Malignant Pleural Mesothelioma: From Pathophysiology to Innovative Actionable Targets

 , , ,
, , , 
Simple Summary
Abstract
1. Introduction
1.1. Glucose-Regulated Protein 78 (GRP78)
1.2. Fibulin-3 (EFEMP1)
1.3. Signal Transducer and Activator of Transcription-3 (STAT-3)
1.4. Osteopontin (OPN)
1.5. Mesothelin (MSLN)
1.6. Programmed Death-Ligand (PD-1/PD-L1)
2. Genetic Mutations in MPM
2.1. BAP1
2.2. CDKN2A
2.3. NF2
3. MicroRNAs in Pleural Mesothelioma
3.1. miR-126
3.2. miR-31
3.3. miR-34a
4. Epigenetic Modifications
5. Progress in Clinical Trials
5.1. Tazemetostat (EZH2 Inhibitor)
5.2. Rucaparib (PARP Inhibitor)
5.3. Abemaciclib (CDK4/6 Inhibitor)
5.4. CheckMate 743
5.5. DREAM3R
5.6. BEAT-Meso
5.7. AtezoMeso
5.8. eVOLVE-Meso
5.9. INFINITE
5.10. DENIM
5.11. ATOMIC-Meso
5.12. LUME-Meso
5.13. PROMISE-Meso
5.14. MesomiR 1
5.15. HITOC
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abbott, D.M.; Bortolotto, C.; Benvenuti, S.; Lancia, A.; Filippi, A.R.; Stella, G.M. Malignant Pleural Mesothelioma: Genetic and Microenviromental Heterogeneity as an Unexpected Reading Frame and Therapeutic Challenge. Cancers 2020, 12, 1186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cai, Y.; Ou, T.; Zhou, H.; Li, H.; Wang, Z.; Cai, K. Global Burden of Mesothelioma Attributable to Occupational Asbestos Exposure in 204 Countries and Territories: 1990–2019. J. Cancer Res. Clin. Oncol. 2024, 150, 282. [Google Scholar] [CrossRef] [PubMed]
- Allena, N.; Venkatram, S.; Diaz-Fuentes, G. Malignant Pleural Mesothelioma. In Challenges in Pleural Pathology; Strumfa, I., Uljanovs, R., Strumfs, B., Eds.; IntechOpen: Rijeka, Croatia, 2024. [Google Scholar]
- Batirel, H.F.; Metintas, M.; Caglar, H.B.; Ak, G.; Yumuk, P.F.; Yildizeli, B.; Yuksel, M. Adoption of Pleurectomy and Decortication for Malignant Mesothelioma Leads to Similar Survival as Extrapleural Pneumonectomy. J. Thorac. Cardiovasc. Surg. 2016, 151, 478–484. [Google Scholar] [CrossRef]
- Treasure, T.; Lang-Lazdunski, L.; Waller, D.; Bliss, J.M.; Tan, C.; Entwisle, J.; Snee, M.; O’Brien, M.; Thomas, G.; Senan, S.; et al. Extra-Pleural Pneumonectomy versus No Extra-Pleural Pneumonectomy for Patients with Malignant Pleural Mesothelioma: Clinical Outcomes of the Mesothelioma and Radical Surgery (MARS) Randomised Feasibility Study. Lancet Oncol. 2011, 12, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.C.J.; Feld, R.; Leighl, N.; Opitz, I.; Anraku, M.; Tsao, M.-S.; Hwang, D.M.; Hope, A.; de Perrot, M. A Feasibility Study Evaluating Surgery for Mesothelioma After Radiation Therapy: The “SMART” Approach for Resectable Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2014, 9, 397–402. [Google Scholar] [CrossRef]
- Lim, E.; Darlison, L.; Edwards, J.; Elliott, D.; Fennell, D.A.; Popat, S.; Rintoul, R.C.; Waller, D.; Ali, C.; Bille, A.; et al. Mesothelioma and Radical Surgery 2 (MARS 2): Protocol for a Multicentre Randomised Trial Comparing (Extended) Pleurectomy Decortication versus No (Extended) Pleurectomy Decortication for Patients with Malignant Pleural Mesothelioma. BMJ Open 2020, 10, e038892. [Google Scholar] [CrossRef]
- Kok, P.S.; Forde, P.M.; Hughes, B.; Sun, Z.; Brown, C.; Ramalingam, S.; Cook, A.; Lesterhuis, W.J.; Yip, S.; O’Byrne, K.; et al. Protocol of DREAM3R: DuRvalumab with ChEmotherapy as First-Line TreAtment in Advanced Pleural Mesothelioma-a Phase 3 Randomised Trial. BMJ Open 2022, 12, e057663. [Google Scholar] [CrossRef]
- Cedres, S.; Valdivia, A.; Iranzo, P.; Callejo, A.; Pardo, N.; Navarro, A.; Martinez-Marti, A.; Assaf-Pastrana, J.D.; Felip, E.; Garrido, P. Current State-of-the-Art Therapy for Malignant Pleural Mesothelioma and Future Options Centered on Immunotherapy. Cancers 2023, 15, 5787. [Google Scholar] [CrossRef]
- Gifford, J.B.; Hill, R. GRP78 Influences Chemoresistance and Prognosis in Cancer. Curr. Drug Targets 2018, 19, 701–708. [Google Scholar] [CrossRef]
- Pyrko, P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The Unfolded Protein Response Regulator GRP78/BiP as a Novel Target for Increasing Chemosensitivity in Malignant Gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER Stress Sensor, Glucose Regulatory Protein 78 (GRP78) Regulates Redox Status in Pancreatic Cancer Thereby Maintaining “Stemness”. Cell Death Dis. 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Luján, H.D.; Mowatt, M.R.; Conrad, J.T.; Nash, T.E. Increased Expression of the Molecular Chaperone BiP/GRP78 during the Differentiation of a Primitive Eukaryote. Biol. Cell 1996, 86, 11–18. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the Endoplasmic Reticulum: Atypical GRP78 in Cell Viability, Signalling and Therapeutic Targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Akinyemi, A.O.; Simpson, K.E.; Oyelere, S.F.; Nur, M.; Ngule, C.M.; Owoyemi, B.C.D.; Ayarick, V.A.; Oyelami, F.F.; Obaleye, O.; Esoe, D.-P.; et al. Unveiling the Dark Side of Glucose-Regulated Protein 78 (GRP78) in Cancers and Other Human Pathology: A Systematic Review. Mol. Med. 2023, 29, 112. [Google Scholar] [CrossRef]
