

Effect of Tasurgratinib as an Orally Available FGFR1–3 Inhibitor on Resistance to a CDK4/6 Inhibitor and Endocrine Therapy in ER+/HER2− Breast Cancer Preclinical Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Test Agents
2.2. Animal Studies
- dT/C =
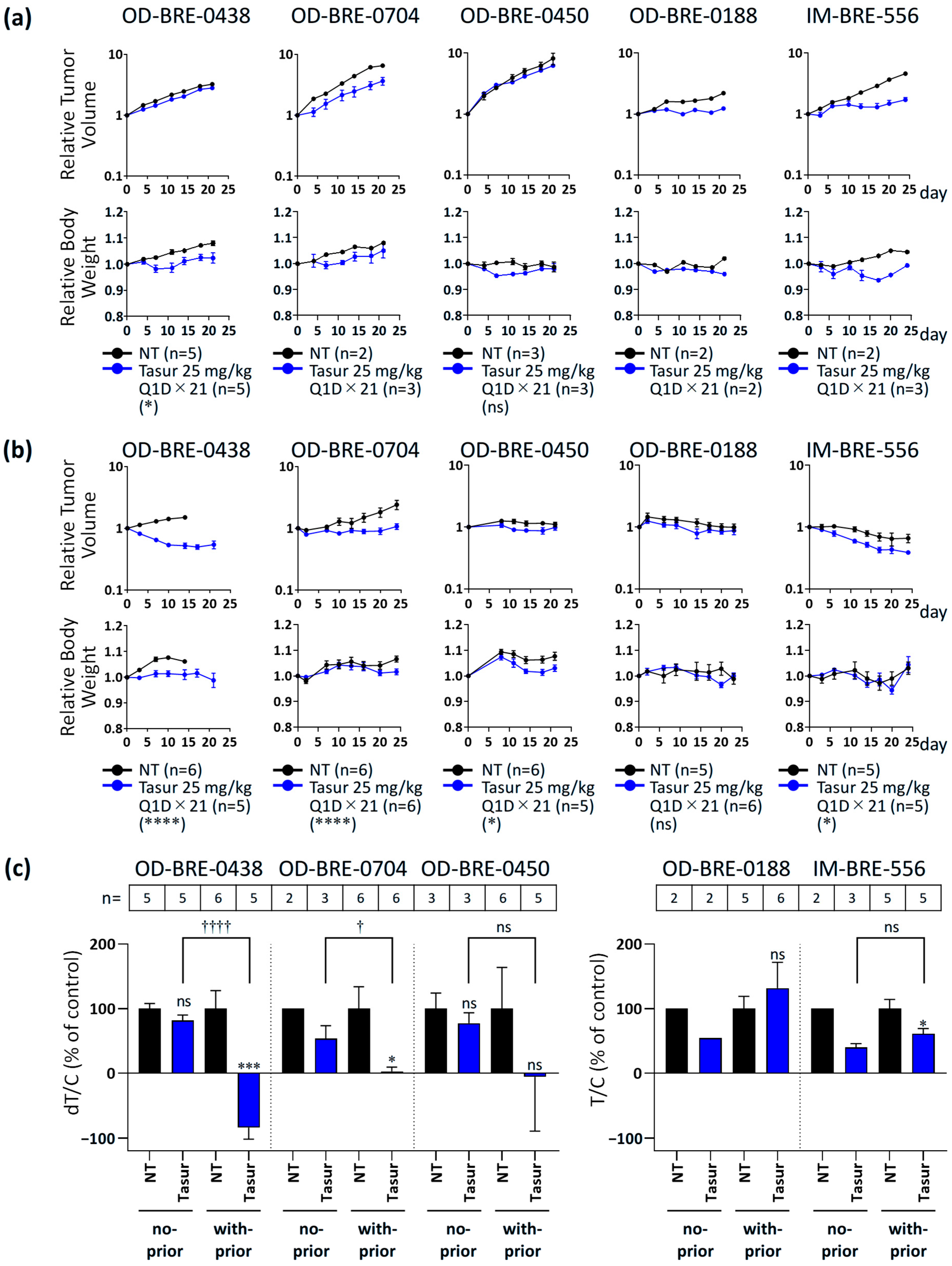
- T/C =where Day X means Day 14 for the OD-BRE-0438 model in the with-prior-treatment cohort; Day 21 for the OD-BRE-0438 model in the no-prior-treatment cohort, the OD-BRE-0704 model in the no-prior-treatment cohort, the OD-BRE-0450 model in both the no-prior-treatment cohort and with-prior-treatment cohort, and the OD-BRE-0188 model in the no-prior-treatment cohort; Day 23 for the OD-BRE-0188 model in the with-prior-treatment cohort; and Day 24 for the OD-BRE-0704 model in the with-prior-treatment cohort and the IM-BRE-556 model in both the no-prior-treatment cohort and with-prior-treatment cohort. The days were selected because of the first measurement after the end of the administration period or the final measurement in the control group. The values of T/C (% of control) were calculated for the OD-BRE-0188 and IM-BRE-556 models because the control groups for both models showed tumor regression after prior palbociclib + fulvestrant treatment, and the calculation of dT/C (% of control) for tumor growth was not applicable in such cases. The median PFS was assessed as the median time to a 1.5-fold increase in tumor volume from treatment initiation.
