
Cancer Cell-Intrinsic Type I Interferon Signaling Promotes Antitumor Immunity in Head and Neck Squamous Cell Carcinoma


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Viral Transduction of Murine Cancer Cells
2.3. Cell Viability and Proliferation Assay
2.4. Bone Marrow Cell Derived Dendric Cells (BMDCs) Induction
2.5. Flow Cytometric Analysis
2.6. Enzyme-Linked Immunosorbent Assay (ELISA) for Cytokine Detection
2.7. Nucleic Acid Extraction and PCR Assay
2.8. Western Blot
2.9. SIINFEKL Peptide Pulsing of Cells
2.10. Tumor Models in Mice
2.11. Tissue Staining and Immunohistochemical Analysis
2.12. Statistics
3. Results
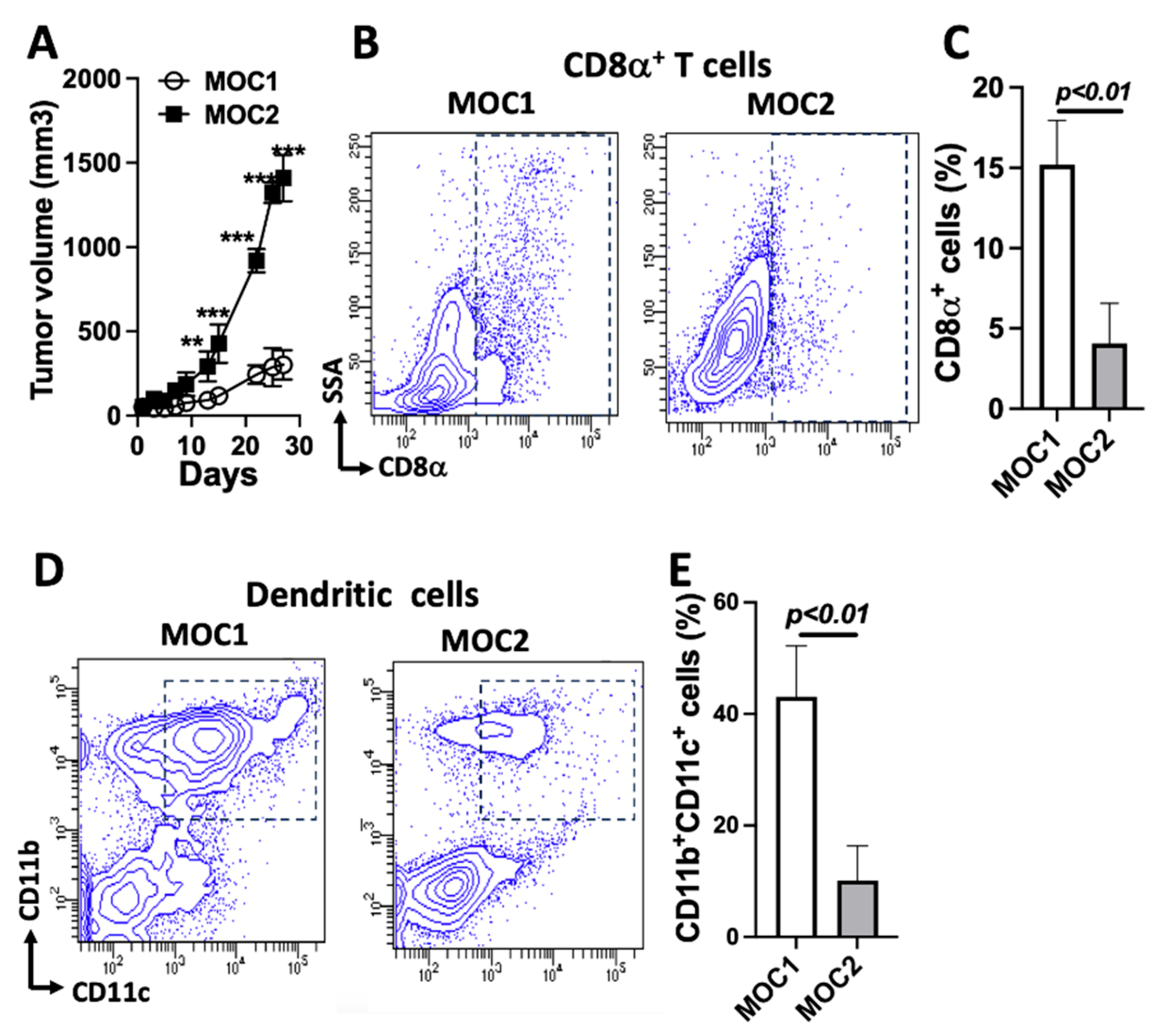
3.1. Deficient Immune Cell Populations in MOC2 Tumors Compared to MOC1 Tumors in Immunocompetent Mice
3.2. Reduced cGAS-STING-IFN-I Signaling in MOC2 Tumors Compared to MOC1 Tumors
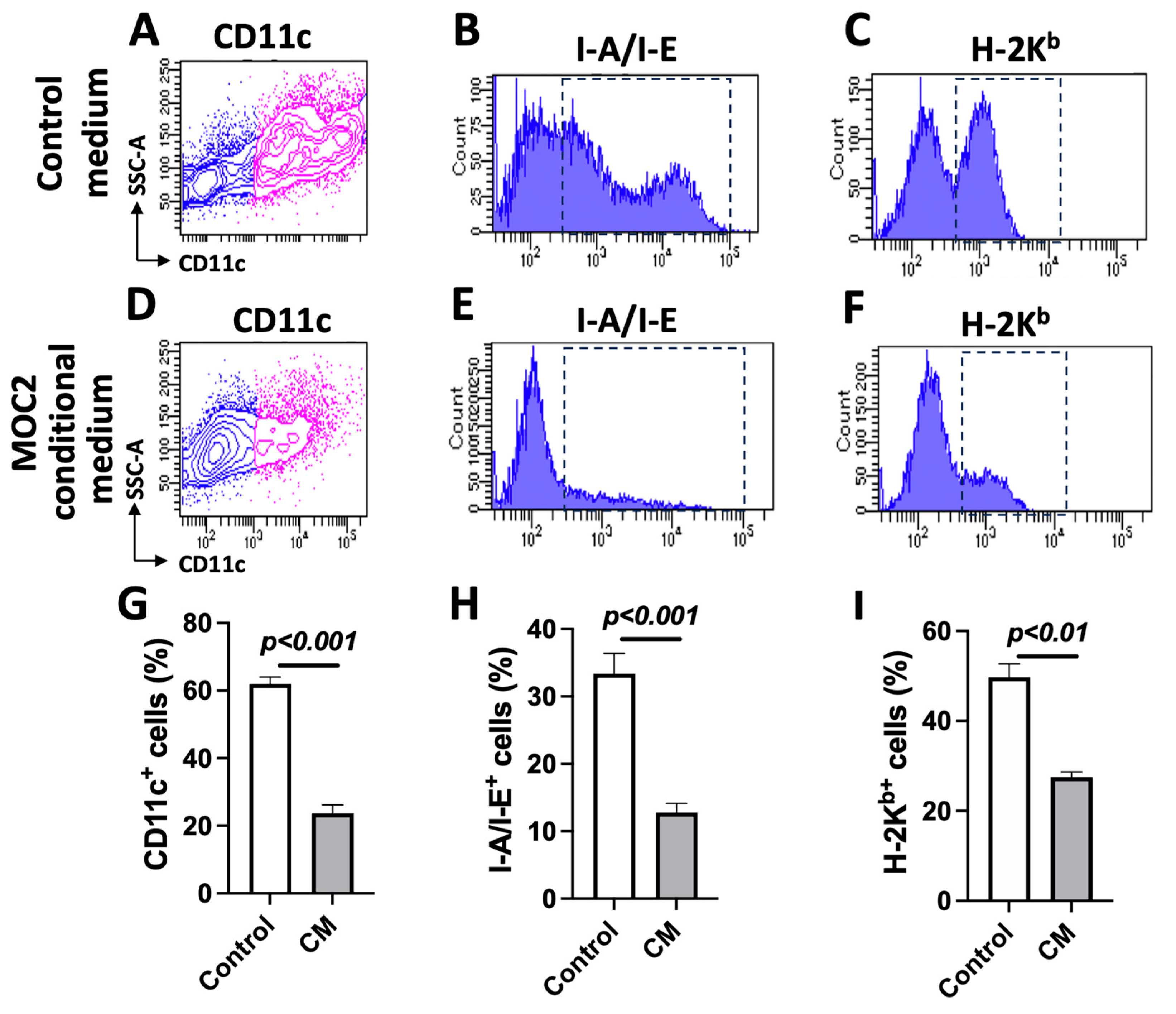
3.3. MOC2 Cells Inhibit Bone-Marrow-Cell-Derived DC Differentiation
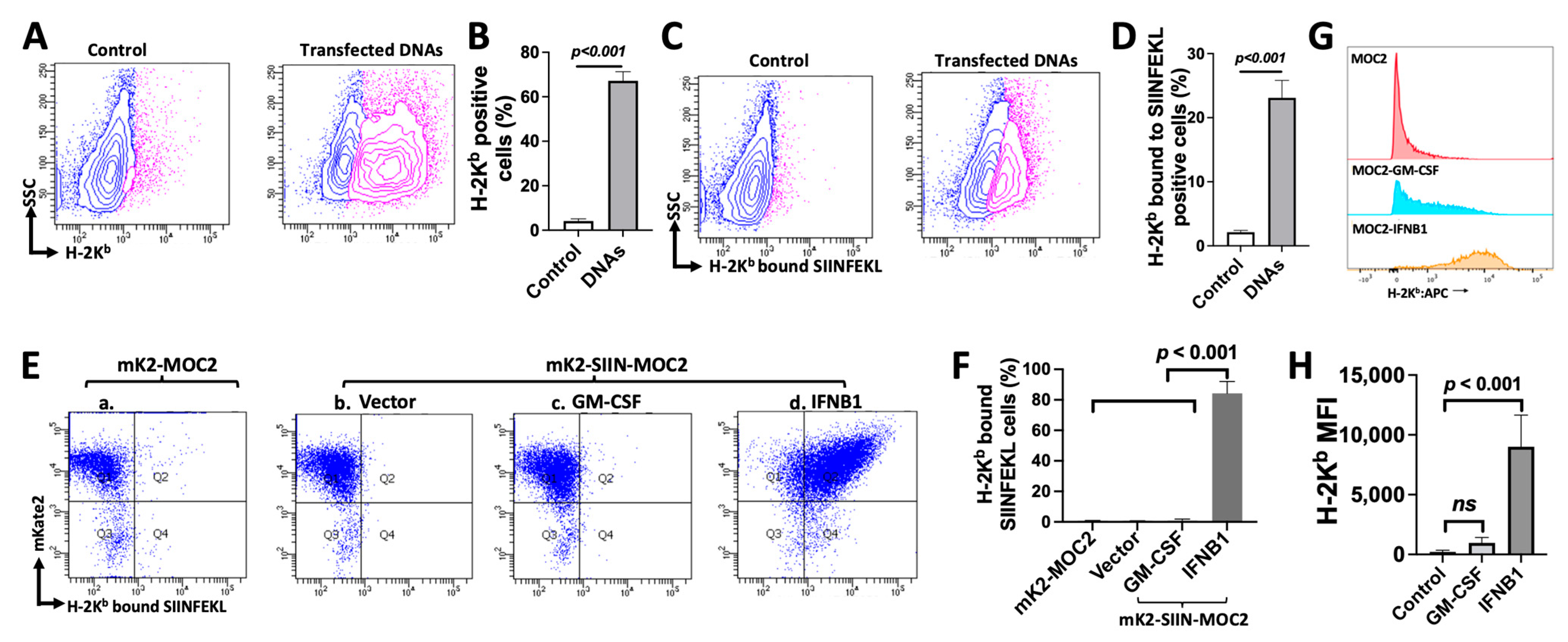
3.4. MOC2 Cells Retain the Ability to Present Antigen in Response to Transfected DNA or IFNB1 Expression
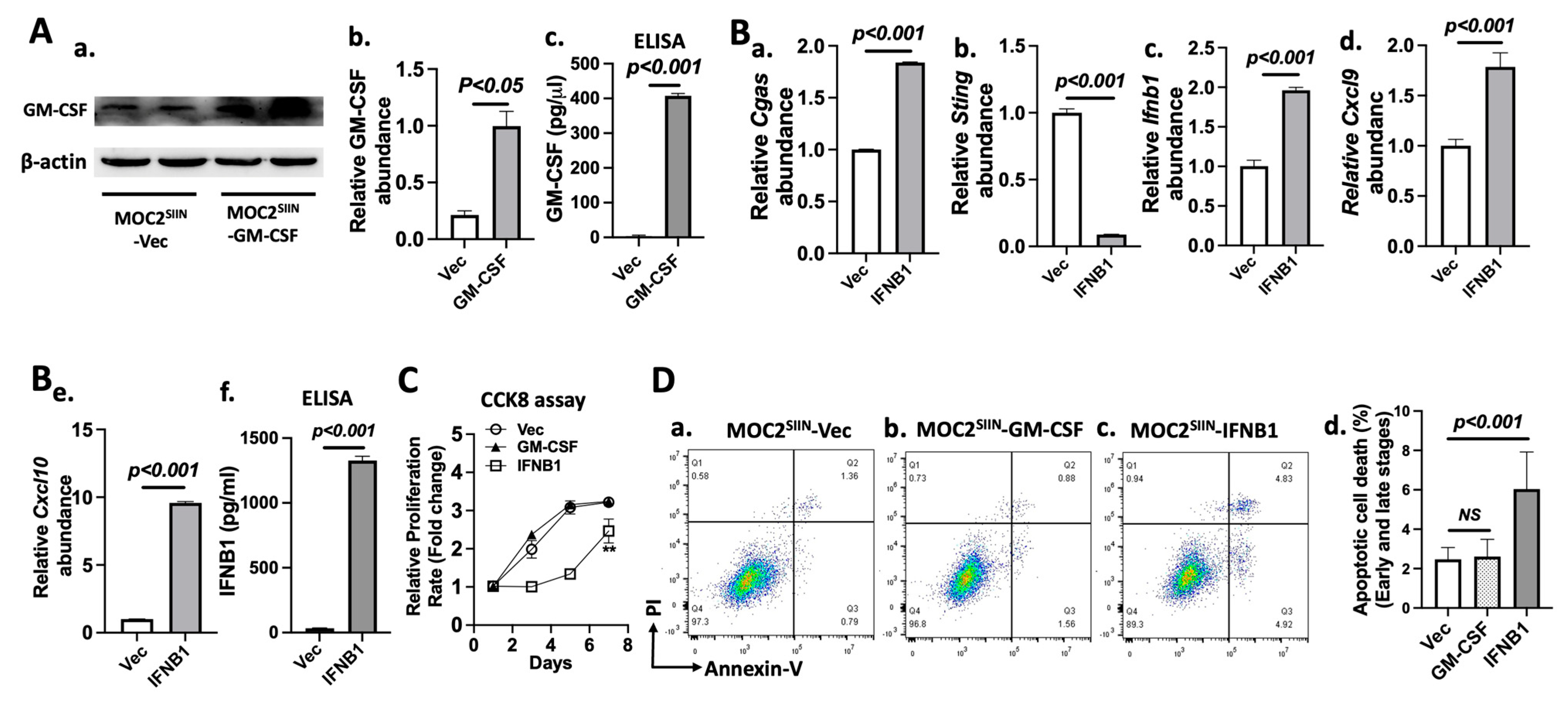
3.5. Effects of IFNB1 Expression on Gene Expression, Cell Proliferation, and Apoptotic Cell Death in MOC2 Subline Cells
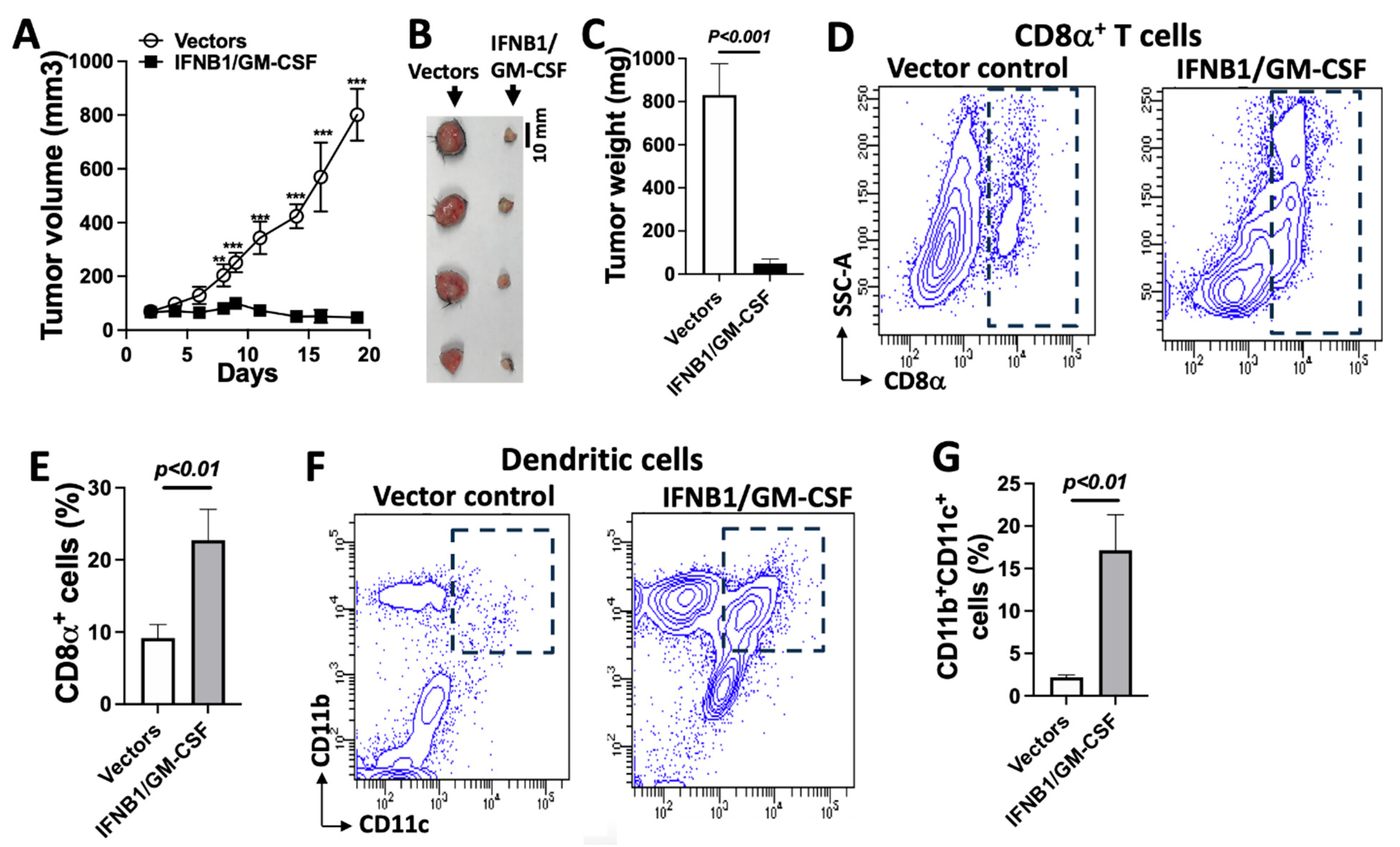
3.6. IFN-I Induces Adaptive Antitumor Immunity-Dependent Tumor Regression, Which Is Enhanced Using GM-CSF