- dos Santos, N.S.; Gonçalves, D.R.; Balbinot, B.; Visioli, F. Is GRP78 (Glucose-Regulated Protein 78) a Prognostic Biomarker in Differents Types of Cancer? A Systematic Review and Meta-Analysis. Pathol.—Res. Pract. 2023, 242, 154301. [Google Scholar] [CrossRef]
- Baier, D.; Schoenhacker-Alte, B.; Rusz, M.; Pirker, C.; Mohr, T.; Mendrina, T.; Kirchhofer, D.; Meier-Menches, S.M.; Hohenwallner, K.; Schaier, M.; et al. The Anticancer Ruthenium Compound BOLD-100 Targets Glycolysis and Generates a Metabolic Vulnerability towards Glucose Deprivation. Pharmaceutics 2022, 14, 238. [Google Scholar] [CrossRef]
- Ninkovic, S.; Harrison, S.J.; Quach, H. Glucose-Regulated Protein 78 (GRP78) as a Potential Novel Biomarker and Therapeutic Target in Multiple Myeloma. Expert Rev. Hematol. 2020, 13, 1201–1210. [Google Scholar] [CrossRef]
- Ranzato, E.; Bonsignore, G.; Martinotti, S. ER Stress Response and Induction of Apoptosis in Malignant Pleural Mesothelioma: The Achilles Heel Targeted by the Anticancer Ruthenium Drug BOLD-100. Cancers 2022, 14, 4126. [Google Scholar] [CrossRef]
- Sorino, C.; Mondoni, M.; Marchetti, G.; Agati, S.; Inchingolo, R.; Mei, F.; Flamini, S.; Lococo, F.; Feller-Kopman, D. Pleural Mesothelioma: Advances in Blood and Pleural Biomarkers. J. Clin. Med. 2023, 12, 7006. [Google Scholar] [CrossRef]
- Agha, M.A.; El-Habashy, M.M.; El-Shazly, R.A. Role of Fibulin-3 in the Diagnosis of Malignant Mesothelioma. Egypt. J. Chest Dis. Tuberc. 2014, 63, 99–105. [Google Scholar] [CrossRef]
- Roshini, A.; Goparaju, C.; Kundu, S.; Nandhu, M.S.; Longo, S.L.; Longo, J.A.; Chou, J.; Middleton, F.A.; Pass, H.I.; Viapiano, M.S. The Extracellular Matrix Protein Fibulin-3/EFEMP1 Promotes Pleural Mesothelioma Growth by Activation of PI3K/Akt Signaling. Front. Oncol. 2022, 12, 1014749. [Google Scholar] [CrossRef] [PubMed]
- Woodard, D.R.; Daniel, S.; Nakahara, E.; Abbas, A.; DiCesare, S.M.; Collier, G.E.; Hulleman, J.D. A Loss-of-Function Cysteine Mutant in Fibulin-3 (EFEMP1) Forms Aberrant Extracellular Disulfide-Linked Homodimers and Alters Extracellular Matrix Composition. Hum. Mutat. 2022, 43, 1945–1955. [Google Scholar] [CrossRef]
- Kirschner, M.B.; Pulford, E.; Hoda, M.A.; Rozsas, A.; Griggs, K.; Cheng, Y.Y.; Edelman, J.J.B.; Kao, S.C.; Hyland, R.; Dong, Y.; et al. Fibulin-3 Levels in Malignant Pleural Mesothelioma Are Associated with Prognosis but Not Diagnosis. Br. J. Cancer 2015, 113, 963–969. [Google Scholar] [CrossRef]
- Jiang, Z.; Shen, W.; Ying, S.; Gao, Z.; He, X.; Chen, R.; Xia, H.; Guo, X.; Fang, Y.; Zhang, Y.; et al. Overexpression of Fibulin-3 in Tumor Tissue Predicts Poor Survival of Malignant Mesothelioma Patients from Hand-Spinning Asbestos Exposed Area in Eastern China. Sci. Rep. 2020, 10, 20373. [Google Scholar] [CrossRef] [PubMed]
- Nandhu, M.S.; Behera, P.; Bhaskaran, V.; Longo, S.L.; Barrera-Arenas, L.M.; Sengupta, S.; Rodriguez-Gil, D.J.; Chiocca, E.A.; Viapiano, M.S. Development of a Function-Blocking Antibody Against Fibulin-3 as a Targeted Reagent for Glioblastoma. Clin. Cancer Res. 2018, 24, 821–833. [Google Scholar] [CrossRef]
- Lapidot, M.; Case, A.E.; Larios, D.; Gandler, H.I.; Meng, C.; Tošić, I.; Weisberg, E.L.; Poitras, M.J.; Gokhale, P.C.; Paweletz, C.P.; et al. Inhibitors of the Transcription Factor STAT3 Decrease Growth and Induce Immune Response Genes in Models of Malignant Pleural Mesothelioma (MPM). Cancers 2020, 13, 7. [Google Scholar] [CrossRef]
- Tošić, I.; Frank, D.A. STAT3 as a Mediator of Oncogenic Cellular Metabolism: Pathogenic and Therapeutic Implications. Neoplasia 2021, 23, 1167–1178. [Google Scholar] [CrossRef]
- Lapidot, M.; Saladi, S.V.; Salgia, R.; Sattler, M. Novel Therapeutic Targets and Immune Dysfunction in Malignant Pleural Mesothelioma. Front. Pharmacol. 2022, 12, 806570. [Google Scholar] [CrossRef]
- Paleari, L.; Rotolo, N.; Imperatori, A.; Puzone, R.; Sessa, F.; Franzi, F.; Meacci, E.; Camplese, P.; Cesario, A.; Paganuzzi, M. Osteopontin Is Not a Specific Marker in Malignant Pleural Mesothelioma. Int. J. Biol. Markers 2009, 24, 112–117. [Google Scholar] [CrossRef]
- Betta, P.; Libener, R.; Orecchia, S.; Salvio, M.; Arnolfo, E.; Bottero, G.; Botta, M.; Piccolini, E.; Pavesi, M. Tissue Expression of Osteopontin (OPN) and Mesothelin (MES) in Malignant Pleural Mesothelioma (MPM). An Immunohistochemical Study of 70 Consecutive Cases. J. Clin. Oncol. 2009, 27, e22102. [Google Scholar] [CrossRef]
- Sun, H.H.; Vaynblat, A.; Pass, H.I. Diagnosis and Prognosis-Review of Biomarkers for Mesothelioma. Ann. Transl. Med. 2017, 5, 244. [Google Scholar] [CrossRef] [PubMed]
