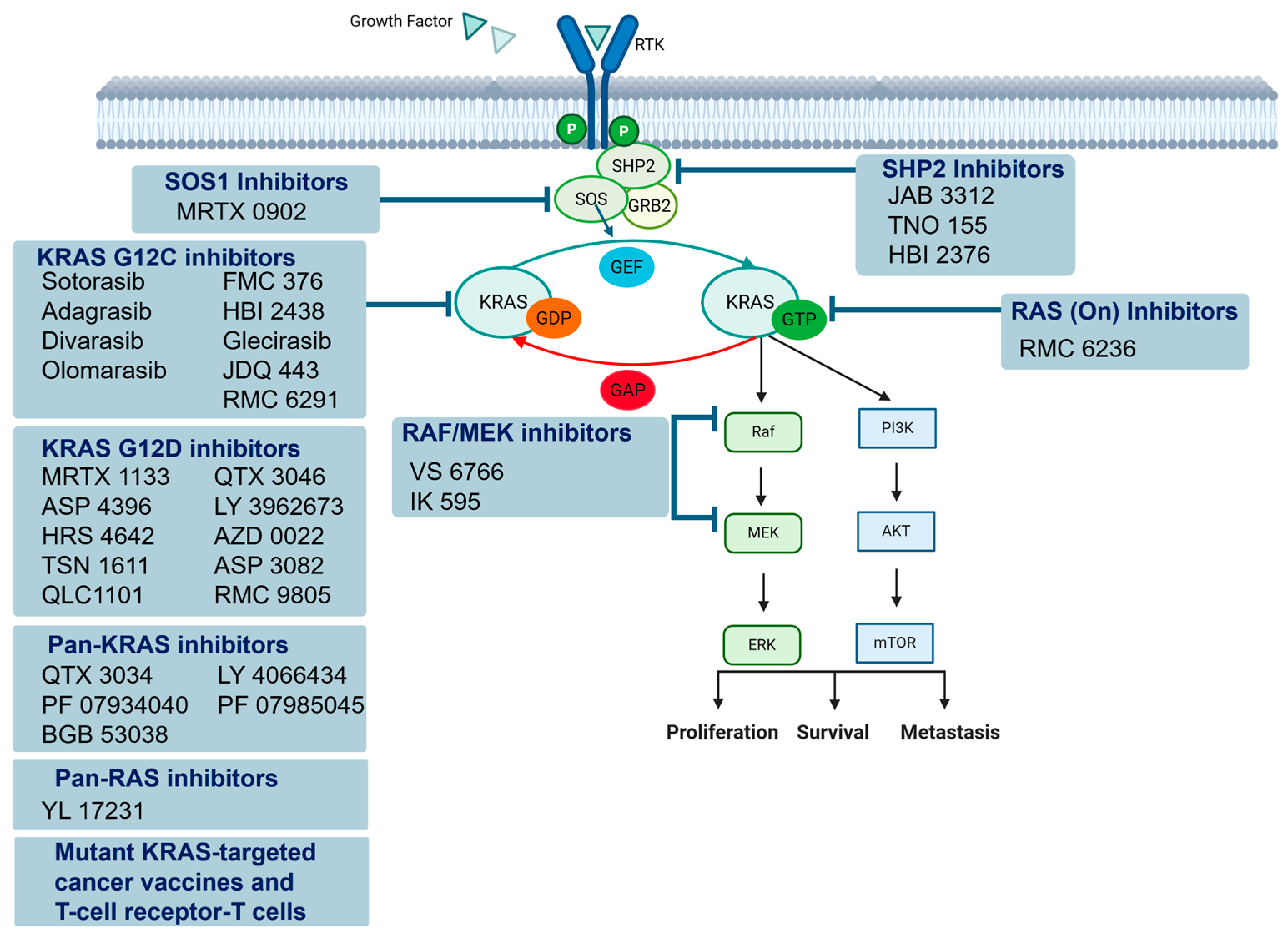
4. Advances in Therapeutics
4.1. KRAS G12C Inhibitors
4.2. KRAS G12C Inhibitors Combined with Anti-EGFR Therapy
4.3. Targeting Other KRAS Mutations
4.3.1. KRAS G12D
4.3.2. Pan-KRAS Inhibitors
4.4. Targeting KRAS-Signaling Pathways
4.5. Targeting Other Pathways
4.6. Mutant-KRAS-Targeted Cancer Vaccines
4.7. Adoptive T-Cell Therapy
5. Challenges and Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Deming, D.A. Development of KRAS Inhibitors and Their Role for Metastatic Colorectal Cancer. J. Natl. Compr. Canc. Netw. 2025, 23, e247067. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bui, T.A.; Yang, X.; Hutvagner, G.; Deng, W. Advancements in gene therapies targeting mutant KRAS in cancers. Cancer Metastasis Rev. 2025, 44, 24. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Tran, N.H.; Cavalcante, L.L.; Lubner, S.J.; Mulkerin, D.L.; LoConte, N.K.; Clipson, L.; Matkowskyj, K.A.; Deming, D.A. Precision medicine in colorectal cancer: The molecular profile alters treatment strategies. Ther. Adv. Med. Oncol. 2015, 7, 252–262. [Google Scholar] [CrossRef]
- Punekar, S.R.; Velcheti, V.; Neel, B.G.; Wong, K.K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. [Google Scholar] [CrossRef]
- Boileve, A.; Smolenschi, C.; Lambert, A.; Boige, V.; Delaye, M.; Camilleri, G.M.; Tarabay, A.; Valery, M.; Fuerea, A.; Pudlarz, T.; et al. KRAS, a New Target for Precision Medicine in Colorectal Cancer? Cancers 2024, 16, 3455. [Google Scholar] [CrossRef]
- Rappaport, A.R.; Kyi, C.; Lane, M.; Hart, M.G.; Johnson, M.L.; Henick, B.S.; Liao, C.Y.; Mahipal, A.; Shergill, A.; Spira, A.I.; et al. A shared neoantigen vaccine combined with immune checkpoint blockade for advanced metastatic solid tumors: Phase 1 trial interim results. Nat. Med. 2024, 30, 1013–1022. [Google Scholar] [CrossRef]
- Pant, S.; Wainberg, Z.A.; Weekes, C.D.; Furqan, M.; Kasi, P.M.; Devoe, C.E.; Leal, A.D.; Chung, V.; Basturk, O.; VanWyk, H.; et al. Lymph-node-targeted, mKRAS-specific amphiphile vaccine in pancreatic and colorectal cancer: The phase 1 AMPLIFY-201 trial. Nat. Med. 2024, 30, 531–542. [Google Scholar] [CrossRef]
- Devoe, C.E.; Pant, S.; Wainberg, Z.A.; Chung, V.; George, T.J.; Kasi, P.M.; VanWyk, H.; Tavares, A.; Perry, J.; Kheoh, T.; et al. AMPLIFY-7P, a first-in-human safety and efficacy trial of adjuvant mKRAS-specific lymph node targeted amphiphile ELI-002 7P vaccine in patients with minimal residual disease–positive pancreatic and colorectal cancer. J. Clin. Oncol. 2024, 42, 2636. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Guo, B. RNA-based therapeutics for colorectal cancer: Updates and future directions. Pharmacol. Res. 2020, 152, 104550. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, K.; Balan, D.J.; Devi, K.P.; Nabavi, S.F.; Reshadat, S.; Khayatkashani, M.; Mahmoodifar, S.; Filosa, R.; Amirkhalili, N.; Pishvaei, S.; et al. Short interfering RNA in colorectal cancer: Is it wise to shoot the messenger? Eur. J. Pharmacol. 2023, 949, 175699. [Google Scholar] [CrossRef]
- Jang, G.; Kweon, J.; Kim, Y. CRISPR prime editing for unconstrained correction of oncogenic KRAS variants. Commun. Biol. 2023, 6, 681. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Kim, H.S.; Song, M.; Cha, Y.H.; Kim, Y.H.; Shin, J.; Lee, E.S.; Joo, Y.; Song, J.J.; et al. Targeting mutant KRAS with CRISPR-Cas9 controls tumor growth. Genome Res. 2018, 28, 374–382. [Google Scholar] [CrossRef]
- Chen, L.; Li, Q. Nanomaterials in the diagnosis and treatment of gastrointestinal tumors: New clinical choices and treatment strategies. Mater. Today Bio 2025, 32, 101782. [Google Scholar] [CrossRef]
- Kasi, P.B.; Mallela, V.R.; Ambrozkiewicz, F.; Trailin, A.; Liska, V.; Hemminki, K. Theranostics Nanomedicine Applications for Colorectal Cancer and Metastasis: Recent Advances. Int. J. Mol. Sci. 2023, 24, 7922. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Beck, H.; Härter, M.; Haß, B.; Schmeck, C.; Baerfacker, L. Small molecules and their impact in drug discovery: A perspective on the occasion of the 125th anniversary of the Bayer Chemical Research Laboratory. Drug Discov. Today 2022, 27, 1560–1574. [Google Scholar] [CrossRef]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef]