3.7. IFN-I Signaling Engages Dendritic Cells to Induce Antitumor Immunity and Control Tumor Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Mourad, M.; Jetmore, T.; Jategaonkar, A.A.; Moubayed, S.; Moshier, E.; Urken, M.L. Epidemiological trends of head and neck cancer in the United States: A SEER population study. J. Oral Maxillofac. Surg. 2017, 75, 2562–2572. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Canning, M.; Guo, G.; Yu, M.; Myint, C.; Groves, M.W.; Byrd, J.K.; Cui, Y. Heterogeneity of the head and neck squamous cell carcinoma immune landscape and its impact on immunotherapy. Front. Cell Dev. Biol. 2019, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. New Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune checkpoint inhibitors for the treatment of cancer: Clinical impact and mechanisms of response and resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, R.W.; Borson, S.; Tsagianni, A.; Zandberg, D.P. Immunotherapy in recurrent/metastatic squamous cell carcinoma of the head and neck. Front. Oncol. 2021, 11, 705614. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Ahn, J.; Xia, T.; Rabasa Capote, A.; Betancourt, D.; Barber, G.N. Extrinsic phagocyte-dependent STING signaling dictates the immunogenicity of dying cells. Cancer Cell 2018, 33, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Naderi Yeganeh, P.; Zhang, H.; Wang, S.Y.; Li, Z.; Gu, B.; Lee, D.J.; Zhang, Z.; Ploumakis, A.; Shi, M.; et al. Tumor editing suppresses innate and adaptive antitumor immunity and is reversed by inhibiting DNA methylation. Nat. Immunol. 2024, 25, 1858–1870. [Google Scholar] [CrossRef]