- Psallidas, I.; Magkouta, S.; Pappas, A.; Moschos, C.; Vazakidou, M.-E.; Roussos, C.; Stathopoulos, G.; Kalomenidis, I. Icmt inhibition exerts anti-angiogenic and anti-hyperpermeability activities impeding malignant pleural effusion. Oncotarget 2016, 7, 20249–20259. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Marin-Muller, C.; Li, M.; Chen, C.; Yao, Q. Mesothelin Confers Pancreatic Cancer Cell Resistance to TNF-α-Induced Apoptosis through Akt/PI3K/NF-ΚB Activation and IL-6/Mcl-1 Overexpression. Mol. Cancer 2011, 10, 106. [Google Scholar] [CrossRef]
- Vizcaya, D.; Farahmand, B.; Walter, A.O.; Kneip, C.; Jöhrens, K.; Tukiainen, M.; Schmitz, A.A. Prognosis of Patients with Malignant Mesothelioma by Expression of Programmed Cell Death 1 Ligand 1 and Mesothelin in a Contemporary Cohort in Finland. Cancer Treat. Res. Commun. 2020, 25, 100260. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Tsunoda, T.; Riku, M.; Ito, H.; Inoko, A.; Murakami, H.; Ebi, M.; Ogasawara, N.; Pastan, I.; Kasugai, K.; et al. Diffuse Mesothelin Expression Leads to Worse Prognosis through Enhanced Cellular Proliferation in Colorectal Cancer. Oncol. Lett. 2020, 19, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Qualiotto, A.N.; Baldavira, C.M.; Balancin, M.; Ab’Saber, A.; Takagaki, T.; Capelozzi, V.L. Mesothelin Expression Remodeled the Immune-Matrix Tumor Microenvironment Predicting the Risk of Death in Patients with Malignant Pleural Mesothelioma. Front. Immunol. 2023, 14, 1268927. [Google Scholar] [CrossRef]
- Chen, S.-H.; Hung, W.-C.; Wang, P.; Paul, C.; Konstantopoulos, K. Mesothelin Binding to CA125/MUC16 Promotes Pancreatic Cancer Cell Motility and Invasion via MMP-7 Activation. Sci. Rep. 2013, 3, 1870. [Google Scholar] [CrossRef]
- Cao, Y. Lack of Basic Rationale in Epithelial-Mesenchymal Transition and Its Related Concepts. Cell Biosci. 2024, 14, 104. [Google Scholar] [CrossRef]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-Targeted CAR-T Cell Therapy for Solid Tumors. Expert Opin. Biol. Ther. 2021, 21, 473–486. [Google Scholar] [CrossRef]
- Hassan, R.; Fennell, D.; Scherpereel, A.; Nowak, A.K.; Von Pawel, J.; Novello, S.; McLaughlin, M.; Hoffman, K.; Wallin, B.A. A Randomized, Placebo-Controlled Study of Amatuximab in Combination with Pemetrexed and Cisplatin (P/C) as Front-Line Therapy for Subjects with Malignant Pleural Mesothelioma (MPM). J. Clin. Oncol. 2016, 34, TPS8577. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, J.; Wada, Y.; Matsumoto, K.; Azuma, M.; Kikuchi, K.; Ueda, S. Overexpression of B7-H1 (PD-L1) Significantly Associates with Tumor Grade and Postoperative Prognosis in Human Urothelial Cancers. Cancer Immunol. Immunother. 2007, 56, 1173–1182. [Google Scholar] [CrossRef]
- van Ingen, J.; Aksamit, T.; Andrejak, C.; Böttger, E.C.; Cambau, E.; Daley, C.L.; Griffith, D.E.; Guglielmetti, L.; Holland, S.M.; Huitt, G.A.; et al. Treatment Outcome Definitions in Nontuberculous Mycobacterial Pulmonary Disease: An NTM-NET Consensus Statement. Eur. Respir. J. 2018, 51, 1800170. [Google Scholar] [PubMed]
- Mansfield, A.S.; Roden, A.C.; Peikert, T.; Sheinin, Y.M.; Harrington, S.M.; Krco, C.J.; Dong, H.; Kwon, E.D. B7-H1 Expression in Malignant Pleural Mesothelioma Is Associated with Sarcomatoid Histology and Poor Prognosis. J. Thorac. Oncol. 2014, 9, 1036–1040. [Google Scholar] [CrossRef]
- Cedrés, S.; Ponce-Aix, S.; Pardo-Aranda, N.; Navarro-Mendivil, A.; Martinez-Marti, A.; Zugazagoitia, J.; Sansano, I.; Montoro, M.A.; Enguita, A.; Felip, E. Analysis of Expression of PTEN/PI3K Pathway and Programmed Cell Death Ligand 1 (PD-L1) in Malignant Pleural Mesothelioma (MPM). Lung Cancer 2016, 96, 1–6. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-Line Nivolumab plus Ipilimumab in Unresectable Malignant Pleural Mesothelioma (CheckMate 743): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Popat, S.; Curioni-Fontecedro, A.; Dafni, U.; Shah, R.; O’Brien, M.; Pope, A.; Fisher, P.; Spicer, J.; Roy, A.; Gilligan, D.; et al. A Multicentre Randomised Phase III Trial Comparing Pembrolizumab versus Single-Agent Chemotherapy for Advanced Pre-Treated Malignant Pleural Mesothelioma: The European Thoracic Oncology Platform (ETOP 9-15) PROMISE-Meso Trial. Ann. Oncol. 2020, 31, 1734–1745. [Google Scholar] [CrossRef]
- Fennell, D.A.; Ewings, S.; Ottensmeier, C.; Califano, R.; Hanna, G.G.; Hill, K.; Danson, S.; Steele, N.; Nye, M.; Johnson, L.; et al. Nivolumab versus Placebo in Patients with Relapsed Malignant Mesothelioma (CONFIRM): A Multicentre, Double-Blind, Randomised, Phase 3 Trial. Lancet. Oncol. 2021, 22, 1530–1540. [Google Scholar] [CrossRef]
- Yap, T.A.; Nakagawa, K.; Fujimoto, N.; Kuribayashi, K.; Guren, T.K.; Calabrò, L.; Shapira-Frommer, R.; Gao, B.; Kao, S.; Matos, I.; et al. Efficacy and Safety of Pembrolizumab in Patients with Advanced Mesothelioma in the Open-Label, Single-Arm, Phase 2 KEYNOTE-158 Study. Lancet Respir. Med. 2021, 9, 613–621. [Google Scholar] [CrossRef]