- Shibata, D.; Schaeffer, J.; Li, Z.H.; Capella, G.; Perucho, M. Genetic heterogeneity of the c-K-ras locus in colorectal adenomas but not in adenocarcinomas. J. Natl. Cancer Inst. 1993, 85, 1058–1063. [Google Scholar] [CrossRef]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Xu, M.; Zhao, X.; Wen, T.; Qu, X. Unveiling the role of KRAS in tumor immune microenvironment. Biomed. Pharmacother. 2024, 171, 116058. [Google Scholar] [CrossRef]
- Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers 2021, 13, 5059. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Ferreira, A.; Pereira, F.; Reis, C.; Oliveira, M.J.; Sousa, M.J.; Preto, A. Crucial Role of Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation: Therapeutic Implications. Cells 2022, 11, 2183. [Google Scholar] [CrossRef]
- Lemieux, E.; Cagnol, S.; Beaudry, K.; Carrier, J.; Rivard, N. Oncogenic KRAS signalling promotes the Wnt/beta-catenin pathway through LRP6 in colorectal cancer. Oncogene 2015, 34, 4914–4927. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Gao, Z.; Bao, Y.; Chen, L.; Huang, Y.; Liu, Y.; Dong, Q.; Wei, X. Wnt/beta-catenin signaling pathway in carcinogenesis and cancer therapy. J. Hematol. Oncol. 2024, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, P.; Knoblaugh, S.; Washington, M.K.; Munoz, N.M.; Tsuchiya, K.D.; Rojas, A.; Song, X.; Ulrich, C.M.; Sasazuki, T.; Shirasawa, S.; et al. TGF-beta receptor inactivation and mutant Kras induce intestinal neoplasms in mice via a beta-catenin-independent pathway. Gastroenterology 2009, 136, 1680–1688.e7. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Ma, Q.; Zhang, W.; Wu, K.; Shi, L. The roles of KRAS in cancer metabolism, tumor microenvironment and clinical therapy. Mol. Cancer 2025, 24, 14. [Google Scholar] [CrossRef]
- Jinesh, G.G.; Sambandam, V.; Vijayaraghavan, S.; Balaji, K.; Mukherjee, S. Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene 2018, 37, 839–846. [Google Scholar] [CrossRef]
- Grabocka, E.; Commisso, C.; Bar-Sagi, D. Molecular pathways: Targeting the dependence of mutant RAS cancers on the DNA damage response. Clin. Cancer Res. 2015, 21, 1243–1247. [Google Scholar] [CrossRef]
- Neumann, J.; Zeindl-Eberhart, E.; Kirchner, T.; Jung, A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol. Res. Pract. 2009, 205, 858–862. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS oncogene substitutions on protein behavior: Implications for signaling and clinical outcome. J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef] [PubMed]
- McFall, T.; Diedrich, J.K.; Mengistu, M.; Littlechild, S.L.; Paskvan, K.V.; Sisk-Hackworth, L.; Moresco, J.J.; Shaw, A.S.; Stites, E.C. A systems mechanism for KRAS mutant allele-specific responses to targeted therapy. Sci. Signal. 2019, 12, eaaw8288. [Google Scholar] [CrossRef] [PubMed]
- McFall, T.; Stites, E.C. Identification of RAS mutant biomarkers for EGFR inhibitor sensitivity using a systems biochemical approach. Cell Rep. 2021, 37, 110096. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Thiagalingam, S.; Lengauer, C.; Leach, F.S.; Schutte, M.; Hahn, S.A.; Overhauser, J.; Willson, J.K.; Markowitz, S.; Hamilton, S.R.; Kern, S.E.; et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat. Genet. 1996, 13, 343–346. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Duong, H.Q. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef]
- Janssen, J.B.E.; Medema, J.P.; Gootjes, E.C.; Tauriello, D.V.F.; Verheul, H.M.W. Mutant RAS and the tumor microenvironment as dual therapeutic targets for advanced colorectal cancer. Cancer Treat. Rev. 2022, 109, 102433. [Google Scholar] [CrossRef]
- Figueras, A.; Arbos, M.A.; Quiles, M.T.; Vinals, F.; Germa, J.R.; Capella, G. The impact of KRAS mutations on VEGF-A production and tumour vascular network. BMC Cancer 2013, 13, 125. [Google Scholar] [CrossRef]
- Parikh, K.; Banna, G.; Liu, S.V.; Friedlaender, A.; Desai, A.; Subbiah, V.; Addeo, A. Drugging KRAS: Current perspectives and state-of-art review. J. Hematol. Oncol. 2022, 15, 152. [Google Scholar] [CrossRef]
- Tripathi, K.; Garg, M. Mechanistic regulation of epithelial-to-mesenchymal transition through RAS signaling pathway and therapeutic implications in human cancer. J. Cell Commun. Signal. 2018, 12, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.N.; Du, W.; Brekken, R.A. Behind the Wheel of Epithelial Plasticity in KRAS-Driven Cancers. Front. Oncol. 2019, 9, 1049. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kuang, Y.; Wang, C.; Yu, Y.; Pan, L.; Hu, X. Impact of KRAS mutation on the tumor microenvironment in colorectal cancer. Int. J. Biol. Sci. 2024, 20, 1947–1964. [Google Scholar] [CrossRef] [PubMed]