- Tan, Y.S.; Sansanaphongpricha, K.; Xie, Y.; Donnelly, C.R.; Luo, X.; Heath, B.R.; Zhao, X.; Bellile, E.; Hu, H.; Chen, H.; et al. Mitigating SOX2-potentiated immune escape of head and neck squamous cell carcinoma with a STING-inducing nanosatellite vaccine. Clin. Cancer Res. 2018, 24, 4242–4255. [Google Scholar] [CrossRef] [PubMed]
- Judd, N.P.; Allen, C.T.; Winkler, A.E.; Uppaluri, R. Comparative analysis of tumor-infiltrating lymphocytes in a syngeneic mouse model of oral cancer. Otolaryngol.—Head. Neck Surg. 2012, 147, 493–500. [Google Scholar] [CrossRef]
- Judd, N.P.; Winkler, A.E.; Murillo-Sauca, O.; Brotman, J.J.; Law, J.H.; Lewis, J.S., Jr.; Dunn, G.P.; Bui, J.D.; Sunwoo, J.B.; Uppaluri, R. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012, 72, 365–374. [Google Scholar] [CrossRef]
- Kono, M.; Saito, S.; Egloff, A.M.; Allen, C.T.; Uppaluri, R. The mouse oral carcinoma (MOC) model: A 10-year retrospective on model development and head and neck cancer investigations. Oral. Oncol. 2022, 132, 106012. [Google Scholar] [CrossRef]
- Moore, E.; Clavijo, P.E.; Davis, R.; Cash, H.; Van Waes, C.; Kim, Y.; Allen, C. Established T cell-inflamed tumors rejected after adaptive resistance was reversed by combination STING activation and PD-1 pathway blockade. Cancer Immunol. Res. 2016, 4, 1061–1071. [Google Scholar] [CrossRef]
- Lu, S.; Concha-Benavente, F.; Shayan, G.; Srivastava, R.M.; Gibson, S.P.; Wang, L.; Gooding, W.E.; Ferris, R.L. STING activation enhances cetuximab-mediated NK cell activation and DC maturation and correlates with HPV (+) status in head and neck cancer. Oral. Oncol. 2018, 78, 186–193. [Google Scholar] [CrossRef]