2.3. Expression Analyses
2.4. In Vitro Antiproliferation Assay
2.5. Statistical Analyses
3. Results
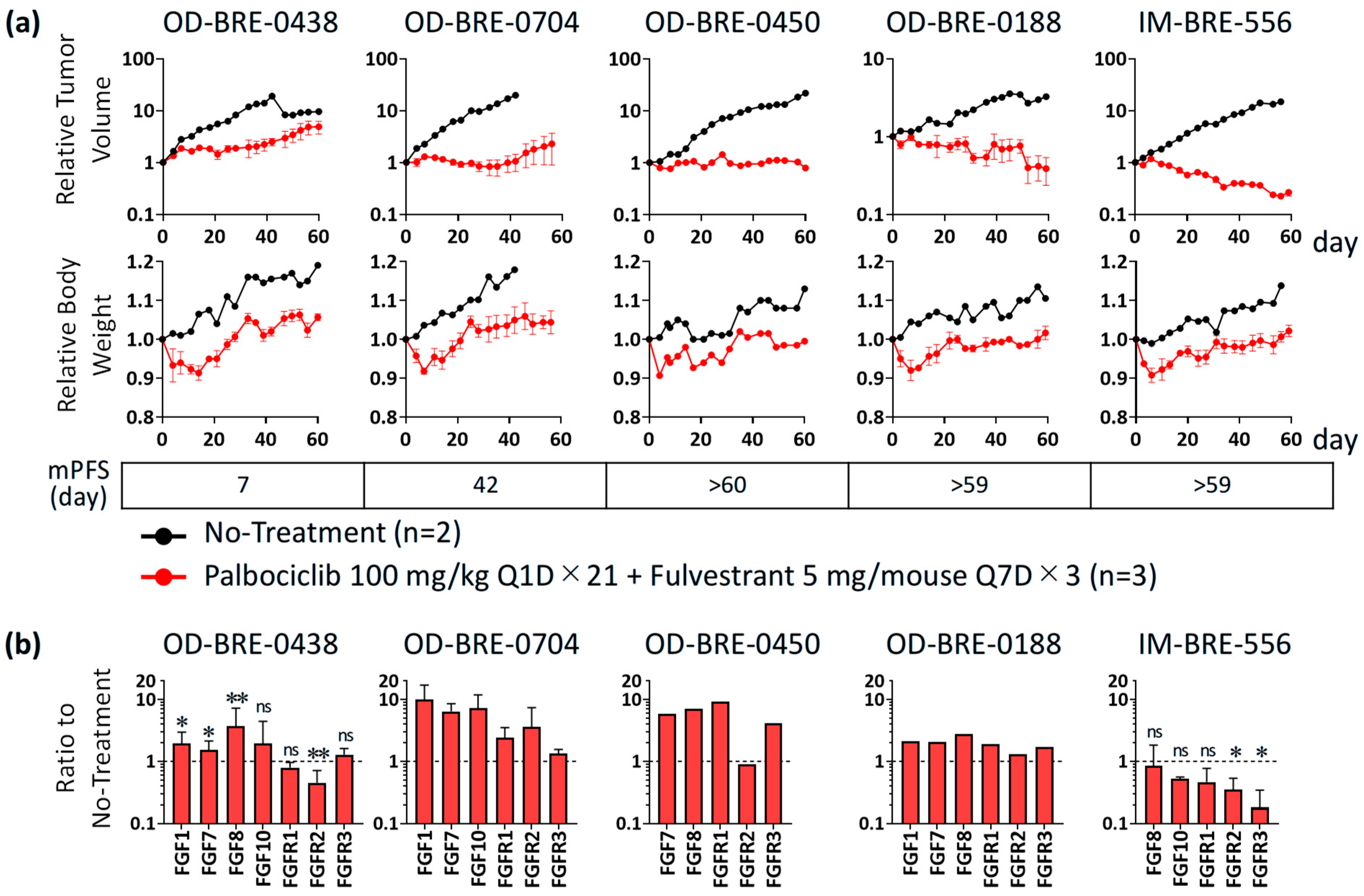
3.1. Antitumor Activity and Alterations of FGF/FGFR Expression with Palbociclib + Fulvestrant Treatments in ER+ Breast Cancer PDX Models