- van Houdt, W.J.; Hoogwater, F.J.; de Bruijn, M.T.; Emmink, B.L.; Nijkamp, M.W.; Raats, D.A.; van der Groep, P.; van Diest, P.; Borel Rinkes, I.H.; Kranenburg, O. Oncogenic KRAS desensitizes colorectal tumor cells to epidermal growth factor receptor inhibition and activation. Neoplasia 2010, 12, 443–452. [Google Scholar] [CrossRef]
- Bellio, H.; Fumet, J.D.; Ghiringhelli, F. Targeting BRAF and RAS in Colorectal Cancer. Cancers 2021, 13, 2201. [Google Scholar] [CrossRef]
- Wong, C.C.; Xu, J.; Bian, X.; Wu, J.L.; Kang, W.; Qian, Y.; Li, W.; Chen, H.; Gou, H.; Liu, D.; et al. In Colorectal Cancer Cells With Mutant KRAS, SLC25A22-Mediated Glutaminolysis Reduces DNA Demethylation to Increase WNT Signaling, Stemness, and Drug Resistance. Gastroenterology 2020, 159, 2163–2180.e6. [Google Scholar] [CrossRef]
- Moss, D.Y.; McCann, C.; Kerr, E.M. Rerouting the drug response: Overcoming metabolic adaptation in KRAS-mutant cancers. Sci. Signal. 2022, 15, eabj3490. [Google Scholar] [CrossRef]
- Strickler, J.H.; Yoshino, T.; Stevinson, K.; Eichinger, C.S.; Giannopoulou, C.; Rehn, M.; Modest, D.P. Prevalence of KRAS G12C Mutation and Co-mutations and Associated Clinical Outcomes in Patients With Colorectal Cancer: A Systematic Literature Review. Oncologist 2023, 28, e981–e994. [Google Scholar] [CrossRef]
- Salem, M.E.; El-Refai, S.M.; Sha, W.; Puccini, A.; Grothey, A.; George, T.J.; Hwang, J.J.; O’Neil, B.; Barrett, A.S.; Kadakia, K.C.; et al. Landscape of KRAS(G12C), Associated Genomic Alterations, and Interrelation with Immuno-Oncology Biomarkers in KRAS-Mutated Cancers. JCO Precis. Oncol. 2022, 6, e2100245. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Fakih, M.G.; Kopetz, S.; Kuboki, Y.; Kim, T.W.; Munster, P.N.; Krauss, J.C.; Falchook, G.S.; Han, S.W.; Heinemann, V.; Muro, K.; et al. Sotorasib for previously treated colorectal cancers with KRAS(G12C) mutation (CodeBreaK100): A prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 2022, 23, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.I.; Janne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRAS(G12C) Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Weiss, J.; Pelster, M.S.; Spira, A.I.; Barve, M.; Ou, S.I.; Leal, T.A.; Bekaii-Saab, T.S.; Paweletz, C.P.; Heavey, G.A.; et al. Adagrasib with or without Cetuximab in Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 388, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Sacher, A.; LoRusso, P.; Patel, M.R.; Miller, W.H., Jr.; Garralda, E.; Forster, M.D.; Santoro, A.; Falcon, A.; Kim, T.W.; Paz-Ares, L.; et al. Single-Agent Divarasib (GDC-6036) in Solid Tumors with a KRAS G12C Mutation. N. Engl. J. Med. 2023, 389, 710–721. [Google Scholar] [CrossRef]
- Desai, J.; Alonso, G.; Kim, S.H.; Cervantes, A.; Karasic, T.; Medina, L.; Shacham-Shmueli, E.; Cosman, R.; Falcon, A.; Gort, E.; et al. Divarasib plus cetuximab in KRAS G12C-positive colorectal cancer: A phase 1b trial. Nat. Med. 2024, 30, 271–278. [Google Scholar] [CrossRef]
- Choi, Y.; Dharia, N.V.; Jun, T.; Chang, J.; Royer-Joo, S.; Yau, K.K.; Assaf, Z.J.; Aimi, J.; Sivakumar, S.; Montesion, M.; et al. Circulating Tumor DNA Dynamics Reveal KRAS G12C Mutation Heterogeneity and Response to Treatment with the KRAS G12C Inhibitor Divarasib in Solid Tumors. Clin. Cancer Res. 2024, 30, 3788–3797. [Google Scholar] [CrossRef]
- Heist, R.S.; Koyama, T.; Murciano-Goroff, Y.R.; Hollebecque, A.; Cassier, P.A.; Han, J.-Y.; Tosi, D.; Sacher, A.G.; Burns, T.F.; Spira, A.I.; et al. Pan-tumor activity of olomorasib (LY3537982), a second-generation KRAS G12C inhibitor (G12Ci), in patients with KRAS G12C-mutant advanced solid tumors. J. Clin. Oncol. 2024, 42, 3007. [Google Scholar] [CrossRef]
- Hollebecque, A.; Kuboki, Y.; Murciano-Goroff, Y.R.; Yaeger, R.; Cassier, P.A.; Heist, R.S.; Fujiwara, Y.; Deming, D.A.; Ammakkanavar, N.; Patnaik, A.; et al. Efficacy and safety of LY3537982, a potent and highly selective KRAS G12C inhibitor in KRAS G12C-mutant GI cancers: Results from a phase 1 study. J. Clin. Oncol. 2024, 42, 94. [Google Scholar] [CrossRef]
- Ryan, M.B.; Coker, O.; Sorokin, A.; Fella, K.; Barnes, H.; Wong, E.; Kanikarla, P.; Gao, F.; Zhang, Y.; Zhou, L.; et al. KRAS(G12C)-independent feedback activation of wild-type RAS constrains KRAS(G12C) inhibitor efficacy. Cell Rep. 2022, 39, 110993. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef]