- Nagaya, T.; Nakamura, Y.; Okuyama, S.; Ogata, F.; Maruoka, Y.; Choyke, P.L.; Allen, C.; Kobayashi, H. Syngeneic mouse models of oral cancer are effectively targeted by Anti-CD44-Based NIR-PIT. Mol. Cancer Res. MCR 2017, 15, 1667–1677. [Google Scholar] [CrossRef]
- Sun, L.; Moore, E.; Berman, R.; Clavijo, P.E.; Saleh, A.; Chen, Z.; Van Waes, C.; Davies, J.; Friedman, J.; Allen, C.T. WEE1 kinase inhibition reverses G2/M cell cycle checkpoint activation to sensitize cancer cells to immunotherapy. Oncoimmunology 2018, 7, e1488359. [Google Scholar] [CrossRef]
- Xie, G.; Shan, L.; Liu, Y.; Wu, T.C.; Gu, X. Antitumor efficacy of EGFR-Targeted recombinant immunotoxin in human head and neck squamous cell carcinoma. Biology 2022, 11, 486. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Shan, L.; Yang, C.; Liu, Y.; Pang, X.; Teng, S.; Wu, T.C.; Gu, X. Recombinant immunotoxin induces tumor intrinsic STING signaling against head and neck squamous cell carcinoma. Sci. Rep. 2023, 13, 18476. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced tumor control with combination mTOR and PD-L1 inhibition in syngeneic oral cavity cancers. Cancer Immunol. Res. 2016, 4, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Caruso, A.; Cash, H.; Waes, C.V.; Allen, C.T. Pools of programmed death-ligand within the oral cavity tumor microenvironment: Variable alteration by targeted therapies. Head Neck 2016, 38, 1176–1186. [Google Scholar] [CrossRef]
