
Identification of Genomic Instability-Associated LncRNAs as Potential Therapeutic Targets in Lung Adenocarcinoma

 ,
,  and
and
Simple Summary
Abstract
1. Introduction
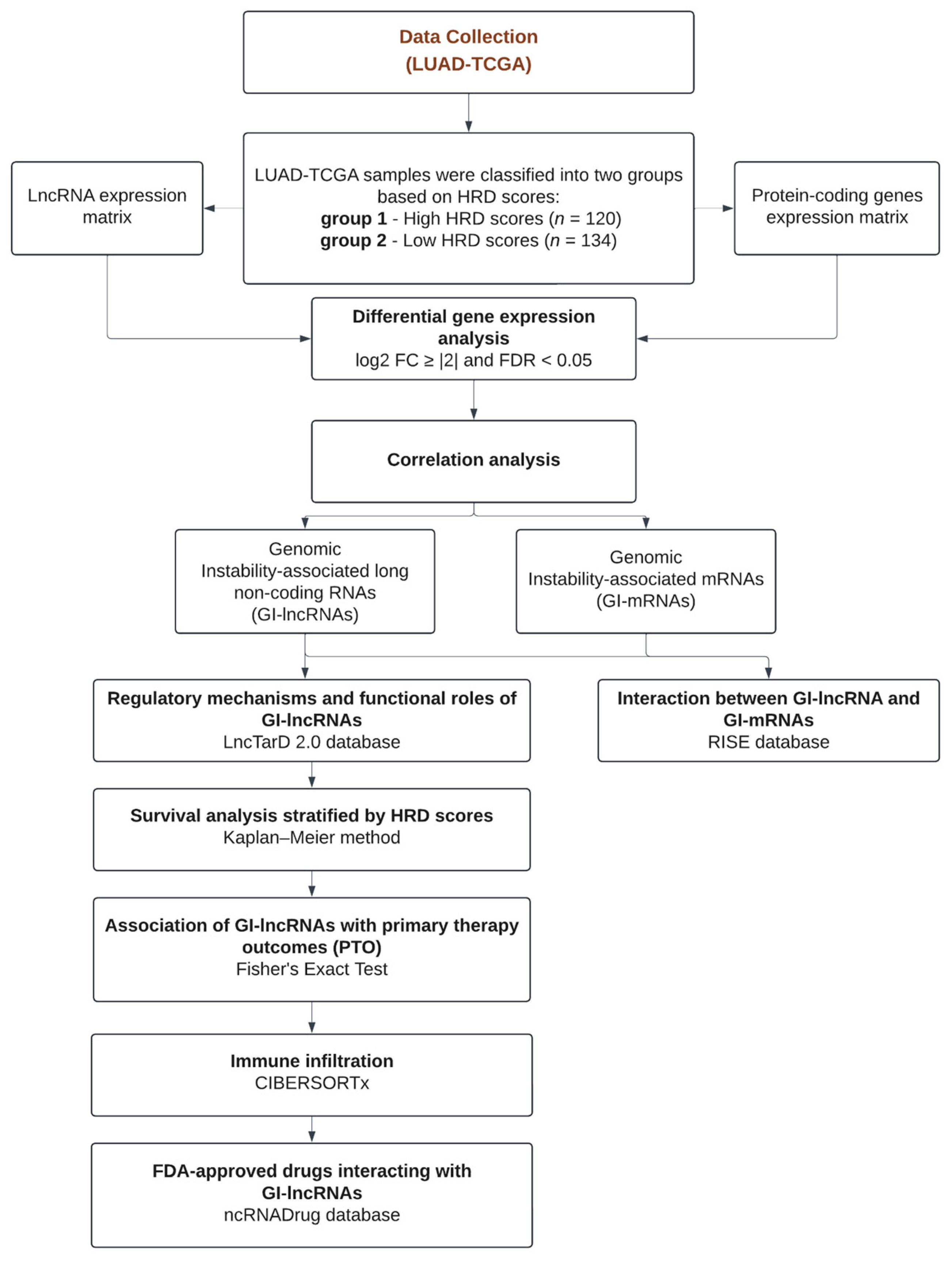
2. Materials and Methods
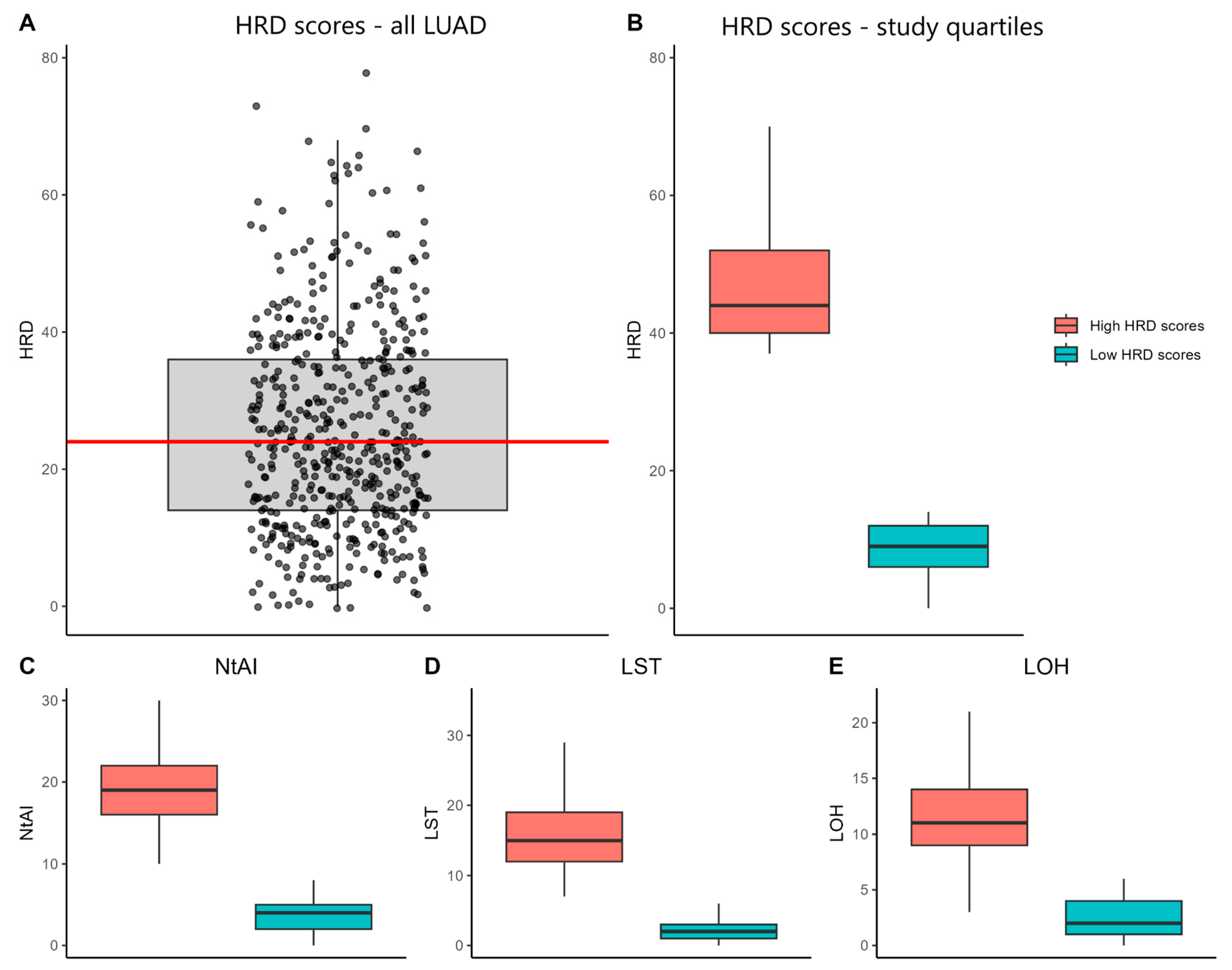
2.1. Data Collection and HRD Scores
2.2. Analysis of Gene Mutational Status and HRD Scores in Lung Adenocarcinoma
2.3. Identification of Genomic Instability-Associated Long Non-Coding RNAs (GI-lncRNAs) and mRNAs (GI-mRNAs), and Differentially Expressed miRNAs (DE-miRNAs)
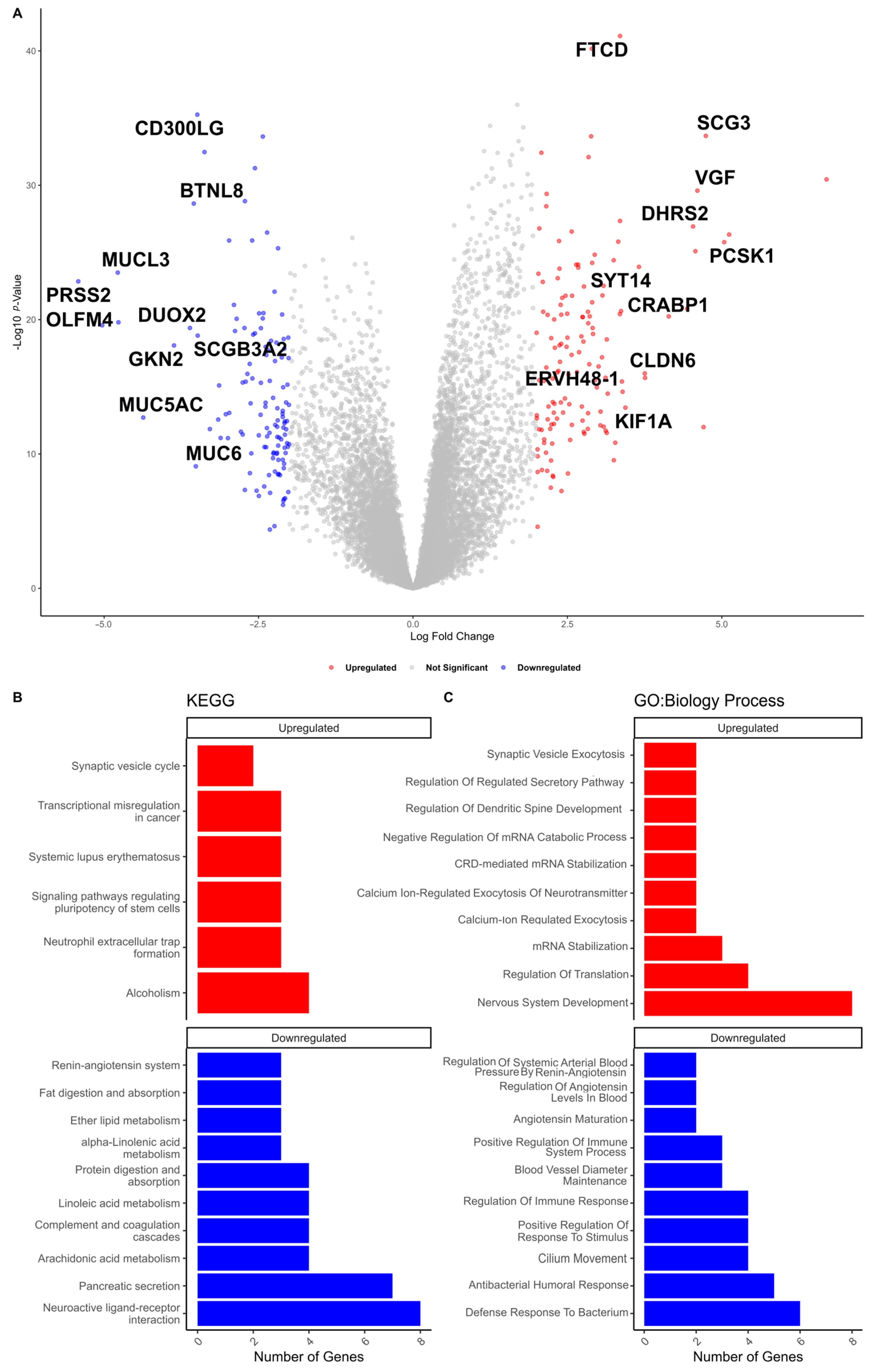