- Kuboki, Y.; Fakih, M.; Strickler, J.; Yaeger, R.; Masuishi, T.; Kim, E.J.; Bestvina, C.M.; Kopetz, S.; Falchook, G.S.; Langer, C.; et al. Sotorasib with panitumumab in chemotherapy-refractory KRAS(G12C)-mutated colorectal cancer: A phase 1b trial. Nat. Med. 2024, 30, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.G.; Salvatore, L.; Esaki, T.; Modest, D.P.; Lopez-Bravo, D.P.; Taieb, J.; Karamouzis, M.V.; Ruiz-Garcia, E.; Kim, T.W.; Kuboki, Y.; et al. Sotorasib plus Panitumumab in Refractory Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 389, 2125–2139. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.; Salvatore, L.; Esaki, T.; Modest, D.P.; Lopez-Bravo, D.P.; Taieb, J.; Karamouzis, M.; Ruiz-Garcia, E.; Kim, T.W.; Kuboki, Y.; et al. Overall survival (OS) of phase 3 CodeBreaK 300 study of sotorasib plus panitumumab (soto+pani) versus investigator’s choice of therapy for KRAS G12C-mutated metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2024, 42, LBA3510. [Google Scholar] [CrossRef]
- Kim, T.W.; Price, T.; Grasselli, J.; Strickler, J.H.; Masuishi, T.; Kwok, G.W.; Yalcin, S.; Obiozor, C.C.; Chan, E.; Gokani, P.; et al. A phase 3 study of first-line sotorasib, panitumumab, and FOLFIRI versus FOLFIRI with or without bevacizumab-awwb for patients with KRAS G12C–mutated metastatic colorectal cancer (CodeBreaK 301). J. Clin. Oncol. 2025, 43, TPS326. [Google Scholar] [CrossRef]
- Yaeger, R.; Uboha, N.V.; Pelster, M.S.; Bekaii-Saab, T.S.; Barve, M.; Saltzman, J.; Sabari, J.K.; Peguero, J.A.; Paulson, A.S.; Janne, P.A.; et al. Efficacy and Safety of Adagrasib plus Cetuximab in Patients with KRASG12C-Mutated Metastatic Colorectal Cancer. Cancer Discov. 2024, 14, 982–993. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Kopetz, S.; Yoshino, T.; Van Cutsem, E.; Eng, C.; Kim, T.W.; Wasan, H.S.; Desai, J.; Ciardiello, F.; Yaeger, R.; Maughan, T.S.; et al. Encorafenib, cetuximab and chemotherapy in BRAF-mutant colorectal cancer: A randomized phase 3 trial. Nat. Med. 2025, 31, 901–908. [Google Scholar] [CrossRef]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef]
- Frank, R.G.; Lave, J.R. The impact of Medicaid benefit design on length of hospital stay and patient transfers. Psychiatr. Serv. 1985, 36, 749–753. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, W.; Song, Z.; Zhang, Y.; Zhang, Y.; Huang, D.; Yang, Z.; Zhou, M.; Mao, R.; Huang, C.; et al. LBA33 A first-in-human phase I study of a novel KRAS G12D inhibitor HRS-4642 in patients with advanced solid tumors harboring KRAS G12D mutation. Ann. Oncol. 2023, 34, S1273. [Google Scholar] [CrossRef]
- Gong, X.; Gao, H.; Bender, M.H.; Ming, W.; Zhang, Y.; Stewart, T.R.; Yu, C.P.; Xu, W.G.; You, A.X.; Bian, W.T.; et al. Abstract 3316: LY3962673, an oral, highly potent, mutant-selective, and non-covalent KRAS G12D inhibitor demonstrates robust anti-tumor activity in KRAS G12D models. Cancer Res. 2024, 84, 3316. [Google Scholar] [CrossRef]
- Knox, J.E.; Burnett, G.L.; Weller, C.; Jiang, L.; Zhang, D.; Vita, N.; Marquez, A.; Seamon, K.J.; Gould, A.; Menard, M.; et al. Abstract ND03: Discovery of RMC-9805, an oral, covalent tri-complex KRASG12D(ON) inhibitor. Cancer Res. 2024, 84, ND03. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.P.; Xie, H.; Ding, J. From bench to bedside: Current development and emerging trend of KRAS-targeted therapy. Acta Pharmacol. Sin. 2024, 45, 686–703. [Google Scholar] [CrossRef]
- He, M.; Cao, C.; Ni, Z.; Liu, Y.; Song, P.; Hao, S.; He, Y.; Sun, X.; Rao, Y. PROTACs: Great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target. Ther. 2022, 7, 181. [Google Scholar] [CrossRef]
- Ji, X.; Li, H.; Wu, G.; Zhang, Q.; He, X.; Wu, Y.; Zong, B.; Xu, X.; Liang, C.; Wang, B.; et al. Abstract 6050: Targeting KRAS G12D mutant tumors with the PROTAC degrader RP03707. Cancer Res. 2024, 84, 6050. [Google Scholar] [CrossRef]
- Li, D.; Geng, K.; Hao, Y.; Gu, J.; Kumar, S.; Olson, A.T.; Kuismi, C.C.; Kim, H.M.; Pan, Y.; Sherman, F.; et al. Targeted degradation of oncogenic KRASG12V triggers antitumor immunity in lung cancer models. J. Clin. Investig. 2024, 135, e174249. [Google Scholar] [CrossRef]