3.2. Antitumor Activity of Tasurgratinib with or Without Prior Palbociclib + Fulvestrant Treatment in ER+ Breast Cancer PDX Models
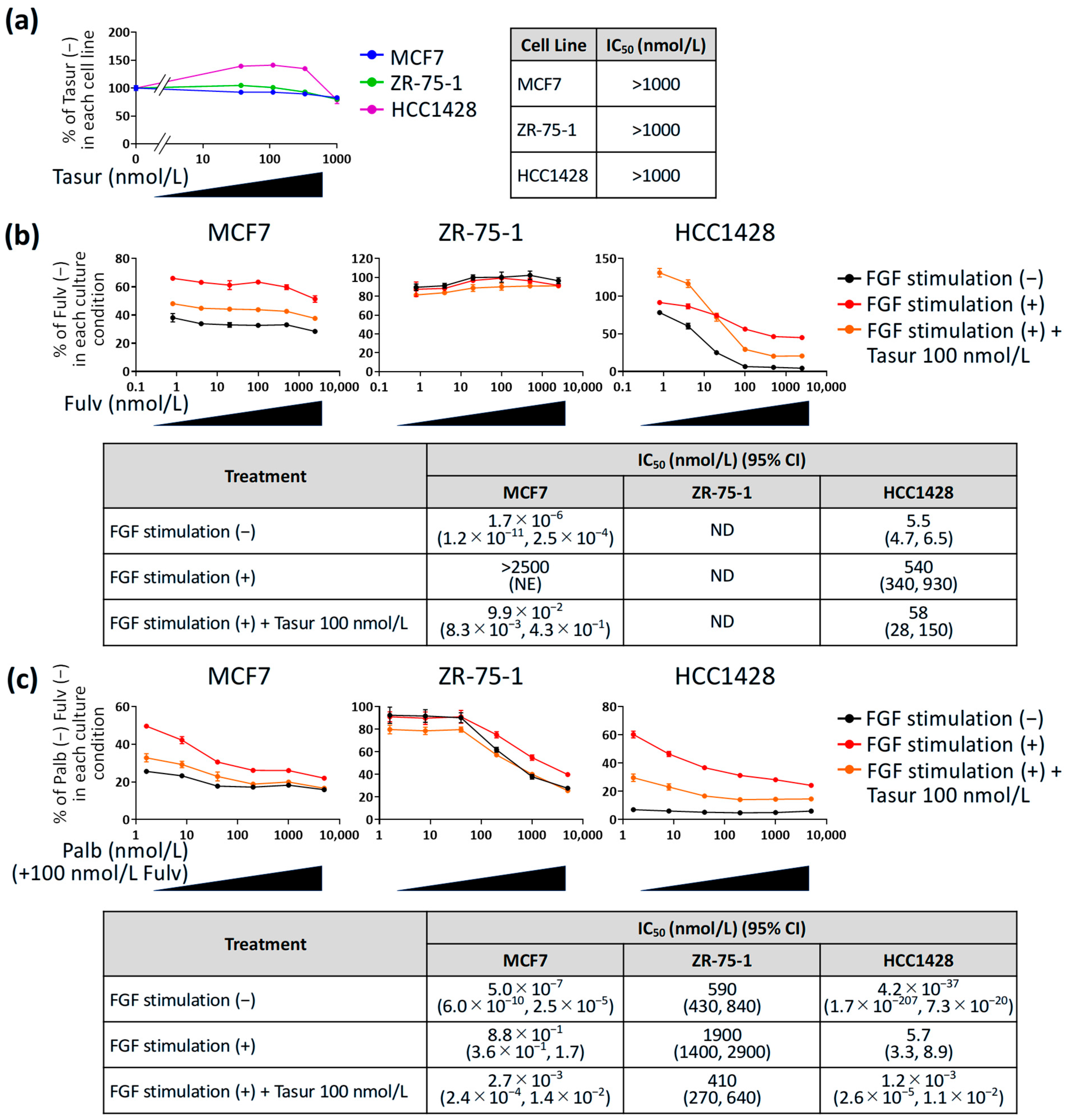
3.3. Effect of Tasurgratinib on Anti-Proliferative Activity of Fulvestrant or Palbociclib + Fulvestrant