- Fu, C.; Jiang, A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, L.; Kroemer, G.; Kepp, O. Conventional type 1 dendritic cells (cDC1) in cancer immunity. Biol. Direct 2023, 18, 71. [Google Scholar] [CrossRef]
- Duong, E.; Fessenden, T.B.; Lutz, E.; Dinter, T.; Yim, L.; Blatt, S.; Bhutkar, A.; Wittrup, K.D.; Spranger, S. Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity 2022, 55, 308–323. [Google Scholar] [CrossRef]
- Samson, N.; Ablasser, A. The cGAS-STING pathway and cancer. Nat. Cancer 2022, 3, 1452–1463. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Katlinskaya, Y.V.; Katlinski, K.V.; Yu, Q.; Ortiz, A.; Beiting, D.P.; Brice, A.; Davar, D.; Sanders, C.; Kirkwood, J.M.; Rui, H.; et al. Suppression of type I interferon signaling overcomes oncogene-induced senescence and mediates melanoma development and progression. Cell Rep. 2016, 15, 171–180. [Google Scholar] [CrossRef]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I downregulation in cancer: Underlying Mechanisms and potential targets for cancer immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Borowska, D.; Sives, S.; Vervelde, L.; Sutton, K.M. Chicken CSF2 and IL-4-, and CSF2-dependent bone marrow cultures differentiate into macrophages over time. Front. Immunol. 2022, 13, 1064084. [Google Scholar] [CrossRef]