- Park, W.; Kasi, A.; Spira, A.I.; Berlin, J.D.; Wang, J.S.; Herzberg, B.; Kuboki, Y.; Kitano, S.; Pelster, M.; Goldman, J.W.; et al. 608O Preliminary safety and clinical activity of ASP3082, a first-in-class, KRAS G12D selective protein degrader in adults with advanced pancreatic (PC), colorectal (CRC), and non-small cell lung cancer (NSCLC). Ann. Oncol. 2024, 35, S486–S487. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Rominger, D.; Vo, E.D.; Silva, J.M.; Zhang, Y.J.; Lee, G.; Micozzi, J.; Reid, B.; McDonough, B.; Hospital, A.; et al. Abstract LB320: Discovery and characterization of QTX3034, a potent, selective, and orally bioavailable allosteric KRAS inhibitor. Cancer Res. 2023, 83, LB320. [Google Scholar] [CrossRef]
- Patnaik, A.; Pelster, M.; Hong, D.S.; Strickler, J.H.; Garrido-Laguna, I.; Aguirre, A.; Curran, D.; Woo, T.; Spira, A.I. A phase 1 trial evaluating the safety, tolerability, PK, and preliminary efficacy of QTX3034, an oral G12D-preferring multi-KRAS inhibitor, in patients with solid tumors with KRASG12D mutation. J. Clin. Oncol. 2024, 42, TPS3172. [Google Scholar] [CrossRef]
- Cregg, J.; Edwards, A.V.; Chang, S.; Lee, B.J.; Knox, J.E.; Tomlinson, A.C.A.; Marquez, A.; Liu, Y.; Freilich, R.; Aay, N.; et al. Discovery of Daraxonrasib (RMC-6236), a Potent and Orally Bioavailable RAS(ON) Multi-selective, Noncovalent Tri-complex Inhibitor for the Treatment of Patients with Multiple RAS-Addicted Cancers. J. Med. Chem. 2025, 68, 6064–6083. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Jiang, L.; Maldonato, B.J.; Wang, Y.; Holderfield, M.; Aronchik, I.; Winters, I.P.; Salman, Z.; Blaj, C.; Menard, M.; et al. Translational and Therapeutic Evaluation of RAS-GTP Inhibition by RMC-6236 in RAS-Driven Cancers. Cancer Discov. 2024, 14, 994–1017. [Google Scholar] [CrossRef]
- Sudhakar, N.; Yan, L.; Qiryaqos, F.; Engstrom, L.D.; Laguer, J.; Calinisan, A.; Hebbert, A.; Waters, L.; Moya, K.; Bowcut, V.; et al. The SOS1 Inhibitor MRTX0902 Blocks KRAS Activation and Demonstrates Antitumor Activity in Cancers Dependent on KRAS Nucleotide Loading. Mol. Cancer Ther. 2024, 23, 1418–1430. [Google Scholar] [CrossRef]
- Drilon, A.; Sharma, M.R.; Johnson, M.L.; Yap, T.A.; Gadgeel, S.; Nepert, D.; Feng, G.; Reddy, M.B.; Harney, A.S.; Elsayed, M.; et al. SHP2 Inhibition Sensitizes Diverse Oncogene-Addicted Solid Tumors to Re-treatment with Targeted Therapy. Cancer Discov. 2023, 13, 1789–1801. [Google Scholar] [CrossRef]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef]
- Zhao, J.; Fang, J.; Yu, Y.; Chu, Q.; Li, X.; Chen, J.; Liu, Z.; Zhang, L.; Wu, L.; Zhuang, W.; et al. Updated safety and efficacy data of combined KRAS G12C inhibitor (glecirasib, JAB-21822) and SHP2 inhibitor (JAB-3312) in patients with KRAS p.G12C mutated solid tumors. J. Clin. Oncol. 2024, 42, 3008. [Google Scholar] [CrossRef]
- van Geel, R.; van Brummelen, E.M.J.; Eskens, F.; Huijberts, S.; de Vos, F.; Lolkema, M.; Devriese, L.A.; Opdam, F.L.; Marchetti, S.; Steeghs, N.; et al. Phase 1 study of the pan-HER inhibitor dacomitinib plus the MEK1/2 inhibitor PD-0325901 in patients with KRAS-mutation-positive colorectal, non-small-cell lung and pancreatic cancer. Br. J. Cancer 2020, 122, 1166–1174. [Google Scholar] [CrossRef]
- van Brummelen, E.M.J.; Huijberts, S.; van Herpen, C.; Desar, I.; Opdam, F.; van Geel, R.; Marchetti, S.; Steeghs, N.; Monkhorst, K.; Thijssen, B.; et al. Phase I Study of Afatinib and Selumetinib in Patients with KRAS-Mutated Colorectal, Non-Small Cell Lung, and Pancreatic Cancer. Oncologist 2021, 26, 290-e545. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Tseng, C.; Tran, H.T.; Gao, M.; Karp, D.D.; Subbiah, V.; Tsimberidou, A.M.; Kawedia, J.D.; Fu, S.; Pant, S.; et al. A phase I trial of the pan-ERBB inhibitor neratinib combined with the MEK inhibitor trametinib in patients with advanced cancer with EGFR mutation/amplification, HER2 mutation/amplification, HER3/4 mutation or KRAS mutation. Cancer Chemother. Pharmacol. 2023, 92, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Do, K.T.; Kim, J.E.; Cleary, J.M.; Parikh, A.R.; Yeku, O.O.; Xiong, N.; Weekes, C.D.; Veneris, J.; Ahronian, L.G.; et al. Phase I/II Study of Combined BCL-xL and MEK Inhibition with Navitoclax and Trametinib in KRAS or NRAS Mutant Advanced Solid Tumors. Clin. Cancer Res. 2024, 30, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Parseghian, C.M.; Sanchez, E.V.; Sun, R.; Eluri, M.; Morris, V.K.; Johnson, B.; Morelli, M.P.; Overman, M.J.; Willis, J.; Huey, R.; et al. Phase 2 study of anti-EGFR rechallenge therapy with panitumumab with or without trametinib in advanced colorectal cancer. J. Clin. Oncol. 2022, 40, 3520. [Google Scholar] [CrossRef]