2.4. Pathway Enrichment Analysis
2.5. Interactions Between GI-lncRNA and GI-mRNAs, Along with the Regulatory Mechanisms and Functional Roles of GI-lncRNAs
2.6. Patient Survival Analysis
2.7. Patient Treatment Response
2.8. Immune-Related Analysis
2.9. Identification of Candidate Drugs Targeting GI-lncRNAs
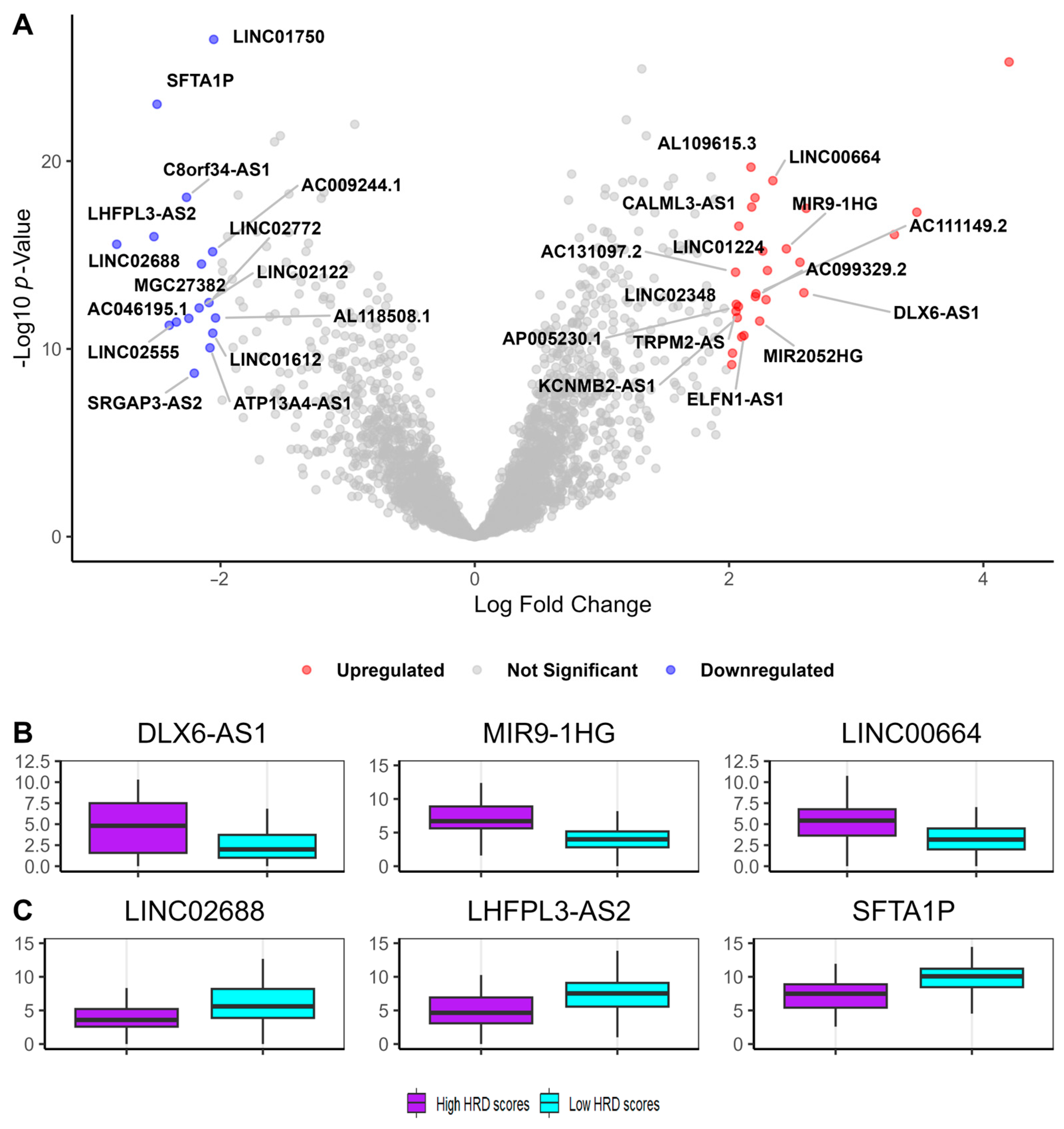
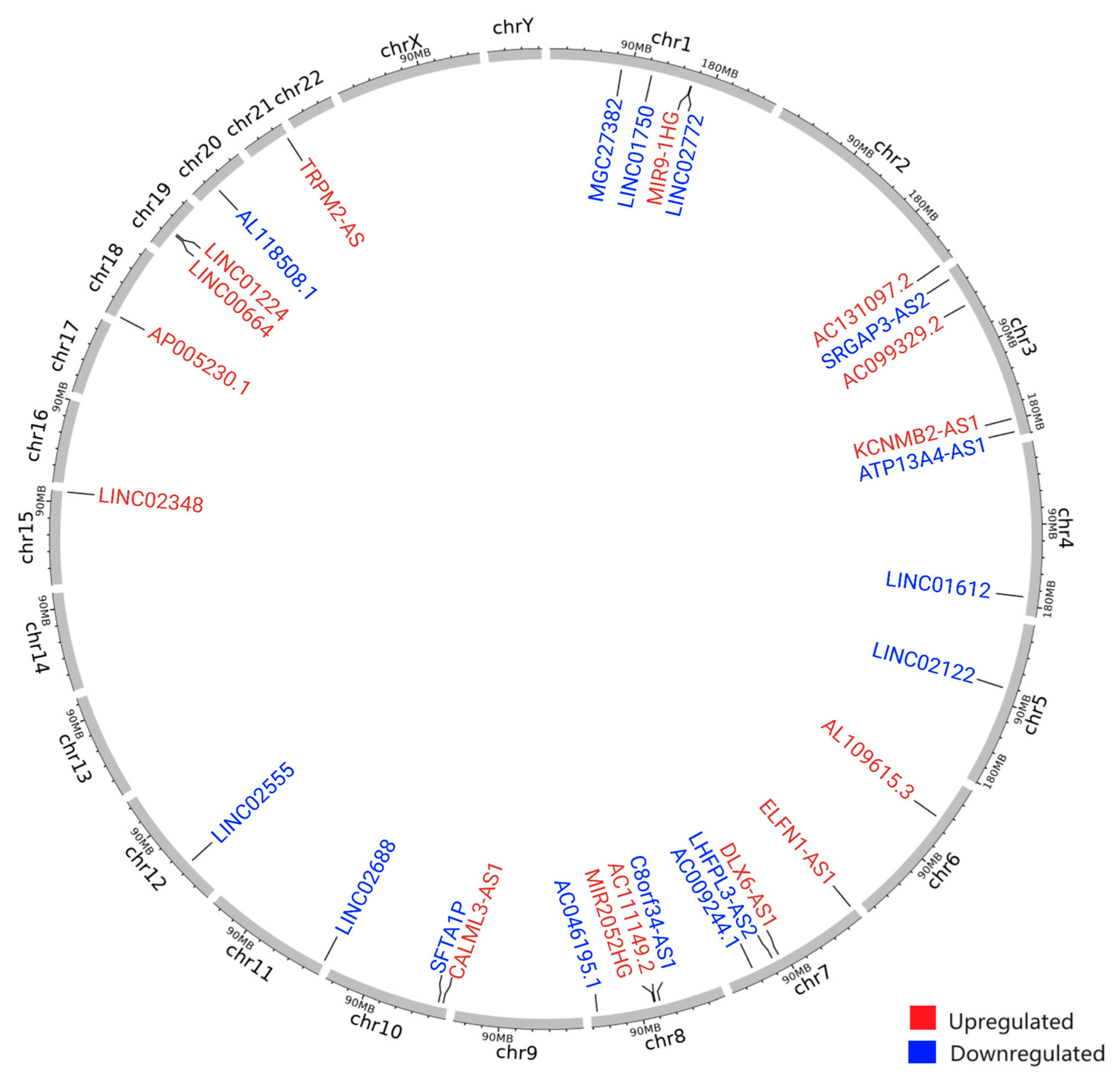
3. Results
3.1. Characterization of HRD Scores and Their Association with Clinicopathological Features in LUAD-TCGA Samples
3.2. Identification of Genomic Instability-Associated Long Non-Coding RNAs (GI-lncRNAs) in Lung Adenocarcinoma