Against ER+ Breast Cancer Cell Lines with FGF Stimulation In Vitro
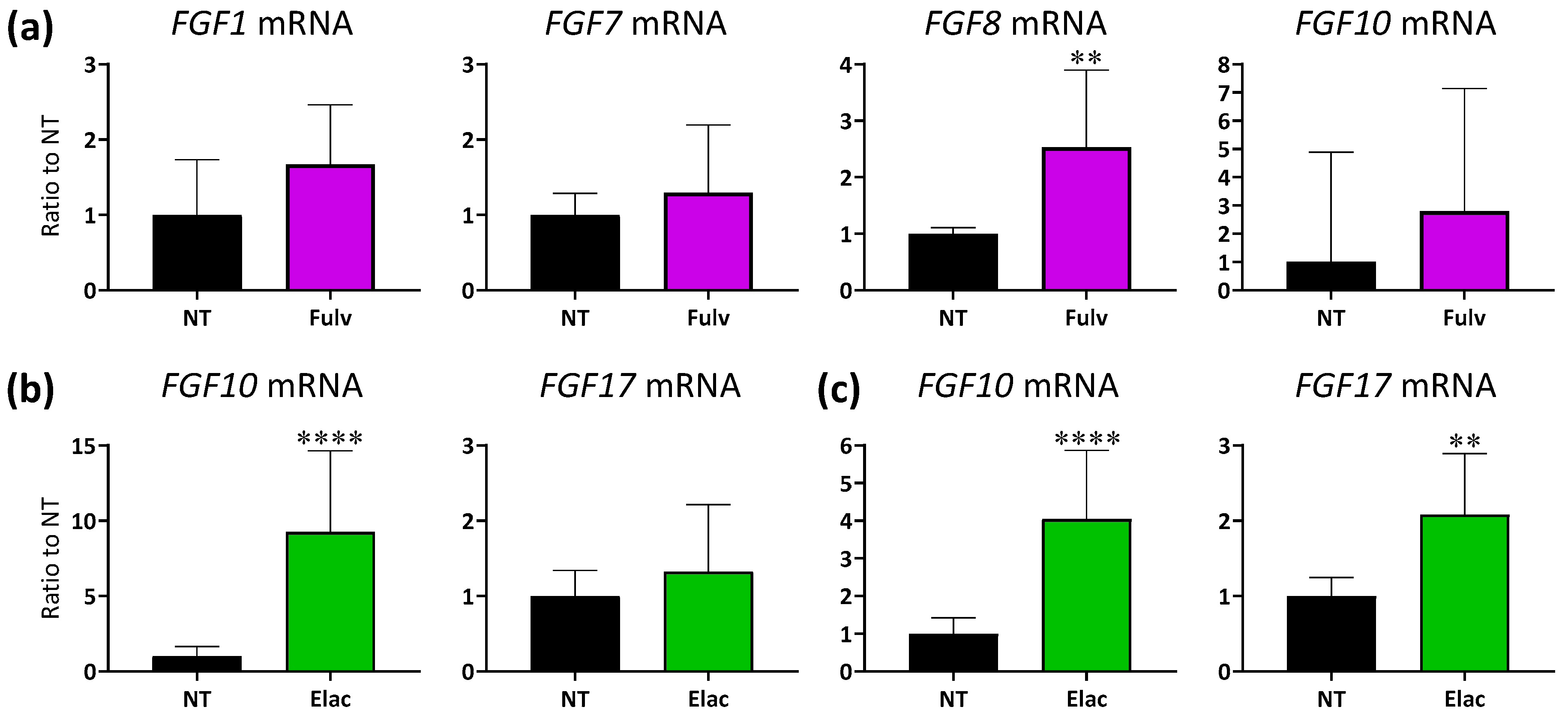
3.4. Expression Levels of FGF Ligand mRNA in ER+ Breast Cancer PDX Tumors with ET Treatment
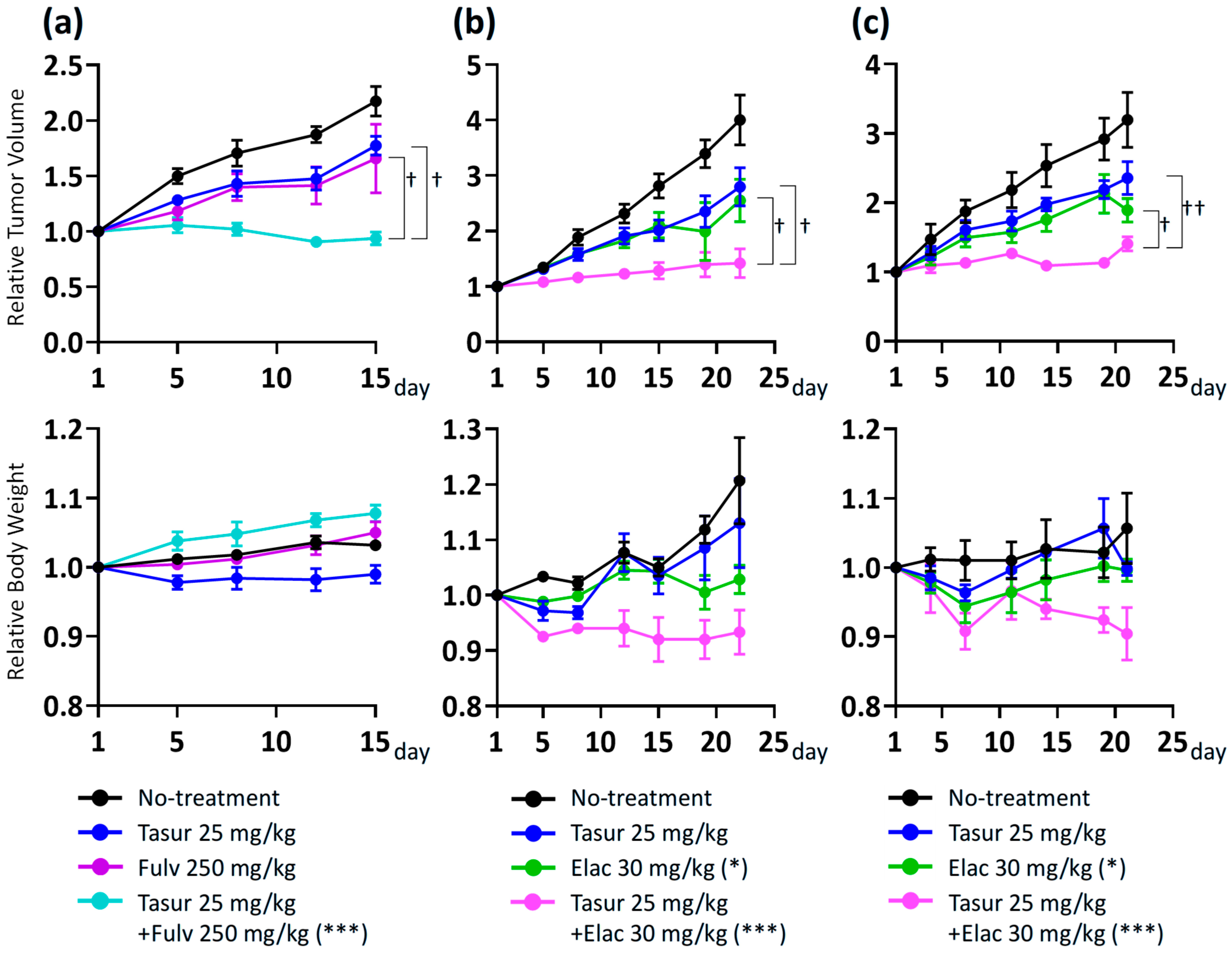
3.5. Combination Activity of Tasurgratinib on Fulvestrant or Elacestrant in ER+ Breast Cancer PDX Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Surveillance, Epidemiology and End Results Program. Available online: https://seer.cancer.gov/statfacts/html/breast-subtypes.html (accessed on 18 December 2024).