- Lee, M.S.; Zemla, T.J.; Ciombor, K.K.; McRee, A.J.; Akce, M.; Dakhil, S.R.; Jaszewski, B.L.; Ou, F.-S.; Bekaii-Saab, T.S.; Kopetz, S. A randomized phase II trial of MEK and CDK4/6 inhibitors vesus tipiracil/trifluridine (TAS-102) in metastatic KRAS/NRAS mutant (mut) colorectal cancer (CRC). J. Clin. Oncol. 2022, 40, 116. [Google Scholar] [CrossRef]
- Akhave, N.S.; Biter, A.B.; Hong, D.S. Mechanisms of Resistance to KRAS(G12C)-Targeted Therapy. Cancer Discov. 2021, 11, 1345–1352. [Google Scholar] [CrossRef]
- Aslam, R.; Richards, C.E.; Fay, J.; Hudson, L.; Workman, J.; Lee, C.L.; Murphy, A.; O’Neill, B.; Toomey, S.; Hennessy, B.T. Synergistic Effects of the Combination of Alpelisib (PI3K Inhibitor) and Ribociclib (CDK4/6 Inhibitor) in Preclinical Colorectal Cancer Models. Int. J. Mol. Sci. 2024, 25, 13264. [Google Scholar] [CrossRef]
- Liu, X.; Xu, W.; Li, L.; Zhang, Z.; Lu, M.; Xia, X. Dual PI3K/mTOR Inhibitor BEZ235 combined with BMS-1166 Promoting Apoptosis in Colorectal Cancer. Int. J. Med. Sci. 2024, 21, 1814–1823. [Google Scholar] [CrossRef]
- Kitai, H.; Choi, P.H.; Yang, Y.C.; Boyer, J.A.; Whaley, A.; Pancholi, P.; Thant, C.; Reiter, J.; Chen, K.; Markov, V.; et al. Combined inhibition of KRAS(G12C) and mTORC1 kinase is synergistic in non-small cell lung cancer. Nat. Commun. 2024, 15, 6076. [Google Scholar] [CrossRef]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin. Cancer Res. 2019, 25, 796–807. [Google Scholar] [CrossRef]
- Karimpour, M.; Totonchi, M.; Behmanesh, M.; Montazeri, H. Pathway-driven analysis of synthetic lethal interactions in cancer using perturbation screens. Life Sci. Alliance 2024, 7, e202302268. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Hahn, W.C. Synthetic Lethal Vulnerabilities in KRAS-Mutant Cancers. Cold Spring Harb. Perspect. Med. 2018, 8, a031518. [Google Scholar] [CrossRef] [PubMed]
- Hendifar, A.E.; Rosen, L.S.; Cercek, A.; McRee, A.J.; Mallick, A.B.; Spigel, D.R.; Tavazoie, S.F.; Rowinsky, E.K.; Szarek, M.; Gonsalves, F.; et al. Phase 1b study of RGX-202-01, a first-in-class oral inhibitor of the SLC6A8/CKB pathway, in combination with FOLFIRI and bevacizumab (BEV) in second-line advanced colorectal cancer (CRC). J. Clin. Oncol. 2022, 40, 3579. [Google Scholar] [CrossRef]
- Ahn, D.H.; Ridinger, M.; Cannon, T.L.; Mendelsohn, L.; Starr, J.S.; Hubbard, J.M.; Kasi, A.; Barzi, A.; Samuelsz, E.; Karki, A.; et al. Onvansertib in Combination With Chemotherapy and Bevacizumab in Second-Line Treatment of KRAS-Mutant Metastatic Colorectal Cancer: A Single-Arm, Phase II Trial. J. Clin. Oncol. 2025, 43, 840–851. [Google Scholar] [CrossRef]
- Ahn, D.H.; Barzi, A.; Ridinger, M.; Samuelsz, E.; Subramanian, R.A.; Croucher, P.J.P.; Smeal, T.; Kabbinavar, F.F.; Lenz, H.J. Onvansertib in Combination with FOLFIRI and Bevacizumab in Second-Line Treatment of KRAS-Mutant Metastatic Colorectal Cancer: A Phase Ib Clinical Study. Clin. Cancer Res. 2024, 30, 2039–2047. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Zhao, X.; Pan, X.; Wang, Y.; Zhang, Y. Targeting neoantigens for cancer immunotherapy. Biomark. Res. 2021, 9, 61. [Google Scholar] [CrossRef]
- Baulu, E.; Gardet, C.; Chuvin, N.; Depil, S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci. Adv. 2023, 9, eadf3700. [Google Scholar] [CrossRef]
- Li, J.; Xiao, Z.; Wang, D.; Jia, L.; Nie, S.; Zeng, X.; Hu, W. The screening, identification, design and clinical application of tumor-specific neoantigens for TCR-T cells. Mol. Cancer 2023, 22, 141. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Huang, H.Y.; Lin, Z.; Ranieri, M.; Li, S.; Sahu, S.; Liu, Y.; Ban, Y.; Guidry, K.; Hu, H.; et al. Genome-Wide CRISPR Screens Identify Multiple Synthetic Lethal Targets That Enhance KRASG12C Inhibitor Efficacy. Cancer Res. 2023, 83, 4095–4111. [Google Scholar] [CrossRef]
- Dilly, J.; Hoffman, M.T.; Abbassi, L.; Li, Z.; Paradiso, F.; Parent, B.D.; Hennessey, C.J.; Jordan, A.C.; Morgado, M.; Dasgupta, S.; et al. Mechanisms of Resistance to Oncogenic KRAS Inhibition in Pancreatic Cancer. Cancer Discov. 2024, 14, 2135–2161. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Downward, J. Exploiting the therapeutic implications of KRAS inhibition on tumor immunity. Cancer Cell 2024, 42, 338–357. [Google Scholar] [CrossRef] [PubMed]
- Isermann, T.; Sers, C.; Der, C.J.; Papke, B. KRAS inhibitors: Resistance drivers and combinatorial strategies. Trends Cancer 2025, 11, 91–116. [Google Scholar] [CrossRef] [PubMed]