3.3. Identification of Genomic Instability-Associated mRNAs (GI-mRNAs) in LUAD
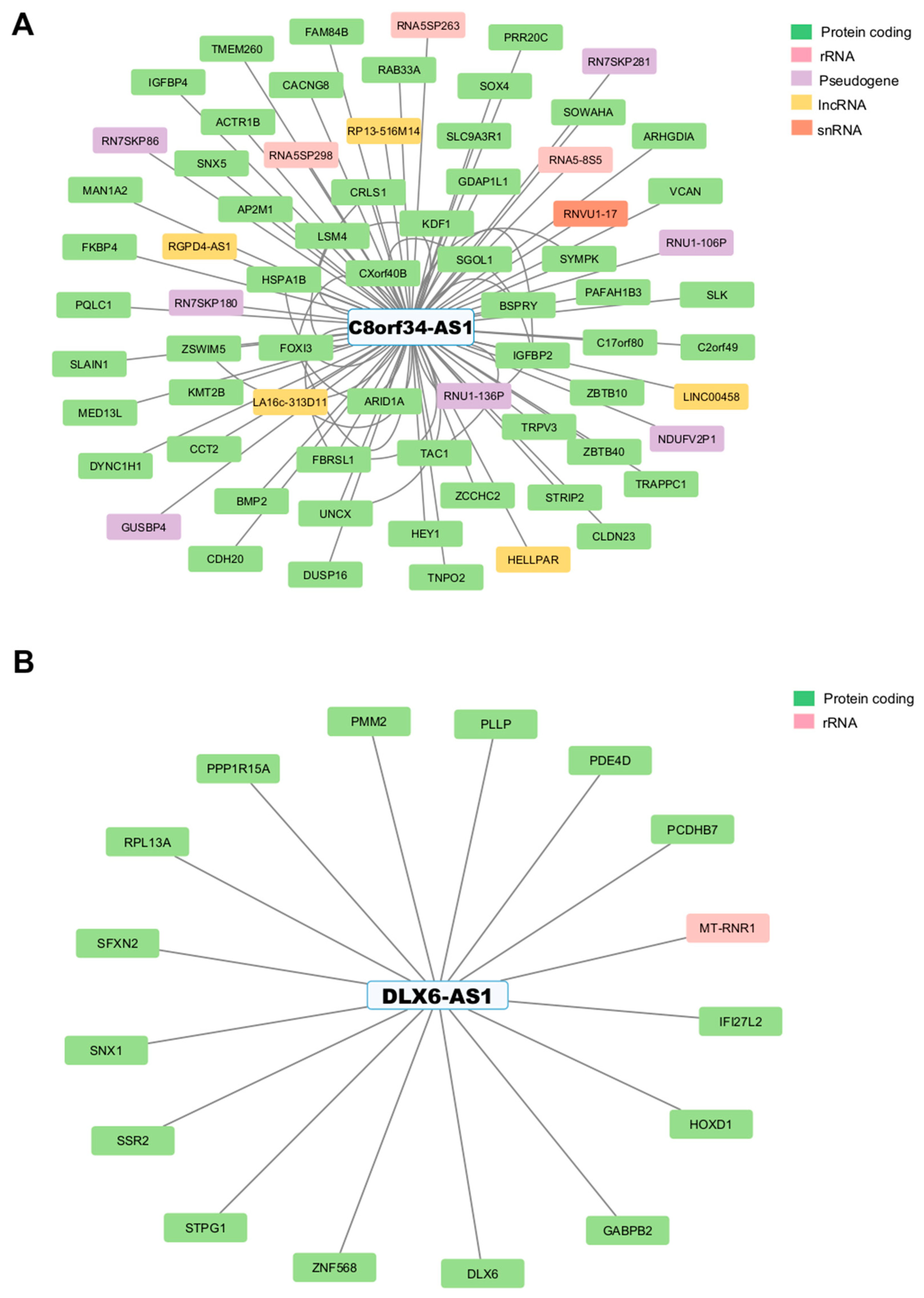
3.4. Identifying Interactions Between GI-lncRNAs and GI-mRNAs
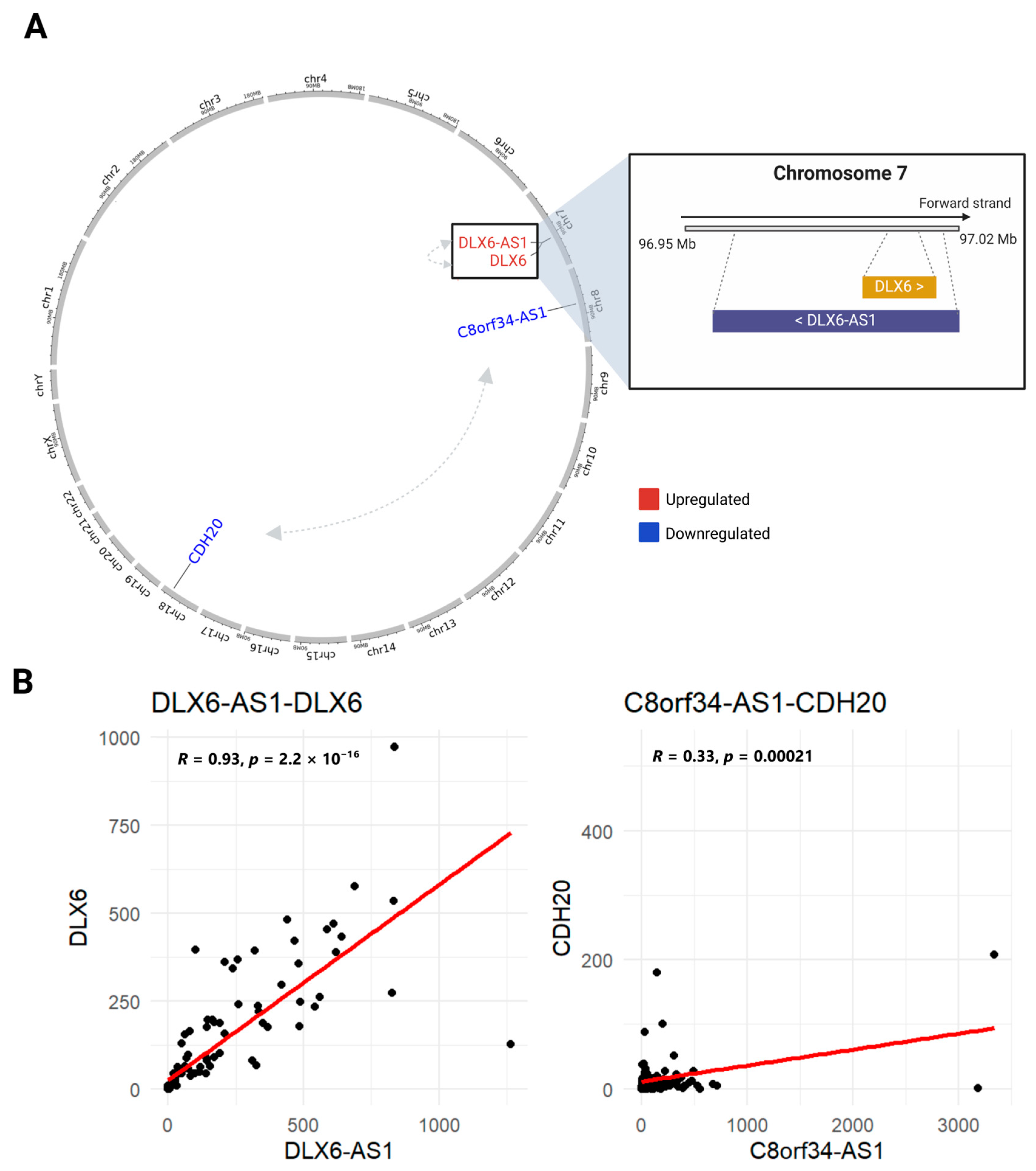
3.5. Regulatory Mechanisms and Functional Roles of GI-lncRNAs in LUAD and Other Diseases
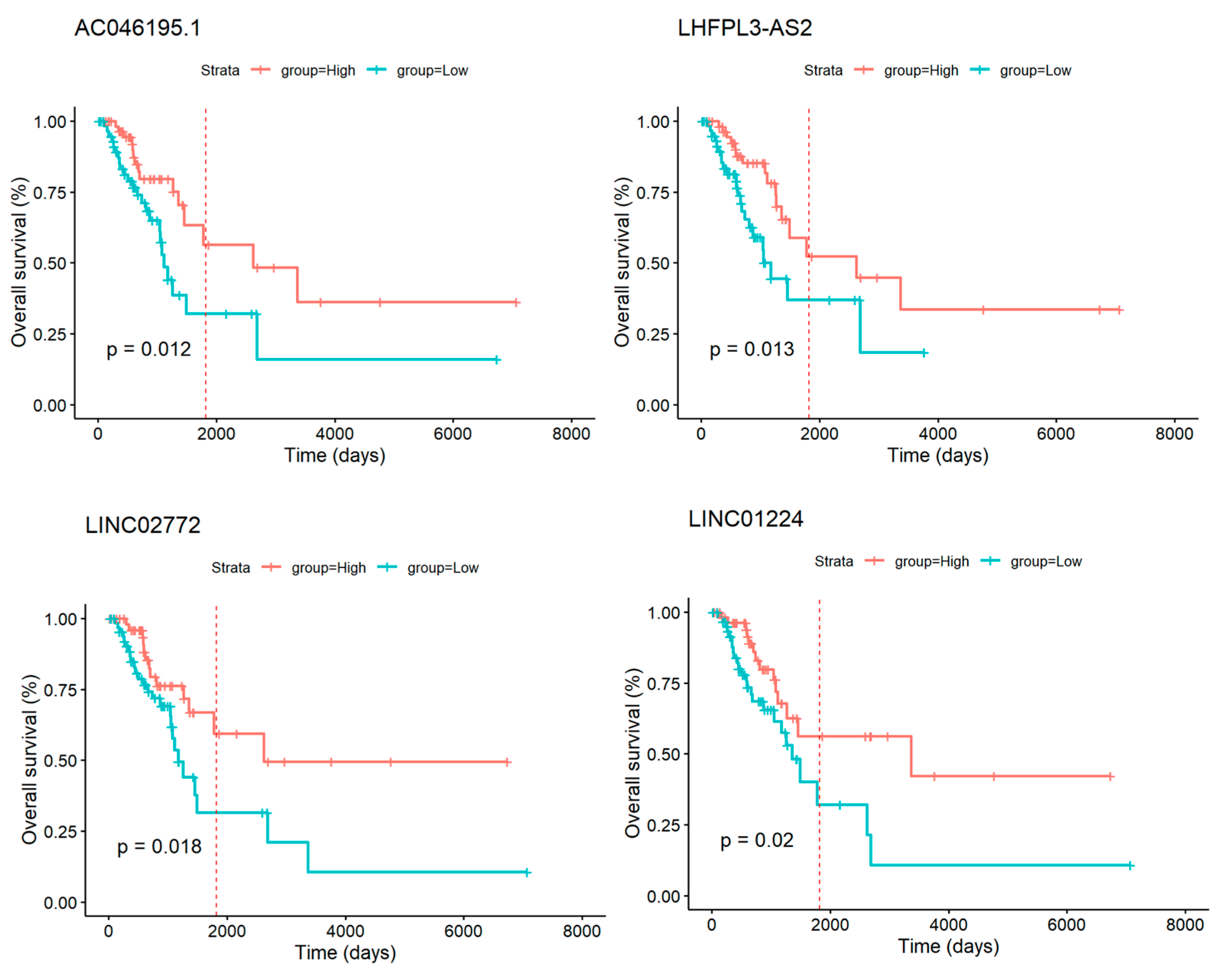
3.6. GI-lncRNAs as Potential Prognostic Biomarkers in LUAD
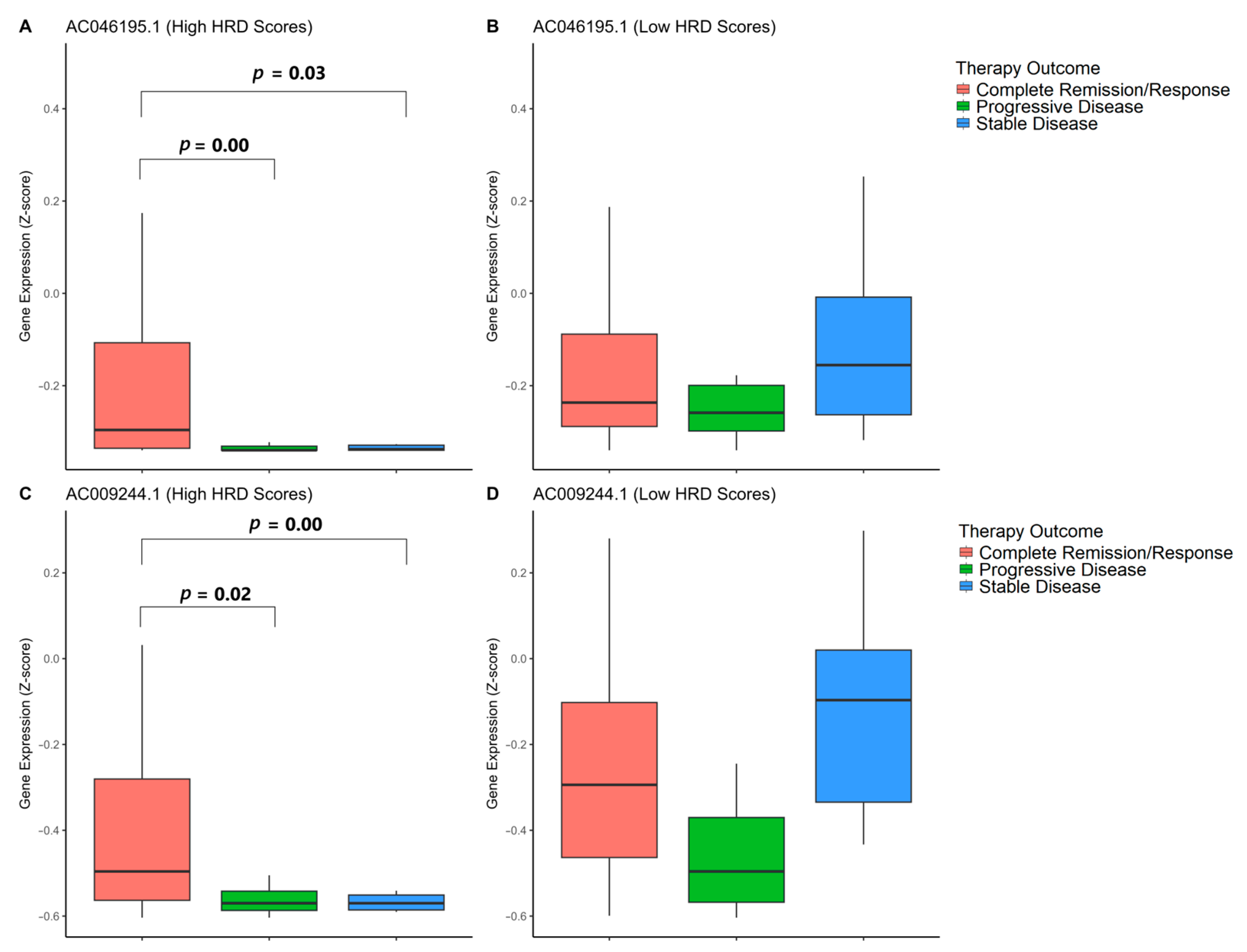
3.7. Association of GI-lncRNAs with Primary Therapy Outcomes
3.8. Analysis of Immune Cell Infiltration in Tumors with High and Low HRD Scores
3.9. Exploring Drugs Targeting GI-lncRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Agency for Research on Cancer (IARC) Lung Cancer Fact Sheet. Available online: https://gco.iarc.who.int/media/globocan/factsheets/cancers/15-trachea-bronchus-and-lung-fact-sheet.pdf (accessed on 7 March 2025).
- Society, A.C. Cancer Facts & Figures 2025. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2025/2025-cancer-facts-and-figures-acs.pdf (accessed on 7 March 2025).
- Tamura, T.; Kurishima, K.; Nakazawa, K.; Kagohashi, K.; Ishikawa, H.; Satoh, H.; Hizawa, N. Specific Organ Metastases and Survival in Metastatic Non-Small-Cell Lung Cancer. Mol. Clin. Oncol. 2015, 3, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Deng, J.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Analysis of Metastases in Non-Small Cell Lung Cancer Patients with Epidermal Growth Factor Receptor Mutation. Ann. Transl. Med. 2021, 9, 206. [Google Scholar] [CrossRef] [PubMed]
- Lung Cancer Survival Rates|5-Year Survival Rates for Lung Cancer. Available online: https://www.cancer.org/cancer/types/lung-cancer/detection-diagnosis-staging/survival-rates.html (accessed on 7 March 2025).