- Hoch, T.; Schulz, D.; Eling, N.; Gomez, J.M.; Levesque, M.P.; Bodenmiller, B. Multiplexed imaging mass cytometry of the chemokine milieus in melanoma characterizes features of the response to immunotherapy. Sci. Immunol. 2022, 7, eabk1692. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.S.; Reinshagen, C.; Van Schaik, T.A.; Rossignoli, F.; Borges, P.; Mendonca, N.C.; Abdi, R.; Simon, B.; Reardon, D.A.; Wakimoto, H.; et al. Bifunctional cancer cell-based vaccine concomitantly drives direct tumor killing and antitumor immunity. Sci. Transl. Med. 2023, 15, eabo4778. [Google Scholar] [CrossRef]
- Gong, W.; Donnelly, C.R.; Heath, B.R.; Bellile, E.; Donnelly, L.A.; Taner, H.F.; Broses, L.; Brenner, J.C.; Chinn, S.B.; Ji, R.R.; et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. Oncoimmunology 2021, 10, 1997385. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse models for cancer immunotherapy research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Winkler, A.E.; Kanchi, K.L.; Chalivendra, V.; Law, J.H.; Rickert, C.G.; Kallogjeri, D.; Judd, N.P.; Dunn, G.P.; Piccirillo, J.F.; et al. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin. Cancer Res. 2014, 20, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Hayman, T.J.; Baro, M.; MacNeil, T.; Phoomak, C.; Aung, T.N.; Cui, W.; Leach, K.; Iyer, R.; Challa, S.; Sandoval-Schaefer, T.; et al. STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nat. Commun. 2021, 12, 2327. [Google Scholar] [CrossRef]
- Wang-Bishop, L.; Wehbe, M.; Shae, D.; James, J.; Hacker, B.C.; Garland, K.; Chistov, P.P.; Rafat, M.; Balko, J.M.; Wilson, J.T. Potent STING activation stimulates immunogenic cell death to enhance antitumor immunity in neuroblastoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The cytosolic DNA-Sensing cGAS-STING pathway in cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mirlekar, B.; Johnson, B.M.; Brickey, W.J.; Wrobel, J.A.; Yang, N.; Song, D.; Entwistle, S.; Tan, X.; Deng, M.; et al. STING-induced regulatory B cells compromise NK function in cancer immunity. Nature 2022, 610, 373–380. [Google Scholar] [CrossRef]
- Cho, M.G.; Kumar, R.J.; Lin, C.C.; Boyer, J.A.; Shahir, J.A.; Fagan-Solis, K.; Simpson, D.A.; Fan, C.; Foster, C.E.; Goddard, A.M.; et al. MRE11 liberates cGAS from nucleosome sequestration during tumorigenesis. Nature 2024, 625, 585–592. [Google Scholar] [CrossRef]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 2019, 29, 1236–1248. [Google Scholar] [CrossRef]
- Zhang, C.X.; Ye, S.B.; Ni, J.J.; Cai, T.T.; Liu, Y.N.; Huang, D.J.; Mai, H.Q.; Chen, Q.Y.; He, J.; Zhang, X.S.; et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ. 2019, 26, 2314–2328. [Google Scholar] [CrossRef]
- Meiser, P.; Knolle, M.A.; Hirschberger, A.; de Almeida, G.P.; Bayerl, F.; Lacher, S.; Pedde, A.M.; Flommersfeld, S.; Honninger, J.; Stark, L.; et al. A distinct stimulatory cDC1 subpopulation amplifies CD8(+) T cell responses in tumors for protective anti-cancer immunity. Cancer Cell 2023, 41, 1498–1515. [Google Scholar] [CrossRef]
- Shapir Itai, Y.; Barboy, O.; Salomon, R.; Bercovich, A.; Xie, K.; Winter, E.; Shami, T.; Porat, Z.; Erez, N.; Tanay, A.; et al. Bispecific dendritic-T cell engager potentiates anti-tumor immunity. Cell 2024, 187, 375–389. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| Cgas | GTG AGG ACC AAT CTA AGA CGA G | AGC ATG TTT TCT CTA TCC CGT G |