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.-A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S.; et al. MONARCH 3 final PFS: A randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 2019, 5, 5. [Google Scholar] [CrossRef]
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor–Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 4760–4766. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, W. Selective estrogen receptor degraders (SERDs): A promising strategy for estrogen receptor positive endocrine-resistant breast cancer. J. Med. Chem. 2020, 63, 15094–15114. [Google Scholar] [CrossRef]
- Agostini, M.; Mandrioli, A.; Zamagni, C. Fulvestrant monotherapy after CDK4/6 inhibitors in metastatic breast cancer patients: A real-life experience. Cancers 2024, 16, 4179. [Google Scholar] [CrossRef] [PubMed]
- Lambert, V.; Kane, S.; Howidi, B.; Nguyen, B.-N.; Chandiwana, D.; Wu, Y.; Edwards, M.; Samjoo, I.A. Systematic literature review of real-world evidence for treatments in HR+/HER2- second-line LABC/mBC after first-line treatment with CDK4/6i. BMC Cancer 2024, 24, 631. [Google Scholar] [CrossRef]
- Mittal, A.; Molto Valiente, C.; Tamimi, F.; Schlam, I.; Sammons, S.; Tolaney, S.M.; Tarantino, P. Filling the gap after CDK4/6 inhibitors: Novel endocrine and biologic treatment options for metastatic hormone receptor positive breast cancer. Cancers 2023, 15, 2015. [Google Scholar] [CrossRef]
- Neupane, N.; Bawek, S.; Gurusinghe, S.; Ghaffary, E.M.; Mirmosayyeb, O.; Thapa, S.; Falkson, C.; O’Regan, R.; Dhakal, A. Oral SERD, a novel endocrine therapy for estrogen receptor-positive breast cancer. Cancers 2024, 16, 619. [Google Scholar] [CrossRef]
- Hoy, S.M. Elacestrant: First Approval. Drugs 2023, 83, 555–561. [Google Scholar] [CrossRef]
- Liu, Q.; Huang, J.; Yan, W.; Liu, Z.; Liu, S.; Fang, W. FGFR families: Biological functions and therapeutic interventions in tumors. MedComm 2023, 4, e367. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Cohen, O.; Kowalski, K.J.; Kusiel, J.; Buendia-Buendia, J.E.; Cuoco, M.S.; Exman, P.; Wander, S.A.; Waks, A.G.; Nayar, U.; et al. Acquired FGFR and FGF alterations confer resistance to estrogen receptor (ER) targeted therapy in ER+ metastatic breast cancer. Clin. Cancer Res. 2020, 26, 5974–5989. [Google Scholar] [CrossRef]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Solovieff, N.; Su, F.; Bardia, A.; Neven, P.; Yap, Y.S.; Tripathy, D.; Lu, Y.S.; Slamon, D.; Chia, S.; et al. Acquired gene alterations in patients treated with ribociclib plus endocrine therapy or endocrine therapy alone using baseline and end-of-treatment circulating tumor DNA samples in the MONALEESA-2, -3, and -7 trials. Ann. Oncol. 2025, 36, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Ikemori-Kawada, M.; Jestel, A.; von König, K.; Funahashi, Y.; Matsushima, T.; Tsuruoka, A.; Inoue, A.; Matsui, J. Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med. Chem. Lett. 2015, 6, 89–94. [Google Scholar] [CrossRef]
- Watanabe Miyano, S.; Yamamoto, Y.; Kodama, K.; Miyajima, Y.; Mikamoto, M.; Nakagawa, T.; Kuramochi, H.; Funasaka, S.; Nagao, S.; Sugi, N.H.; et al. E7090, a novel selective inhibitor of fibroblast growth factor receptors, displays potent antitumor activity and prolongs survival in preclinical models. Mol. Cancer Ther. 2016, 15, 2630–2639. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Kawada, M.I.; Fukushima, S.; Arai, Y.; Shibata, T.; Miyano, S.W. Antitumor activity of tasurgratinib as an orally available FGFR1-3 inhibitor in cholangiocarcinoma models with FGFR2-fusion. Anticancer Res. 2024, 44, 2393. [Google Scholar] [CrossRef]
- Furuse, J.; Jiang, B.; Kuwahara, T.; Satoh, T.; Ma, X.; Yan, S.; Zhao, H.-T.; Ikeda, M.; Cui, T.; Sasaki, T.; et al. Pivotal single-arm, phase 2 trial of tasurgratinib for patients with fibroblast growth factor receptor (FGFR)-2 gene fusion-positive cholangiocarcinoma (CCA). J. Clin. Oncol. 2024, 42, 471. [Google Scholar] [CrossRef]
- Matsumura, N.; Mandai, M. PMDA regulatory update on approval and revision of the precautions for use of anticancer drugs; approval of sacituzumab govitecan for breast cancer, fruquintinib for colorectal cancer, amivantamab for lung cancer, repotrectinib for lung cancer, tasurgratinib for biliary tract cancer, enfortumab vedotin plus pembrolizumab for urothelial cancer, and dabrafenib plus trametinib for glioma in Japan. Int. J. Clin. Oncol. 2024, 29, 1775–1776. [Google Scholar] [CrossRef]