- Alexander, M.; Kim, S.Y.; Cheng, H. Update 2020: Management of Non-Small Cell Lung Cancer. Lung 2020, 198, 897–907. [Google Scholar] [CrossRef]
- Mountzios, G.; Naidoo, J.; Wang, C.; Creelan, B.C.; Trotier, D.C.; Campbell, T.C.; Peters, S. Beyond Chemoimmunotherapy in Advanced Non-Small Cell Lung Cancer: New Frontiers, New Challenges. Am. Soc. Clin. Oncol. Educ. Book 2024, 44, e432526. [Google Scholar] [CrossRef] [PubMed]
- Garassino, M.C.; Gadgeel, S.; Speranza, G.; Felip, E.; Esteban, E.; Dómine, M.; Hochmair, M.J.; Powell, S.F.; Bischoff, H.G.; Peled, N.; et al. Pembrolizumab Plus Pemetrexed and Platinum in Nonsquamous Non-Small-Cell Lung Cancer: 5-Year Outcomes From the Phase 3 KEYNOTE-189 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2023, 41, 1992–1998. [Google Scholar] [CrossRef]
- Borghaei, H.; Ciuleanu, T.-E.; Lee, J.-S.; Pluzanski, A.; Caro, R.B.; Gutierrez, M.; Ohe, Y.; Nishio, M.; Goldman, J.; Ready, N.; et al. Long-Term Survival with First-Line Nivolumab plus Ipilimumab in Patients with Advanced Non-Small-Cell Lung Cancer: A Pooled Analysis. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2023, 34, 173–185. [Google Scholar] [CrossRef]
- Desai, A.; Peters, S. Immunotherapy-Based Combinations in Metastatic NSCLC. Cancer Treat. Rev. 2023, 116, 102545. [Google Scholar] [CrossRef]
- Xu, Y.; Li, H.; Fan, Y. Progression Patterns, Treatment, and Prognosis Beyond Resistance of Responders to Immunotherapy in Advanced Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 642883. [Google Scholar] [CrossRef]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; de Roos, J.A.D.M.; Dürrbaum, M.; Kuffer, C.; Müller, S.; Zaman, G.J.R.; Kloosterman, W.P.; et al. Chromosomal Instability, Tolerance of Mitotic Errors and Multidrug Resistance Are Promoted by Tetraploidization in Human Cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef]
- Replogle, J.M.; Zhou, W.; Amaro, A.E.; McFarland, J.M.; Villalobos-Ortiz, M.; Ryan, J.; Letai, A.; Yilmaz, O.; Sheltzer, J.; Lippard, S.J.; et al. Aneuploidy Increases Resistance to Chemotherapeutics by Antagonizing Cell Division. Proc. Natl. Acad. Sci. USA 2020, 117, 30566–30576. [Google Scholar] [CrossRef]
- Hosea, R.; Hillary, S.; Naqvi, S.; Wu, S.; Kasim, V. The Two Sides of Chromosomal Instability: Drivers and Brakes in Cancer. Signal Transduct. Target. Ther. 2024, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Lukow, D.A.; Sheltzer, J.M. Chromosomal Instability and Aneuploidy as Causes of Cancer Drug Resistance. Trends Cancer 2022, 8, 43–53. [Google Scholar] [CrossRef]
- Santaguida, S.; Richardson, A.; Iyer, D.R.; M’Saad, O.; Zasadil, L.; Knouse, K.A.; Wong, Y.L.; Rhind, N.; Desai, A.; Amon, A. Chromosome Mis-Segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes That Are Eliminated by the Immune System. Dev. Cell 2017, 41, 638–651.e5. [Google Scholar] [CrossRef]
- Rempel, E.; Kluck, K.; Beck, S.; Ourailidis, I.; Kazdal, D.; Neumann, O.; Volckmar, A.L.; Kirchner, M.; Goldschmid, H.; Pfarr, N.; et al. Pan-Cancer Analysis of Genomic Scar Patterns Caused by Homologous Repair Deficiency (HRD). NPJ Precis. Oncol. 2022, 6, 36. [Google Scholar] [CrossRef]
- Lin, P.; Chen, Y.; Xu, J.; Huang, X.; Wen, W.; Zhang, L.; Kong, W.; Zhao, Z.; Ye, Y.; Bao, Z.; et al. A Multicenter-Retrospective Cohort Study of Chromosome Instability in Lung Cancer: Clinical Characteristics and Prognosis of Patients Harboring Chromosomal Instability Detected by Metagenomic next-Generation Sequencing. J. Thorac. Dis. 2023, 15, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Xu, C.; Farooqui, M.; Zhang, Y.; Asch, A.S.; Yamada, H.Y. Survival-Critical Genes Associated with Copy Number Alterations in Lung Adenocarcinoma. Cancers 2021, 13, 2586. [Google Scholar] [CrossRef]
- Rao, C.V.; Xu, C.; Zhang, Y.; Asch, A.S.; Yamada, H.Y. Genomic Instability Genes in Lung and Colon Adenocarcinoma Indicate Organ Specificity of Transcriptomic Impact on Copy Number Alterations. Sci. Rep. 2022, 12, 11739. [Google Scholar] [CrossRef]
- How, C.; Bruce, J.; So, J.; Pintilie, M.; Haibe-Kains, B.; Hui, A.; Clarke, B.A.; Hedley, D.W.; Hill, R.P.; Milosevic, M.; et al. Chromosomal Instability as a Prognostic Marker in Cervical Cancer. BMC Cancer 2015, 15, 361. [Google Scholar] [CrossRef] [PubMed]
- Habermann, J.K.; Doering, J.; Hautaniemi, S.; Roblick, U.J.; Bündgen, N.K.; Nicorici, D.; Kronenwett, U.; Rathnagiriswaran, S.; Mettu, R.K.R.; Ma, Y.; et al. The Gene Expression Signature of Genomic Instability in Breast Cancer Is an Independent Predictor of Clinical Outcome. Int. J. Cancer 2009, 124, 1552–1564. [Google Scholar] [CrossRef]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and Temporal Diversity in Genomic Instability Processes Defines Lung Cancer Evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef]
- Varella-Garcia, M. Chromosomal and Genomic Changes in Lung Cancer. Cell Adhes. Migr. 2010, 4, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lan, Y.; Liu, F.; Yuan, Y.; Ge, J.; Wei, S.; Luo, H.; Li, J.; Luo, T.; Bian, X. Combination of Genomic Instability Score and TP53 Status for Prognosis Prediction in Lung Adenocarcinoma. NPJ Precis. Oncol. 2023, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef]
- Peng, B.; Li, H.; Na, R.; Lu, T.; Li, Y.; Zhao, J.; Zhang, H.; Zhang, L. Identification of a Novel Prognostic Signature of Genome Instability-Related LncRNAs in Early Stage Lung Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 706454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lam, T.-W.; Ting, H.-F. Genome Instability-Derived Genes as a Novel Prognostic Signature for Lung Adenocarcinoma. Front. Cell Dev. Biol. 2023, 11, 1224069. [Google Scholar] [CrossRef]
- Sonugür, F.G.; Akbulut, H. The Role of Tumor Microenvironment in Genomic Instability of Malignant Tumors. Front. Genet. 2019, 10, 1063. [Google Scholar] [CrossRef]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef]