- Cheung, M.; Testa, J.R. BAP1, a Tumor Suppressor Gene Driving Malignant Mesothelioma. Transl. Lung Cancer Res. 2017, 6, 270–278. [Google Scholar] [CrossRef]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 Mutations Predispose to Malignant Mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive Genomic Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Affar, E.B.; Carbone, M. BAP1 Regulates Different Mechanisms of Cell Death. Cell Death Dis. 2018, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.H.; Toyokuni, S. Malignant Mesothelioma as an Oxidative Stress-Induced Cancer: An Update. Free Radic. Biol. Med. 2015, 86, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Louw, A.; Panou, V.; Szejniuk, W.M.; Meristoudis, C.; Chai, S.M.; van Vliet, C.; Lee, Y.C.G.; Dick, I.M.; Firth, T.; Lynggaard, L.A.; et al. BAP1 Loss by Immunohistochemistry Predicts Improved Survival to First-Line Platinum and Pemetrexed Chemotherapy for Patients With Pleural Mesothelioma: A Validation Study. J. Thorac. Oncol. 2022, 17, 921–930. [Google Scholar] [CrossRef]
- Cigognetti, M.; Lonardi, S.; Fisogni, S.; Balzarini, P.; Pellegrini, V.; Tironi, A.; Bercich, L.; Bugatti, M.; Rossi, G.; Murer, B.; et al. BAP1 (BRCA1-Associated Protein 1) Is a Highly Specific Marker for Differentiating Mesothelioma from Reactive Mesothelial Proliferations. Mod. Pathol. 2015, 28, 1043–1057. [Google Scholar] [CrossRef]
- Chapel, D.B.; Hornick, J.L.; Barlow, J.; Bueno, R.; Sholl, L.M. Clinical and Molecular Validation of BAP1, MTAP, P53, and Merlin Immunohistochemistry in Diagnosis of Pleural Mesothelioma. Mod. Pathol. 2022, 35, 1383–1397. [Google Scholar] [CrossRef]
- Ogbue, O.; Unlu, S.; Sorathia, S.; Nanah, A.R.; Abdeljaleel, F.; Haddad, A.S.; Daw, H. Impact of BAP1 Mutational Status on Immunotherapy Outcomes in Advanced Malignant Pleural Mesothelioma: A Single Institution Experience. J. Clin. Oncol. 2023, 41, e20537. [Google Scholar] [CrossRef]
- American Association for Cancer Research. Tazemetostat Shows Antitumor Activity in Malignant Pleural Mesothelioma. Cancer Discov. 2022, 12, 1610. [Google Scholar] [CrossRef]
- Zauderer, M.G.; Szlosarek, P.W.; Le Moulec, S.; Popat, S.; Taylor, P.; Planchard, D.; Scherpereel, A.; Koczywas, M.; Forster, M.; Cameron, R.B.; et al. EZH2 Inhibitor Tazemetostat in Patients with Relapsed or Refractory, BAP1-Inactivated Malignant Pleural Mesothelioma: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2022, 23, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Illei, P.B.; Ladanyi, M.; Rusch, V.W.; Zakowski, M.F. The Use of CDKN2A Deletion as a Diagnostic Marker for Malignant Mesothelioma in Body Cavity Effusions. Cancer 2003, 99, 51–56. [Google Scholar] [CrossRef]
- Vrugt, B.; Kirschner, M.B.; Meerang, M.; Oehl, K.; Wagner, U.; Soltermann, A.; Moch, H.; Opitz, I.; Wild, P.J. Deletions of CDKN2A and MTAP Detected by Copy-Number Variation Array Are Associated with Loss of P16 and MTAP Protein in Pleural Mesothelioma. Cancers 2023, 15, 4978. [Google Scholar] [CrossRef] [PubMed]
- Kettunen, E.; Savukoski, S.; Salmenkivi, K.; Böhling, T.; Vanhala, E.; Kuosma, E.; Anttila, S.; Wolff, H. CDKN2A Copy Number and P16 Expression in Malignant Pleural Mesothelioma in Relation to Asbestos Exposure. BMC Cancer 2019, 19, 507. [Google Scholar] [CrossRef]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-Exome Sequencing Reveals Frequent Genetic Alterations in BAP1, NF2, CDKN2A, and CUL1 in Malignant Pleural Mesothelioma. Cancer Res. 2015, 75, 264–269. [Google Scholar] [CrossRef]
- Fennell, D.A.; King, A.; Mohammed, S.; Greystoke, A.; Anthony, S.; Poile, C.; Nusrat, N.; Scotland, M.; Bhundia, V.; Branson, A.; et al. Abemaciclib in Patients with P16ink4A-Deficient Mesothelioma (MiST2): A Single-Arm, Open-Label, Phase 2 Trial. Lancet Oncol. 2022, 23, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Nardone, V.; Porta, C.; Giannicola, R.; Correale, P.; Mutti, L. Abemaciclib for Malignant Pleural Mesothelioma. Lancet Oncol. 2022, 23, e237. [Google Scholar] [CrossRef]
- Xu, D.; Yin, S.; Shu, Y. NF2: An Underestimated Player in Cancer Metabolic Reprogramming and Tumor Immunity. Npj. Precis. Oncol. 2024, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Sekido, Y.; Sato, T. NF2 Alteration in Mesothelioma. Front. Toxicol. 2023, 5, 1161995. [Google Scholar] [CrossRef]
- Fu, M.; Hu, Y.; Lan, T.; Guan, K.-L.; Luo, T.; Luo, M. The Hippo Signalling Pathway and Its Implications in Human Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 376. [Google Scholar] [CrossRef]
- Yang, H.; Hall, S.R.R.; Sun, B.; Zhao, L.; Gao, Y.; Schmid, R.A.; Tan, S.T.; Peng, R.-W.; Yao, F. NF2 and Canonical Hippo-YAP Pathway Define Distinct Tumor Subsets Characterized by Different Immune Deficiency and Treatment Implications in Human Pleural Mesothelioma. Cancers 2021, 13, 1561. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H. Molecular Pathways: Targeting Mechanisms of Asbestos and Erionite Carcinogenesis in Mesothelioma. Clin. Cancer Res. 2012, 18, 598–604. [Google Scholar] [CrossRef]