- Kogawa, T.; Yamashita, T.; Ishida, T.; Yonemori, K.; Iwata, H.; Shimomura, A.; Aogi, K.; Inoue, K.; Mukohara, T.; Nakayama, T.; et al. Phase Ib trial of tasurgratinib (E7090) with or without endocrine therapies for patients (pts) with ER+, HER2− recurrent/metastatic breast cancer (BC) after receiving a CDK4/6 inhibitor. J. Clin. Oncol. 2024, 42, 3103. [Google Scholar] [CrossRef]
- Khatpe, A.S.; Adebayo, A.K.; Herodotou, C.A.; Kumar, B.; Nakshatri, H. Nexus between PI3K/AKT and Estrogen Receptor Signaling in Breast Cancer. Cancers 2021, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Vives-Usano, M.; García Pelaez, B.; Román Lladó, R.; Garzón Ibañez, M.; Aldeguer, E.; Rodriguez, S.; Aguilar, A.; Pons, F.; Viteri, S.; Cabrera, C.; et al. Analysis of copy number variations in solid tumors using a next generation sequencing custom panel. J. Mol. Pathol. 2021, 2, 123–134. [Google Scholar] [CrossRef]
- Zingg, D.; Bhin, J.; Yemelyanenko, J.; Kas, S.M.; Rolfs, F.; Lutz, C.; Lee, J.K.; Klarenbeek, S.; Silverman, I.M.; Annunziato, S.; et al. Truncated FGFR2 is a clinically actionable oncogene in multiple cancers. Nature 2022, 608, 609–617. [Google Scholar] [CrossRef]
- Lazo De La Vega, L.; Comeau, H.; Sallan, S.; Al-Ibraheemi, A.; Gupta, H.; Li, Y.Y.; Tsai, H.K.; Kang, W.; Ward, A.; Church, A.J.; et al. Rare FGFR oncogenic alterations in sequenced pediatric solid and brain tumors suggest FGFR is a relevant molecular target in childhood cancer. JCO Precis. Oncol. 2022, 6, e2200390. [Google Scholar] [CrossRef]
- Turczyk, L.; Kitowska, K.; Mieszkowska, M.; Mieczkowski, K.; Czaplinska, D.; Piasecka, D.; Kordek, R.; Skladanowski, A.C.; Potemski, P.; Romanska, H.M.; et al. FGFR2-driven signaling counteracts tamoxifen effect on ERα-positive breast cancer cells. Neoplasia 2017, 19, 791–804. [Google Scholar] [CrossRef]
- Formisano, L.; Stauffer, K.M.; Young, C.D.; Bhola, N.E.; Guerrero-Zotano, A.L.; Jansen, V.M.; Estrada, M.M.; Hutchinson, K.E.; Giltnane, J.M.; Schwarz, L.J.; et al. Association of FGFR1 with ERα maintains ligand-independent ER transcription and mediates resistance to estrogen deprivation in ER+ breast cancer. Clin. Cancer Res. 2017, 23, 6138–6150. [Google Scholar] [CrossRef] [PubMed]
- Haines, E.; Chen, T.; Kommajosyula, N.; Chen, Z.; Herter-Sprie, G.S.; Cornell, L.; Wong, K.-K.; Shapiro, G.I. Palbociclib resistance confers dependence on an FGFR-MAP kinase-mTOR-driven pathway in KRAS -mutant non-small cell lung cancer. Oncotarget 2018, 9, 31572. [Google Scholar] [CrossRef]
- Sobhani, N.; Fassl, A.; Mondani, G.; Generali, D.; Otto, T. Targeting aberrant FGFR signaling to overcome CDK4/6 inhibitor resistance in breast cancer. Cells 2021, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.-C.; Kaklamani, V.G.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.-A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; et al. Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor–positive, human epidermal growth factor receptor 2–negative advanced breast cancer: Results from the randomized phase III EMERALD trial. J. Clin. Oncol. 2022, 40, 3246–3256. [Google Scholar] [CrossRef]
- Coombes, R.C.; Badman, P.D.; Lozano-Kuehne, J.P.; Liu, X.; Macpherson, I.R.; Zubairi, I.; Baird, R.D.; Rosenfeld, N.; Garcia-Corbacho, J.; Cresti, N.; et al. Results of the phase IIa RADICAL trial of the FGFR inhibitor AZD4547 in endocrine resistant breast cancer. Nat. Commun. 2022, 13, 3246. [Google Scholar] [CrossRef]
- Gong, J.; Mita, A.C.; Wei, Z.; Cheng, H.H.; Mitchell, E.P.; Wright, J.J.; Ivy, S.P.; Wang, V.; Gray, R.C.; McShane, L.M.; et al. Phase II Study of Erdafitinib in Patients With Tumors With FGFR Amplifications: Results From the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol K1. JCO Precis. Oncol. 2024, 8, e2300406. [Google Scholar] [CrossRef]
- Kaminska, K.; Akrap, N.; Staaf, J.; Alves, C.L.; Ehinger, A.; Ebbesson, A.; Hedenfalk, I.; Beumers, L.; Veerla, S.; Harbst, K.; et al. Distinct mechanisms of resistance to fulvestrant treatment dictate level of ER independence and selective response to CDK inhibitors in metastatic breast cancer. Breast Cancer Res. 2021, 23, 26. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Approves Elacestrant for ER-Positive, HER2-Negative, ESR1-Mutated Advanced or Metastatic Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-elacestrant-er-positive-her2-negative-esr1-mutated-advanced-or-metastatic-breast-cancer (accessed on 14 January 2025).