| Sting | GTC CTC TAT AAG TCC CTA AGC ATG | AAG ATC AAC CGC AAG TAC CC |
| Ifnb | CGA GCA GAG ATC TTC AGG AAC | TCA CTA CCA GTC CCA GAG TC |
| Cxcl9 | AGT CCG CTG TTC TTT TCC TC | TGA GGT CTT TGA GGG ATT TGT AG |
| Cxcl10 | TCA GCA CCA TGA ACC CAA G | CTA TGG CCC TCA TTC TCA CTG |
| Gapdh | TGC CCC CAT GTT TGT GAT GG | AAT GCC AAA GTT GTC ATG GAT GAC C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, G.; Yang, C.; Pang, X.; Wu, T.-C.; Gu, X. Cancer Cell-Intrinsic Type I Interferon Signaling Promotes Antitumor Immunity in Head and Neck Squamous Cell Carcinoma. Cancers 2025, 17, 1279. https://doi.org/10.3390/cancers17081279
Xie G, Yang C, Pang X, Wu T-C, Gu X. Cancer Cell-Intrinsic Type I Interferon Signaling Promotes Antitumor Immunity in Head and Neck Squamous Cell Carcinoma. Cancers. 2025; 17(8):1279. https://doi.org/10.3390/cancers17081279
Chicago/Turabian StyleXie, Guiqin, Cuicui Yang, Xiaowu Pang, Tzyy-Choou Wu, and Xinbin Gu. 2025. "Cancer Cell-Intrinsic Type I Interferon Signaling Promotes Antitumor Immunity in Head and Neck Squamous Cell Carcinoma" Cancers 17, no. 8: 1279. https://doi.org/10.3390/cancers17081279
APA StyleXie, G., Yang, C., Pang, X., Wu, T.-C., & Gu, X. (2025). Cancer Cell-Intrinsic Type I Interferon Signaling Promotes Antitumor Immunity in Head and Neck Squamous Cell Carcinoma. Cancers, 17(8), 1279. https://doi.org/10.3390/cancers17081279