- Ghalavand, M.A.; Asghari, A.; Farhadi, M.; Taghizadeh-Hesary, F.; Garshasbi, M.; Falah, M. The Genetic Landscape and Possible Therapeutics of Neurofibromatosis Type 2. Cancer Cell Int. 2023, 23, 99. [Google Scholar] [CrossRef]
- Hillen, H.; Candi, A.; Vanderhoydonck, B.; Kowalczyk, W.; Sansores-Garcia, L.; Kesikiadou, E.C.; Van Huffel, L.; Spiessens, L.; Nijs, M.; Soons, E.; et al. A Novel Irreversible TEAD Inhibitor, SWTX-143, Blocks Hippo Pathway Transcriptional Output and Causes Tumor Regression in Preclinical Mesothelioma Models. Mol. Cancer Ther. 2024, 23, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, Q.; Han, Y.Q.; Li, P.; Ouyang, P.H.; Wang, M.Y.; Hu, Z. De Diagnostic Accuracy of Circulating MiR-126 for Malignant Pleural Mesothelioma: A Systematic Review and Meta-Analysis. Transl. Cancer Res. 2021, 10, 1856–1862. [Google Scholar] [CrossRef]
- Santarelli, L.; Strafella, E.; Staffolani, S.; Amati, M.; Emanuelli, M.; Sartini, D.; Pozzi, V.; Carbonari, D.; Bracci, M.; Pignotti, E.; et al. Association of MiR-126 with Soluble Mesothelin-Related Peptides, a Marker for Malignant Mesothelioma. PLoS ONE 2011, 6, e18232. [Google Scholar] [CrossRef]
- Monaco, F.; Gaetani, S.; Alessandrini, F.; Tagliabracci, A.; Bracci, M.; Valentino, M.; Neuzil, J.; Amati, M.; Bovenzi, M.; Tomasetti, M.; et al. Exosomal Transfer of MiR-126 Promotes the Anti-Tumour Response in Malignant Mesothelioma: Role of MiR-126 in Cancer-Stroma Communication. Cancer Lett. 2019, 463, 27–36. [Google Scholar] [CrossRef]
- Monaco, F.; De Conti, L.; Vodret, S.; Zanotta, N.; Comar, M.; Manzotti, S.; Rubini, C.; Graciotti, L.; Fulgenzi, G.; Bovenzi, M.; et al. Force-Feeding Malignant Mesothelioma Stem-Cell like with Exosome-Delivered MiR-126 Induces Tumour Cell Killing. Transl. Oncol. 2022, 20, 101400. [Google Scholar] [CrossRef]
- Liu, B.; Peng, X.-C.; Zheng, X.-L.; Wang, J.; Qin, Y.-W. MiR-126 Restoration down-Regulate VEGF and Inhibit the Growth of Lung Cancer Cell Lines in Vitro and in Vivo. Lung Cancer 2009, 66, 169–175. [Google Scholar] [CrossRef]
- De Santi, C.; Melaiu, O.; Bonotti, A.; Cascione, L.; Di Leva, G.; Foddis, R.; Cristaudo, A.; Lucchi, M.; Mora, M.; Truini, A.; et al. Deregulation of MiRNAs in Malignant Pleural Mesothelioma Is Associated with Prognosis and Suggests an Alteration of Cell Metabolism. Sci. Rep. 2017, 7, 3140. [Google Scholar] [CrossRef]
- Tomasetti, M.; Gaetani, S.; Monaco, F.; Neuzil, J.; Santarelli, L. Epigenetic Regulation of MiRNA Expression in Malignant Mesothelioma: MiRNAs as Biomarkers of Early Diagnosis and Therapy. Front. Oncol. 2019, 9, 01293. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Johnson, T.G.; van Zandwijk, N. Manipulating MicroRNAs for the Treatment of Malignant Pleural Mesothelioma: Past, Present and Future. Front. Oncol. 2020, 10, 00105. [Google Scholar] [CrossRef]
- Ivanov, S.V.; Goparaju, C.M.V.; Lopez, P.; Zavadil, J.; Toren-Haritan, G.; Rosenwald, S.; Hoshen, M.; Chajut, A.; Cohen, D.; Pass, H.I. Pro-Tumorigenic Effects of MiR-31 Loss in Mesothelioma. J. Biol. Chem. 2010, 285, 22809–22817. [Google Scholar] [CrossRef] [PubMed]
- Moody, H.L.; Lind, M.J.; Maher, S.G. MicroRNA-31 Regulates Chemosensitivity in Malignant Pleural Mesothelioma. Mol. Ther. Nucleic Acids 2017, 8, 317–329. [Google Scholar] [CrossRef]
- Tanaka, N.; Toyooka, S.; Soh, J.; Tsukuda, K.; Shien, K.; Furukawa, M.; Muraoka, T.; Maki, Y.; Ueno, T.; Yamamoto, H.; et al. Downregulation of MicroRNA-34 Induces Cell Proliferation and Invasion of Human Mesothelial Cells. Oncol. Rep. 2013, 29, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, S.; Alinovi, R.; Corradi, M.; Poli, D.; Cavallo, D.; Pelosi, G.; Ampollini, L.; Goldoni, M.; Mozzoni, P. A Comparison between the Effects of Over-Expression of MiRNA-16 and MiRNA-34a on Cell Cycle Progression of Mesothelioma Cell Lines and on Their Cisplatin Sensitivity. Cancer Treat. Res. Commun. 2021, 26, 100276. [Google Scholar] [CrossRef]
- Andersen, M.; Grauslund, M.; Ravn, J.; Sørensen, J.B.; Andersen, C.B.; Santoni-Rugiu, E. Diagnostic Potential of MiR-126, MiR-143, MiR-145, and MiR-652 in Malignant Pleural Mesothelioma. J. Mol. Diagn. 2014, 16, 418–430. [Google Scholar] [CrossRef]
- Benjamin, H.; Lebanony, D.; Rosenwald, S.; Cohen, L.; Gibori, H.; Barabash, N.; Ashkenazi, K.; Goren, E.; Meiri, E.; Morgenstern, S.; et al. A Diagnostic Assay Based on MicroRNA Expression Accurately Identifies Malignant Pleural Mesothelioma. J. Mol. Diagn. 2010, 12, 771–779. [Google Scholar] [CrossRef]
- Ak, G.; Tomaszek, S.C.; Kosari, F.; Metintas, M.; Jett, J.R.; Metintas, S.; Yildirim, H.; Dundar, E.; Dong, J.; Aubry, M.C.; et al. MicroRNA and MRNA Features of Malignant Pleural Mesothelioma and Benign Asbestos-Related Pleural Effusion. Biomed. Res. Int. 2015, 2015, 635748. [Google Scholar] [CrossRef] [PubMed]
- Augustin, R. Hypersensitivity Diseases of the Lungs Due to Fungi and Organic Dusts. Immunology 1970, 19, 855. [Google Scholar]