- Jhaveri, K.L.; Neven, P.; Casalnuovo, M.L.; Kim, S.-B.; Tokunaga, E.; Aftimos, P.; Saura, C.; O’Shaughnessy, J.; Harbeck, N.; Carey, L.A.; et al. Imlunestrant with or without Abemaciclib in advanced breast cancer. N. Engl. J. Med. 2024. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Abreu, C.; Costa, L.; Casimiro, S. The evolving pathways of the efficacy of and resistance to CDK4/6 inhibitors in breast cancer. Cancers 2023, 15, 4835. [Google Scholar] [CrossRef]
- Raheem, F.; Karikalan, S.A.; Batalini, F.; El Masry, A.; Mina, L. Metastatic ER+ breast cancer: Mechanisms of resistance and future therapeutic approaches. Int. J. Mol. Sci. 2023, 24, 16198. [Google Scholar] [CrossRef]
- Yamashita, T.; Kogawa, T.; Ishida, T.; Yonemori, K.; Kataoka, A.; Shimomura, A.; Aogi, K.; Takai, K.; Mukohara, T.; Nakayama, T.; et al. Potential predictive biomarkers for a fibroblast growth factor receptor (FGFR) inhibitor: Phase 1b trial of tasurgratinib (E7090) with endocrine therapies (ET) for ER+, HER2+ recurrent/metastatic breast cancer (BC) resistant to CDK4/6 inhibitors. In Proceedings of the San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 10–14 December 2024. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawano, S.; Fukushima, S.; Nishibata, K.; Gejima, R.; Miyano, S.W. Effect of Tasurgratinib as an Orally Available FGFR1–3 Inhibitor on Resistance to a CDK4/6 Inhibitor and Endocrine Therapy in ER+/HER2− Breast Cancer Preclinical Models. Cancers 2025, 17, 1084. https://doi.org/10.3390/cancers17071084
Kawano S, Fukushima S, Nishibata K, Gejima R, Miyano SW. Effect of Tasurgratinib as an Orally Available FGFR1–3 Inhibitor on Resistance to a CDK4/6 Inhibitor and Endocrine Therapy in ER+/HER2− Breast Cancer Preclinical Models. Cancers. 2025; 17(7):1084. https://doi.org/10.3390/cancers17071084
Chicago/Turabian StyleKawano, Satoshi, Sayo Fukushima, Kyoko Nishibata, Ryu Gejima, and Saori Watanabe Miyano. 2025. "Effect of Tasurgratinib as an Orally Available FGFR1–3 Inhibitor on Resistance to a CDK4/6 Inhibitor and Endocrine Therapy in ER+/HER2− Breast Cancer Preclinical Models" Cancers 17, no. 7: 1084. https://doi.org/10.3390/cancers17071084
APA StyleKawano, S., Fukushima, S., Nishibata, K., Gejima, R., & Miyano, S. W. (2025). Effect of Tasurgratinib as an Orally Available FGFR1–3 Inhibitor on Resistance to a CDK4/6 Inhibitor and Endocrine Therapy in ER+/HER2− Breast Cancer Preclinical Models. Cancers, 17(7), 1084. https://doi.org/10.3390/cancers17071084