- Parseghian, C.M.; Sun, R.; Woods, M.; Napolitano, S.; Lee, H.M.; Alshenaifi, J.; Willis, J.; Nunez, S.; Raghav, K.P.; Morris, V.K.; et al. Resistance Mechanisms to Anti-Epidermal Growth Factor Receptor Therapy in RAS/RAF Wild-Type Colorectal Cancer Vary by Regimen and Line of Therapy. J. Clin. Oncol. 2023, 41, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, J.C.; Yilmaz, O.H.; Roper, J. Metabolism and Colorectal Cancer. Annu. Rev. Pathol. 2023, 18, 467–492. [Google Scholar] [CrossRef]
- Shi, Y.; Zheng, H.; Wang, T.; Zhou, S.; Zhao, S.; Li, M.; Cao, B. Targeting KRAS: From metabolic regulation to cancer treatment. Mol. Cancer 2025, 24, 9. [Google Scholar] [CrossRef]
- Garassino, M.C.; Theelen, W.S.M.E.; Jotte, R.; Laskin, J.; de Marinis, F.; Aguado, C.; Badin, F.B.; Chmielewska, I.; Hochmair, M.J.; Lu, S.; et al. LBA65 KRYSTAL-7: Efficacy and safety of adagrasib with pembrolizumab in patients with treatment-naive, advanced non-small cell lung cancer (NSCLC) harboring a KRASG12C mutation. Ann. Oncol. 2023, 34, S1309–S1310. [Google Scholar] [CrossRef]
- Li, B.T.; Falchook, G.S.; Durm, G.A.; Burns, T.F.; Skoulidis, F.; Ramalingam, S.S.; Spira, A.; Bestvina, C.M.; Goldberg, S.B.; Veluswamy, R.; et al. OA03.06 CodeBreaK 100/101: First Report of Safety/Efficacy of Sotorasib in Combination with Pembrolizumab or Atezolizumab in Advanced KRAS p.G12C NSCLC. J. Thorac. Oncol. 2022, 17, S10–S11. [Google Scholar] [CrossRef]
- Boumelha, J.; Molina-Arcas, M.; Downward, J. Facts and Hopes on RAS Inhibitors and Cancer Immunotherapy. Clin. Cancer Res. 2023, 29, 5012–5020. [Google Scholar] [CrossRef]
- Leidner, R.; Sanjuan Silva, N.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Patelli, G.; Mauri, G.; Tosi, F.; Amatu, A.; Bencardino, K.; Bonazzina, E.; Pizzutilo, E.G.; Villa, F.; Calvanese, G.; Agostara, A.G.; et al. Circulating Tumor DNA to Drive Treatment in Metastatic Colorectal Cancer. Clin. Cancer Res. 2023, 29, 4530–4539. [Google Scholar] [CrossRef]
- Mauri, G.; Vitiello, P.P.; Sogari, A.; Crisafulli, G.; Sartore-Bianchi, A.; Marsoni, S.; Siena, S.; Bardelli, A. Liquid biopsies to monitor and direct cancer treatment in colorectal cancer. Br. J. Cancer 2022, 127, 394–407. [Google Scholar] [CrossRef]
- Urbini, M.; Marisi, G.; Azzali, I.; Bartolini, G.; Chiadini, E.; Capelli, L.; Tedaldi, G.; Angeli, D.; Canale, M.; Molinari, C.; et al. Dynamic Monitoring of Circulating Tumor DNA in Patients With Metastatic Colorectal Cancer. JCO Precis. Oncol. 2023, 7, e2200694. [Google Scholar] [CrossRef]
- Chong, W.; Zhu, X.; Ren, H.; Ye, C.; Xu, K.; Wang, Z.; Jia, S.; Shang, L.; Li, L.; Chen, H. Integrated multi-omics characterization of KRAS mutant colorectal cancer. Theranostics 2022, 12, 5138–5154. [Google Scholar] [CrossRef]

{kind=link}
| Drug | Target | Clinical Trial | Phase | Intervention |
|---|---|---|---|---|
| Sotorasib (AMG 510) | KRAS G12C | NCT05198934 (CodeBreak 300) | 3 | Sotorasib 960 mg + panitumumab vs. sotorasib 240 mg + panitumumab vs. trifluridine/tipiracil or regorafenib |
| Sotorasib | KRAS G12C | NCT06252649 (CodeBreaK 301) | 3 | Sotorasib + panitumumab + FOLFIRI vs. FOLFIRI +/− bevacizumab |
| Adagrasib (MRTX849) | KRAS G12C | NCT03785249 (KRYSTAL-1) | 1/2 | Adagrasib +/− cetuximab |
| Adagrasib | KRAS G12C | NCT04793958 (KRYSTAL-10) | 3 | Adagrasib + cetuximab vs. mFOLFOX or FOLFIRI |
| Adagrasib | KRAS G12C | NCT05722327 | 1 | Adagrasib + cetuximab + irinotecan |
| Adagrasib | KRAS G12C | NCT06412198 | 1/2 | Adagrasib + cetuximab + cemiplimab |
| Adagrasib + TNO155 | KRAS G12C, SHP2 | NCT04330664 (KRYSTAL 2) | 1 | Adagrasib + TNO155 |
| INCB099280 + adagrasib | PD-L1, KRAS G12C | NCT06039384 | 1 | INCB099280 + adagrasib |
| KO-2806 +/- adagrasib | farnesyl transferase, KRAS G12C | NCT06026410 | 1 | KO-2806 +/− adagrasib |
| MRTX0902 +/- adagrasib | SOS1, KRAS G12C | NCT05578092 | 1/2 | MRTX0902 +/− adagrasib |
| Divarasib (GDC-6036) | KRAS G12C | NCT04449874 | 1 | Divarasib +/− cetuximab |
| Olomorasib (LY3537982) | KRAS G12C | NCT04956640 | 1/2 | Olomorasib +/− cetuximab or pembrolizumab |
| FMC-376 | KRAS G12C | NCT06244771 (PROSPER) | 1/2 | FMC-376 |
| HBI-2438 | KRAS G12C | NCT05485974 | 1 | HBI-2438 |
| Glecirasib (JAB-21822) | KRAS G12C | NCT05194995 | 1/2 | JAB-21822 + cetuximab |
| Glecirasib (JAB-21822) + JAB-3312 | KRAS G12C, SHP2 | NCT05288205 | 1/2 | JAB-21822 + JAB-3312 |
| JDQ443 | KRAS G12C | NCT05358249 | 1/2 | JDQ443 + trametinib, ribociclib, or cetuximab |