- Piber, P.; Vavpetic, N.; Goricar, K.; Dolzan, V.; Kovac, V.; Franko, A. The Influence of Genetic Variability in IL1B and MIR146A on the Risk of Pleural Plaques and Malignant Mesothelioma. Radiol. Oncol. 2020, 54, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Martinez, V.D.; Minatel, B.C.; Pewarchuk, M.E.; Marshall, E.A.; MacAulay, G.M.; Hubaux, R.; Pearson, D.D.; Goodarzi, A.A.; Dellaire, G.; et al. Genomics and Epigenetics of Malignant Mesothelioma. High-Throughput 2018, 7, 20. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Kuribayashi, K.; Minami, T.; Ohmuraya, M.; Kijima, T. Epigenetic Alterations and Biomarkers for Immune Checkpoint Inhibitors-Current Standards and Future Perspectives in Malignant Pleural Mesothelioma Treatment. Front. Oncol. 2020, 10, 554570. [Google Scholar] [CrossRef]
- Ferrari, L.; Carugno, M.; Mensi, C.; Pesatori, A.C. Circulating Epigenetic Biomarkers in Malignant Pleural Mesothelioma: State of the Art and Critical Evaluation. Front. Oncol. 2020, 10, 445. [Google Scholar] [CrossRef]
- Reardon, E.S.; Shukla, V.; Xi, S.; Gara, S.K.; Liu, Y.; Straughan, D.; Zhang, M.; Hong, J.A.; Payabyab, E.C.; Kumari, A.; et al. UHRF1 Is a Novel Druggable Epigenetic Target in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2021, 16, 89–103. [Google Scholar] [CrossRef]
- Baird, A.-M.; Finn, S.P.; Gray, S.G.; Sheils, O. Epigenetic Modifier UHRF1 May Be a Potential Target in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2021, 16, 14–16. [Google Scholar] [CrossRef]
- Yang, G.; Li, C.; Tao, F.; Liu, Y.; Zhu, M.; Du, Y.; Fei, C.; She, Q.; Chen, J. The Emerging Roles of Lysine-Specific Demethylase 4A in Cancer: Implications in Tumorigenesis and Therapeutic Opportunities. Genes Dis. 2024, 11, 645–663. [Google Scholar] [CrossRef]
- Lapidot, M.; Case, A.E.; Weisberg, E.L.; Meng, C.; Walker, S.R.; Garg, S.; Ni, W.; Podar, K.; Hung, Y.P.; Carrasco, R.D.; et al. Essential Role of the Histone Lysine Demethylase KDM4A in the Biology of Malignant Pleural Mesothelioma (MPM). Br. J. Cancer 2021, 125, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, A.; Tajima, K.; Takahashi, F.; Mitsuishi, Y.; Winardi, W.; Hidayat, M.; Hayakawa, D.; Matsumoto, N.; Izumi, K.; Asao, T.; et al. A Novel Therapeutic Strategy Targeting the Mesenchymal Phenotype of Malignant Pleural Mesothelioma by Suppressing LSD1. Mol. Cancer Res. 2022, 20, 127–138. [Google Scholar] [CrossRef]
- Straining, R.; Eighmy, W. Tazemetostat: EZH2 Inhibitor. J. Adv. Pract. Oncol. 2022, 13, 158–163. [Google Scholar] [CrossRef]
- Fennell, D.A.; King, A.; Mohammed, S.; Branson, A.; Brookes, C.; Darlison, L.; Dawson, A.G.; Gaba, A.; Hutka, M.; Morgan, B.; et al. Rucaparib in Patients with BAP1-Deficient or BRCA1-Deficient Mesothelioma (MiST1): An Open-Label, Single-Arm, Phase 2a Clinical Trial. Lancet Respir. Med. 2021, 9, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Cerbone, L.; del Rio, B.; Delfanti, S.; Boccuzzi, F.; Barbieri, P.; De Angelis, A.M.; Meda, S.; Savi, L.; Righi, L.; Sculco, M.; et al. Rucaparib in Refractory Pleural Mesothelioma Harboring Somatic Pathogenic BRCA1 and BRCA2 Mutation. A Report of Two Cases. Lung Cancer Manag. 2025, 14, 2424133. [Google Scholar] [CrossRef]
- Costa, A.; Forte, I.M.; Pentimalli, F.; Iannuzzi, C.A.; Alfano, L.; Capone, F.; Camerlingo, R.; Calabrese, A.; von Arx, C.; Benot Dominguez, R.; et al. Pharmacological Inhibition of CDK4/6 Impairs Diffuse Pleural Mesothelioma 3D Spheroid Growth and Reduces Viability of Cisplatin-Resistant Cells. Front. Oncol. 2024, 14, 1418951. [Google Scholar] [CrossRef] [PubMed]
- Terenziani, R.; Galetti, M.; La Monica, S.; Fumarola, C.; Zoppi, S.; Alfieri, R.; Digiacomo, G.; Cavazzoni, A.; Cavallo, D.; Corradi, M.; et al. CDK4/6 Inhibition Enhances the Efficacy of Standard Chemotherapy Treatment in Malignant Pleural Mesothelioma Cells. Cancers 2022, 14, 5925. [Google Scholar] [CrossRef]
- Fennell, D.A.; Nusrat, N. Abemaciclib for Malignant Pleural Mesothelioma—Authors’ Reply. Lancet Oncol. 2022, 23, e238. [Google Scholar] [CrossRef]
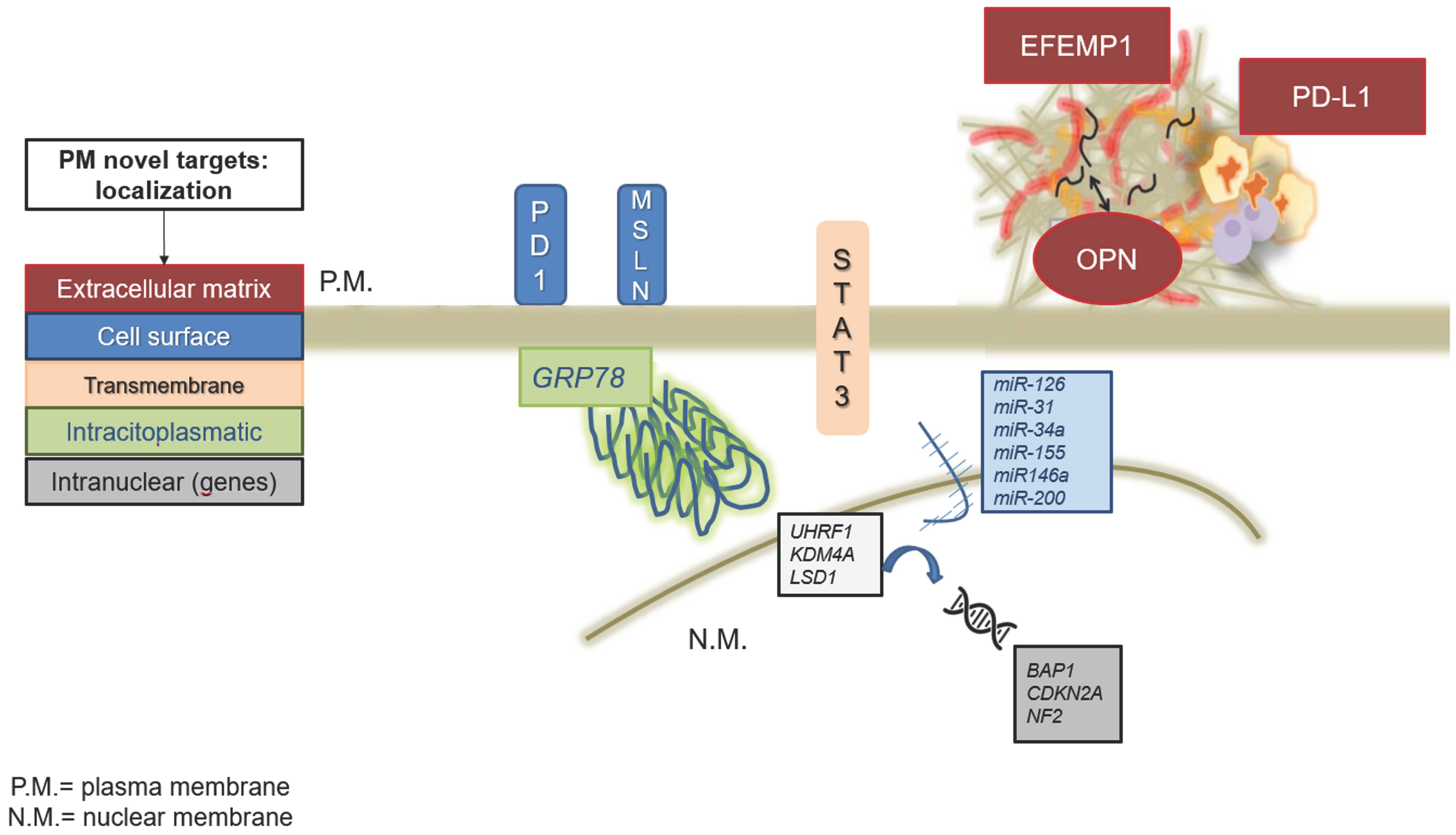

{kind=link}
| Protein | Function | Mechanism of Action | Clinical Implications |
|---|---|---|---|
| Glucose-Regulated Protein 78 (GRP78) | Chaperone protein involved in protein folding and assembly | Regulates the Unfolded Protein Response (UPR) by interacting with PERK, ATF6, and IRE1; promotes tumor survival and chemoresistance by inhibiting caspases | Potential prognostic biomarker; target for GRP78 modulators such as BOLD-100, which disrupts calcium homeostasis and induces UPR-mediated cell death |
| Fibulin-3 (EFEMP1) | Extracellular matrix protein promoting tumor aggressiveness | Activates the PI3K/Akt pathway; knockdown reduces tumor burden and disrupts cell–ECM interactions | Investigated as a diagnostic and therapeutic target; function-blocking antibodies (mAb428.2) have shown promising tumor suppression effects |