- Weaver, B.A.A.; Cleveland, D.W. Does Aneuploidy Cause Cancer? Curr. Opin. Cell Biol. 2006, 18, 658–667. [Google Scholar] [CrossRef]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarró, L.; Couturier, D.-L.; Liu, L.; Schneider, M.; et al. A Pan-Cancer Compendium of Chromosomal Instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 Defects with Genomic Scores Predictive of DNA Damage Repair Deficiency among Breast Cancer Subtypes. Breast Cancer Res. BCR 2014, 16, 475. [Google Scholar] [CrossRef] [PubMed]
- Gou, R.; Dong, H.; Lin, B. Application and Reflection of Genomic Scar Assays in Evaluating the Efficacy of Platinum Salts and PARP Inhibitors in Cancer Therapy. Life Sci. 2020, 261, 118434. [Google Scholar] [CrossRef] [PubMed]
- Tsang, E.S.; Csizmok, V.; Williamson, L.M.; Pleasance, E.; Topham, J.T.; Karasinska, J.M.; Titmuss, E.; Schrader, I.; Yip, S.; Tessier-Cloutier, B.; et al. Homologous Recombination Deficiency Signatures in Gastrointestinal and Thoracic Cancers Correlate with Platinum Therapy Duration. NPJ Precis. Oncol. 2023, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Chen, B.; Han, X.; Gu, W.; Liang, S.; Wu, L. Genomic and Molecular Landscape of Homologous Recombination Deficiency across Multiple Cancer Types. Sci. Rep. 2023, 13, 8899. [Google Scholar] [CrossRef]
- Kawale, A.S.; Sung, P. Mechanism and Significance of Chromosome Damage Repair by Homologous Recombination. Essays Biochem. 2020, 64, 779–790. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of Genomic Loss of Heterozygosity Predict Homologous Recombination Repair Defects in Epithelial Ovarian Cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef]
- Ter Brugge, P.; Moser, S.C.; Bièche, I.; Kristel, P.; Ibadioune, S.; Eeckhoutte, A.; De Bruijn, R.; Van Der Burg, E.; Lutz, C.; Annunziato, S.; et al. Homologous Recombination Deficiency Derived from Whole-Genome Sequencing Predicts Platinum Response in Triple-Negative Breast Cancers. Nat. Commun. 2023, 14, 1958. [Google Scholar] [CrossRef]
- Zhao, E.Y.; Shen, Y.; Pleasance, E.; Kasaian, K.; Leelakumari, S.; Jones, M.; Bose, P.; Ch’ng, C.; Reisle, C.; Eirew, P.; et al. Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer. Clin. Cancer Res. 2017, 23, 7521–7530. [Google Scholar] [CrossRef]
- Park, Y.-B.; Kim, J.-M. Identification of Long Non-Coding RNA-mRNA Interactions and Genome-Wide lncRNA Annotation in Animal Transcriptome Profiling. J. Anim. Sci. Technol. 2023, 65, 293–310. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed]
- Samudyata; Castelo-Branco, G.; Bonetti, A. Birth, Coming of Age and Death: The Intriguing Life of Long Noncoding RNAs. Semin. Cell Dev. Biol. 2018, 79, 143–152. [Google Scholar] [CrossRef]
- Taniue, K.; Akimitsu, N. The Functions and Unique Features of LncRNAs in Cancer Development and Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 632. [Google Scholar] [CrossRef] [PubMed]
- Andonegui-Elguera, M.A.; Cáceres-Gutiérrez, R.E.; Oliva-Rico, D.; Díaz-Chávez, J.; Herrera, L.A. LncRNAs-Associated to Genomic Instability: A Barrier to Cancer Therapy Effectiveness. Front. Genet. 2022, 13, 984329. [Google Scholar] [CrossRef]
- Lee, S.; Kopp, F.; Chang, T.-C.; Sataluri, A.; Chen, B.; Sivakumar, S.; Yu, H.; Xie, Y.; Mendell, J.T. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 2016, 164, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, Y.; Guo, Y.; Xu, J.; Wang, X.; Ning, W.; Ma, L.; Qu, Y.; Zhang, M.; Zhang, H. Mutation-Derived, Genomic Instability-Associated lncRNAs Are Prognostic Markers in Gliomas. PeerJ 2023, 11, e15810. [Google Scholar] [CrossRef]
- Lin, C.; Lin, K.; Li, P.; Yuan, H.; Lin, X.; Dai, Y.; Zhang, Y.; Xie, Z.; Liu, T.; Wei, C. A Genomic Instability-Associated lncRNA Signature for Predicting Prognosis and Biomarkers in Lung Adenocarcinoma. Sci. Rep. 2024, 14, 14460. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-Coding RNAs as Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, M.; Huang, H.; Jin, W.-L. Drug Repurposing for Cancer Therapy. Signal Transduct. Target. Ther. 2024, 9, 92. [Google Scholar] [CrossRef]
- Ghiam, S.; Eslahchi, C.; Shahpasand, K.; Habibi-Rezaei, M.; Gharaghani, S. Identification of Repurposed Drugs Targeting Significant Long Non-Coding RNAs in the Cross-Talk between Diabetes Mellitus and Alzheimer’s Disease. Sci. Rep. 2022, 12, 18332. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, Z.; Hong, M.; Yang, D.; Xie, W. Long-Noncoding RNAs (lncRNAs) in Drug Metabolism and Disposition, Implications in Cancer Chemo-Resistance. Acta Pharm. Sin. B 2020, 10, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Huang, H.-T.; Huang, H.-C.; Juan, H.-F. LncTx: A Network-Based Method to Repurpose Drugs Acting on the Survival-Related lncRNAs in Lung Cancer. Comput. Struct. Biotechnol. J. 2021, 19, 3990–4002. [Google Scholar] [CrossRef]
- Zhao, H.; Yin, X.; Xu, H.; Liu, K.; Liu, W.; Wang, L.; Zhang, C.; Bo, L.; Lan, X.; Lin, S.; et al. LncTarD 2.0: An Updated Comprehensive Database for Experimentally-Supported Functional lncRNA-Target Regulations in Human Diseases. Nucleic Acids Res. 2023, 51, D199–D207. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Shao, D.; Xu, K.; Lu, Z.; Lu, Z.J.; Yang, Y.T.; Zhang, Q.C. RISE: A Database of RNA Interactome from Sequencing Experiments. Nucleic Acids Res. 2018, 46, D194–D201. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Lucid Visual Collaboration Suite. Available online: https://lucid.app/users/login#/login (accessed on 10 November 2024).
- Liang, Y.; Sun, H.-X.; Ma, B.; Meng, Q.-K. Identification of a Genomic Instability-Related Long Noncoding RNA Prognostic Model in Colorectal Cancer Based on Bioinformatic Analysis. Dis. Markers 2022, 2022, 4556585. [Google Scholar] [CrossRef]
- TCGA Research Network. Available online: https://www.cancer.gov/tcga (accessed on 10 August 2024).
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 11 November 2024).