| JDQ443 | KRAS G12C | NCT04699188 (KontRASt-01) | 1/2 | JDQ443 +/− TNO155 and/or tislelizumab |
| RMC-6291 | KRAS G12C | NCT05462717 | 1 | RMC-6291 |
| Drug | Target | Clinical Trial | Phase | Intervention |
|---|---|---|---|---|
| MRTX1133 | KRAS G12D | NCT05737706 | 1/2 | MRTX1133 |
| ASP4396 | KRAS G12D | NCT06364696 | 1 | ASP4396 |
| HRS-4642 | KRAS G12D | NCT06385678 | 1/2 | HRS-4642 + adebrelimab, cetuximab, or SHR-9839 (EGFR/c-Met bispecific antibody) |
| TSN1611 | KRAS G12D | NCT06385925 | 1/2 | TSN1611 |
| QLC1101 | KRAS G12D | NCT06403735 | 1 | QLC1101 |
| QTX3046 | KRAS G12D | NCT06428500 | 1 | QTX3046 +/− cetuximab |
| LY3962673 | KRAS G12D | NCT06586515 (MOONRAY-01) | 1 | LY3962673 +/− cetuximab or chemotherapy |
| AZD0022 | KRAS G12D | NCT06599502 | 1/2 | AZD0022 +/− cetuximab |
| ASP3082 | KRASG12D degrader | NCT05382559 | 1 | ASP3082 +/− cetuximab |
| RMC-9805 | KRAS G12D(ON) inhibitor | NCT06040541 | 1 | RMC-9805 +/− RMC-6236 |
| QTX3034 | Multi-KRAS inhibitor | NCT06227377 | 1 | QTX3034 +/− cetuximab |
| PF-07934040 | Pan-KRAS | NCT06447662 | 1 | PF-07934040 +/− cetuximab or FOLFOX/bevacizumab |
| BGB-53038 | Pan-KRAS | NCT06585488 | 1 | BGB-53038 +/− tislelizumab or cetuximab |
| LY4066434 | Pan-KRAS | NCT06607185 | 1 | LY4066434 +/− cetuximab, chemotherapy, or pembrolizumab |
| PF-07985045, PF-07284892 | Pan-KRAS, SHP2 | NCT06704724 | 1 | PF-07985045 +/− cetuximab, FOLFOX/bevacizumab, or PF-07284892 |
| YL-17231 | Pan-RAS | NCT06078800 | 1 | YL-17231 |
| Daraxonrasib (RMC-6236) | RAS-MULTI(ON) inhibitor | NCT05379985 | 1 | RMC-6236 |
| Daraxonrasib (RMC-6236) | RAS-MULTI(ON) inhibitor | NCT06445062 | 1 | RMC-6236 +/− chemotherapy, cetuximab, bevacizumab and/or RMC-9805 |
| Clinical Trial | Phase | Intervention | Target |
|---|---|---|---|
| NCT05163028 | 1 | HBI-2376 | SHP2 |
| NCT04121286 | 1 | JAB-3312 | SHP2 |
| NCT05786924 | 1 | BDTX-4933 | RAF |
| NCT06194877 | 1 | Brimarafenib (BGB-3245) + panitumumab | RAF, EGFR |
| NCT05200442 | 1/2 | Avutometinib (VS-6766) + cetuximab | RAF/MEK, EGFR |
| NCT06270082 | 1 | IK-595 | RAF/MEK |
| NCT06634875 | 2 | Isunakinra +/− pembrolizumab | IL-1R1 |
| NCT06229340 | 2 | leflunomide +/− MEK inhibitor and hydroxychloroquine +/- bevacizumab | DHODH, MEK |
| NCT03597581 | 1 | Ompenaclid (RGX-202-01) +/− FOLFIRI or FOLFIRI/bevacizumab or FOLFOX/bevacizumab | SLC6A8 |
| NCT03829410 | 1/2 | Onvansertib + FOLFIRI + bevacizumab | PLK1 |
| NCT04599140 (STOPTRAFFIC-1) | 1/2 | SX-682 +/− nivolumab | CXCR1/2 |
| Clinical Trial | Phase | Intervention | Mechanism |
|---|---|---|---|
| NCT04117087 | 1 | KRAS peptide vaccine + nivolumab + ipilimumab | Pooled mutant KRAS long peptide vaccine |
| NCT04853017 (AMPLIFY-201) | 1 | ELI-002 2P | KRAS G12D and G12R peptide vaccine |
| NCT05726864 (AMPLIFY-7P) | 1/2 | ELI-002 7P | KRAS/NRAS (G12D, G12R, G12V, G12A, G12C, G12S, and G13D) peptide vaccines |
| NCT06411691 | 1 | SPL mKRASvax + balstilimab + botensilimab | Mutant KRAS long peptide vaccine |
| NCT06105021 | 1/2 | AFNT-211 | Autologous KRAS G12V-specific transgenic TCR T-cells |
| NCT06218914 | 1 | NT-112 | Autologous KRAS G12D-specific TCR T-cells |
| NCT06253520 | 1 | KRAS TCR-transduced PBL + GRT-C903/GRT-R904 | Autologous KRAS G12D- or KRAS G12V-specific TCR T-cells and KRAS vaccine |
| NCT06487377 | 1 | IX001 | Autologous KRAS G12D- or KRAS G12V-specific TCR T-cells |
| NCT06690281 | 2 | KRAS TCR-transduced PBL | Autologous KRAS G12D- or KRAS G12V-specific TCR T-cells |
| NCT06707896 | 1 | TCR1020-CD8 T-cells | Autologous KRAS G12V-specific TCR T-cells |
| NCT06767046 | 1 | CRTKVA11 | Autologous KRAS G12V-specific TCR T-cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, R.; Yu, J.; Kim, R.D. Targeting the KRAS Oncogene for Patients with Metastatic Colorectal Cancer. Cancers 2025, 17, 1512. https://doi.org/10.3390/cancers17091512
Miao R, Yu J, Kim RD. Targeting the KRAS Oncogene for Patients with Metastatic Colorectal Cancer. Cancers. 2025; 17(9):1512. https://doi.org/10.3390/cancers17091512
Chicago/Turabian StyleMiao, Ruoyu, James Yu, and Richard D. Kim. 2025. "Targeting the KRAS Oncogene for Patients with Metastatic Colorectal Cancer" Cancers 17, no. 9: 1512. https://doi.org/10.3390/cancers17091512
APA StyleMiao, R., Yu, J., & Kim, R. D. (2025). Targeting the KRAS Oncogene for Patients with Metastatic Colorectal Cancer. Cancers, 17(9), 1512. https://doi.org/10.3390/cancers17091512