| Signal Transducer and Activator of Transcription-3 (STAT-3) | Mediates immune evasion, inflammation, and proliferation | Activated by tyrosine phosphorylation (Tyr705); forms a complex with NFkB in chemoresistant PM cells | STAT-3 inhibitors combined with immune checkpoint inhibitors offer a potential therapeutic strategy |
| Osteopontin (OPN) | Matricellular protein involved in invasion, angiogenesis, and inflammation | Activates PI3K/Akt and FAK-Src-Rho pathways; contributes to chronic inflammation and immune cell retention | Potential diagnostic biomarker, but specificity is low; OPN-mediated signaling may promote immunosuppressive environments |
| Mesothelin (MSLN) | Surface glycoprotein overexpressed in PM | Binds to MUC16, facilitating tumor adhesion and metastasis; interacts with PI3K/Akt and MAPK pathways to promote survival and drug resistance | Target for immunotherapies including monoclonal antibodies (amatuximab), antibody-drug conjugates (anetumab ravtansine), and CAR T cell therapy |
| Programmed Death-Ligand (PD-1/PD-L1) | Immune checkpoint regulator | Inhibits T-cell activation and promotes immune evasion in PM | PD-1/PD-L1 inhibitors (nivolumab, pembrolizumab) approved for PM treatment; predictive biomarkers of response remain under investigation |
| Gene | Primary Function | Alteration Frequency | Mechanism of Action | Therapeutic Implications |
|---|---|---|---|---|
| BAP1 | DNA repair, transcription regulation, and cell cycle control | 1–7% (germline), 20–64% (somatic) | Point mutations, copy number loss, rearrangements | Increased sensitivity to platinum-based therapy, potential target for PARP inhibitors (PARPi) and EZH2 inhibitors, possible response to immune checkpoint inhibitors (ICPi) |
| CDKN2A | Cell cycle regulation (encodes p16INK4A and p14ARF) | 61–88% | Homozygous/hemizygous deletion (most common), promoter hypermethylation | CDK4/6 inhibitors (e.g., Abemaciclib), potential synergy with immune checkpoint blockade |
| NF2 | Hippo signaling pathway regulation (encodes Merlin) | 30–40% | Nonsense/missense mutations, deletions, rearrangements | Targeting YAP/TAZ within the Hippo pathway, TEAD inhibitors under clinical investigation |
| MicroRNA | Mechanism of Action | Effects on PM | Potential Therapeutic Approach |
|---|---|---|---|
| miR-126 | Suppresses VEGF and EGFR signaling, inhibiting angiogenesis and tumor proliferation. | Downregulated in PM, leading to increased tumor growth, migration, and invasion. Associated with poorer prognosis. | Restoration using miRNA mimics or gene therapy to inhibit angiogenesis and tumor spread. Potential combination with immunotherapy or chemotherapy. |
| miR-31 | Regulates CDK4/6 and p27, controlling cell cycle progression. Can also downregulate p53 and FAS. | Overexpressed in PM, leading to increased tumor invasiveness, poor prognosis, and resistance to apoptosis. | Inhibition via miR-31 blockers to restore tumor suppressor gene function. Potential combination with chemotherapy to enhance therapeutic response. |
| miR-34a | Direct target of p53; regulates apoptosis (BCL-2), stress response (SIRT1), and cell cycle progression (CDK6, E2F3). | Downregulated in PM, leading to enhanced tumor cell survival, proliferation, and invasion. Correlated with poor survival. | Restoration using miRNA mimics or small molecules to induce miR-34a expression. Potential synergy with chemotherapy and immunotherapy. |
| miR-200 family | Inhibits epithelial-mesenchymal transition (EMT), reducing migratory and invasive tumor properties. | Downregulation leads to EMT activation, increasing metastasis and chemoresistance. | Restoration to reverse EMT and reduce tumor invasiveness. |
| miR-155 | Modulates immune response and inflammation. | Overexpressed in PM, contributing to immune evasion and tumor progression. | Targeting miR-155 to enhance immune surveillance and reduce tumor-associated inflammation. |
| miR-146a | Regulates immune response and inflammatory cytokine levels. | Downregulated in PM, leading to an altered tumor microenvironment and reduced immune response. | Restoration to improve immune regulation and enhance anti-tumor immunity. |
| Target | Mechanism of Action | Effects on PM | Potential Therapeutic Approach |
|---|---|---|---|
| UHRF1 (Ubiquitin-like with PHD and Ring Finger domains 1) | Recruits DNA methyltransferases to newly synthesized DNA, leading to CpG island methylation and tumor-suppressor gene silencing. | Overexpressed in PM cells; associated with poor prognosis and reduced overall survival (OS). | Inhibition of HDM2 or SP1 (e.g., pucacilin) to restore p53 signaling and suppress UHRF1 overexpression. |
| KDM4A (K36 demethylase 4A) | Demethylates histone H3 lysine, regulating cell proliferation, DNA repair, and self-renewal. | Promotes PM progression by facilitating uncontrolled cell division. | KDM4A inhibition reduces tumor cell proliferation and viability. |
| LSD1 (Lysine-specific demethylase 1) | Removes monomethyl and dimethyl marks from histones H3, H4, and H9, repressing target gene transcription. | Induces epithelial-mesenchymal transition (EMT), contributing to chemotherapy resistance and poor prognosis. | LSD1 inhibition to block EMT via the FAK-Akt-GSK3β pathway. |
| Trial Name | Drugs Used | Phase | Clinical Trial Identifier | Trial Duration | Results |
|---|---|---|---|---|---|
| Tazemetostat Study | Tazemetostat (EZH2 inhibitor) | II | Not provided | 12 weeks (primary endpoint) | Disease control rate: 54% (week 12), 28% (week 24). Well-tolerated toxicity profile. |
| MIST-1 | Rucaparib (PARP inhibitor) | II | NCT03654833 | Not provided | Study showed rucaparib was well tolerated, with grade 3–4 adverse events in 35% of patients. No treatment-related deaths. Common adverse events: nausea (69%), fatigue (54%), appetite loss (38%). Grade 3–4: respiratory infections (12%), anemia (12%). Dose reductions in 35% of patients. |
| Abemaciclib Study | Abemaciclib (CDK4/6 inhibitor), Cisplatin, Pemetrexed | Ib | NCT02079636 | Not provided | Study assessed safety and efficacy of abemaciclib in combination therapy for advanced solid tumors, including NSCLC and mesothelioma. Well-tolerated with manageable adverse events. |
| CheckMate 743 | Nivolumab, Ipilimumab | III | NCT02899299 | Not provided | Median OS: 18.1 months (immunotherapy) vs. 14.1 months (chemotherapy). Non-epithelioid histology benefited most. PD-L1 expression had no impact. |
| DREAM3R | Durvalumab, Cisplatin/Carboplatin, Pemetrexed | III | NCT04334759 | Ongoing | OS is the primary endpoint. Secondary: PFS, ORR, QoL. Recruitment target: 480 patients. Awaiting results. |
| BEAT-Meso | Bevacizumab, Atezolizumab, Chemotherapy | III | NCT03762018 | Ongoing | OS is primary endpoint. Secondary: PFS, ORR, disease control at 24 weeks. 400 patients across 45 European clinics. |
| AtezoMeso | Atezolizumab vs. Placebo (adjuvant) | III | NCT04566637 | Maximum 12 months | Primary endpoint: Disease-free survival. Secondary: OS, QoL, safety. Therapy every 21 days until recurrence or toxicity. |
| eVOLVE-Meso | Volrustomig (PD-1/CTLA-4 bispecific mAb), Chemotherapy, Nivolumab, Ipilimumab | III | NCT06097728 | Expected results by 2028 | OS is the primary outcome. Ongoing study. |
| INFINITE | Adenovirus-delivered interferon-alpha-2b, Celecoxib, Gemcitabine | III | NCT03710876 | Expected results by 2024 | OS is the primary endpoint. Intrapleural therapy stimulates immune system response. Awaiting final results. |
| DENIM | Dendritic cell immunization (tumor lysate-loaded autologous cells) | III | NCT03610360 | Not provided | OS is primary endpoint. Secondary: ORR, PFS, 18-month survival rate. Dendritic cell therapy enhances immune response. |
| ATOMIC-Meso | Pegargiminase (ADI-PEG20), Chemotherapy | III | NCT02709512 | Not provided | OS: 9.3 months (pegargiminase + chemo) vs. 7.7 months (chemo alone). PFS improved. |
| LUME-Meso | Nintedanib, Pemetrexed, Cisplatin | III | NCT01907100 | Not provided | Improved PFS but OS benefit limited. Further studies recommended. |
| PROMISE-Meso | Pembrolizumab vs. Chemotherapy | III | NCT02991482 | Not provided | No significant OS improvement, but higher disease control rate. |
| MesomiR 1 | TargomiRs (microRNA-based therapy) | I | NCT02369198 | Not provided | Some patients showed tumor size reduction. |
| HITOC | Hyperthermic Intrathoracic Chemoperfusion (HITOC) with Cisplatin | Not provided | NCT05508555 | Not provided | OS: 28 months (HITOC) vs. 22 months (surgery alone). PFS: 8 months (HITOC) vs. 6 months (surgery alone). HITOC safe with manageable complications. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertuccio, F.R.; Montini, S.; Fusco, M.A.; Di Gennaro, A.; Sciandrone, G.; Agustoni, F.; Galli, G.; Bortolotto, C.; Saddi, J.; Baietto, G.; et al. Malignant Pleural Mesothelioma: From Pathophysiology to Innovative Actionable Targets. Cancers 2025, 17, 1160. https://doi.org/10.3390/cancers17071160
Bertuccio FR, Montini S, Fusco MA, Di Gennaro A, Sciandrone G, Agustoni F, Galli G, Bortolotto C, Saddi J, Baietto G, et al. Malignant Pleural Mesothelioma: From Pathophysiology to Innovative Actionable Targets. Cancers. 2025; 17(7):1160. https://doi.org/10.3390/cancers17071160
Chicago/Turabian StyleBertuccio, Francesco Rocco, Simone Montini, Maria Antonietta Fusco, Antonella Di Gennaro, Gaetano Sciandrone, Francesco Agustoni, Giulia Galli, Chandra Bortolotto, Jessica Saddi, Guido Baietto, and et al. 2025. "Malignant Pleural Mesothelioma: From Pathophysiology to Innovative Actionable Targets" Cancers 17, no. 7: 1160. https://doi.org/10.3390/cancers17071160
APA StyleBertuccio, F. R., Montini, S., Fusco, M. A., Di Gennaro, A., Sciandrone, G., Agustoni, F., Galli, G., Bortolotto, C., Saddi, J., Baietto, G., Melloni, G., D’Ambrosio, G., Corsico, A. G., & Stella, G. M. (2025). Malignant Pleural Mesothelioma: From Pathophysiology to Innovative Actionable Targets. Cancers, 17(7), 1160. https://doi.org/10.3390/cancers17071160