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, L.; Lun, A.T.L.; Baldoni, P.L.; Smyth, G.K. edgeR 4.0: Powerful Differential Analysis of Sequencing Data with Expanded Functionality and Improved Support for Small Counts and Larger Datasets. Nucleic Acids Res. 2024, 53, gkaf018. [Google Scholar] [CrossRef]
- Dillies, M.-A.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, G.; Castel, D.; Estelle, J.; et al. A Comprehensive Evaluation of Normalization Methods for Illumina High-Throughput RNA Sequencing Data Analysis. Brief. Bioinform. 2013, 14, 671–683. [Google Scholar] [CrossRef]
- Seyednasrollah, F.; Laiho, A.; Elo, L.L. Comparison of Software Packages for Detecting Differential Expression in RNA-Seq Studies. Brief. Bioinform. 2015, 16, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Liu, S.; Sun, Z.; Zhang, Y.; Zhang, F.; Zhang, C.; Shang, D.; Yang, H.; Su, F.; Xu, Y.; et al. LncRNAs2Pathways: Identifying the Pathways Influenced by a Set of lncRNAs of Interest Based on a Global Network Propagation Method. Sci. Rep. 2017, 7, 46566. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Sha, K.; Lu, Y.; Zhang, P.; Pei, R.; Shi, X.; Fan, Z.; Chen, L. Identifying a Novel 5-Gene Signature Predicting Clinical Outcomes in Acute Myeloid Leukemia. Clin. Transl. Oncol. 2021, 23, 648–656. [Google Scholar] [CrossRef]
- Therneau, T.M.; Lumley, T.; Elizabeth, A.; Cynthia, C. Survival: Survival Analysis, Version 3.7-0; GitHub, Inc.: San Francisco, CA, USA, 2024.
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Survminer: Drawing Survival Curves Using “Ggplot2”, Version 0.5.0; GitHub, Inc.: San Francisco, CA, USA, 2024.
- Hu, E. YuLab-SMU/GeoTcgaData; GitHub, Inc.: San Francisco, CA, USA, 2024. [Google Scholar]
- Wickham, H. Ggplot2. Available online: https://ggplot2.tidyverse.org/ (accessed on 11 November 2024).
- Cao, X.; Zhou, X.; Hou, F.; Huang, Y.; Yuan, M.; Long, M.; Chen, S.; Lei, W.; Zhu, J.; Chen, J.; et al. ncRNADrug: A Database for Validated and Predicted ncRNAs Associated with Drug Resistance and Targeted by Drugs. Nucleic Acids Res. 2024, 52, D1393–D1399. [Google Scholar] [CrossRef]
- Kratz, J.R.; Li, J.Z.; Tsui, J.; Lee, J.C.; Ding, V.W.; Rao, A.A.; Mann, M.J.; Chan, V.; Combes, A.J.; Krummel, M.F.; et al. Genetic and Immunologic Features of Recurrent Stage I Lung Adenocarcinoma. Sci. Rep. 2021, 11, 23690. [Google Scholar] [CrossRef]
- shinyCircos-V2.0. Available online: https://venyao.xyz/shinycircos/#div2.2.7 (accessed on 19 August 2024).
- Gong, C.; Maquat, L.E. lncRNAs Transactivate STAU1-Mediated mRNA Decay by Duplexing with 3’ UTRs via Alu Elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of Somatic Tissue Differentiation by the Long Non-Coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Panda, A.C.; Kang, M.-J.; Guo, R.; Kim, J.; Grammatikakis, I.; Yoon, J.-H.; Dudekula, D.B.; Noh, J.H.; Yang, X.; et al. 7SL RNA Represses P53 Translation by Competing with HuR. Nucleic Acids Res. 2014, 42, 10099–10111. [Google Scholar] [CrossRef]
- Das, G.; Das, T.; Parida, S.; Ghosh, Z. LncRTPred: Predicting RNA-RNA Mode of Interaction Mediated by lncRNA. IUBMB Life 2024, 76, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, L.; Tian, G.; Dong, Y.; Zhang, X.; Zhou, Z.; Luo, X.; Li, Y.; Yao, W. shinyCircos-V2.0: Leveraging the Creation of Circos Plot with Enhanced Usability and Advanced Features. iMeta 2023, 2, e109. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Yao, C.; Teng, X.; Chai, J.; Yang, X.; Li, B. MiR-140-3p Suppressed Cell Growth and Invasion by Downregulating the Expression of ATP8A1 in Non-Small Cell Lung Cancer. Tumor Biol. 2016, 37, 2973–2985. [Google Scholar] [CrossRef]
- Zhu, X.; Ma, X.; Zhao, S.; Cao, Z. DLX6-AS1 Accelerates Cell Proliferation through Regulating miR-497-5p/SNCG Pathway in Prostate Cancer. Environ. Toxicol. 2021, 36, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef]
- Liu, Y.; Xun, Z.; Ma, K.; Liang, S.; Li, X.; Zhou, S.; Sun, L.; Liu, Y.; Du, Y.; Guo, X.; et al. Identification of a Tumour Immune Barrier in the HCC Microenvironment That Determines the Efficacy of Immunotherapy. J. Hepatol. 2023, 78, 770–782. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, Y.; Wang, D.; Hu, X.; Zhang, Z. Single-Cell Meta-Analyses Reveal Responses of Tumor-Reactive CXCL13+ T Cells to Immune-Checkpoint Blockade. Nat. Cancer 2022, 3, 1123–1136. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1000165. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, K.; Wang, L.; Bi, J. Genome Instability-Related Long Non-Coding RNA in Clear Renal Cell Carcinoma Determined Using Computational Biology. BMC Cancer 2021, 21, 727. [Google Scholar] [CrossRef] [PubMed]
- Dianatpour, A.; Ghafouri-Fard, S. The Role of Long Non Coding RNAs in the Repair of DNA Double Strand Breaks. Int. J. Mol. Cell. Med. 2017, 6, 1–12. [Google Scholar] [PubMed]
- Lou, M.-M.; Tang, X.-Q.; Wang, G.-M.; He, J.; Luo, F.; Guan, M.-F.; Wang, F.; Zou, H.; Wang, J.-Y.; Zhang, Q.; et al. Long Noncoding RNA BS-DRL1 Modulates the DNA Damage Response and Genome Stability by Interacting with HMGB1 in Neurons. Nat. Commun. 2021, 12, 4075. [Google Scholar] [CrossRef]
- Zhu, S.; Zhao, J.; Nie, L.; Yin, W.; Zhang, Y.; Zhao, F.; Ni, Y.; Zhang, X.; Wang, Z.; Dai, J.; et al. Homologous Recombination Deficiency (HRD) Score in Aggressive Prostatic Adenocarcinoma with or without Intraductal Carcinoma of the Prostate (IDC-P). BMC Med. 2022, 20, 237. [Google Scholar] [CrossRef]
- Zhou, Z.; Ding, Z.; Yuan, J.; Shen, S.; Jian, H.; Tan, Q.; Yang, Y.; Chen, Z.; Luo, Q.; Cheng, X.; et al. Homologous Recombination Deficiency (HRD) Can Predict the Therapeutic Outcomes of Immuno-Neoadjuvant Therapy in NSCLC Patients. J. Hematol. Oncol. 2022, 15, 62. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2021, 26, e164–e172. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Li, C.H.; Haider, S.; Boutros, P.C. Age Influences on the Molecular Presentation of Tumours. Nat. Commun. 2022, 13, 208. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Taylor, B.S. Homing in on Genomic Instability as a Therapeutic Target in Cancer. Nat. Commun. 2021, 12, 3663. [Google Scholar] [CrossRef]
- Requesens, M.; Foijer, F.; Nijman, H.W.; de Bruyn, M. Genomic Instability as a Driver and Suppressor of Anti-Tumor Immunity. Front. Immunol. 2024, 15, 1462496. [Google Scholar] [CrossRef]
- Sobral, D.; Martins, M.; Kaplan, S.; Golkaram, M.; Salmans, M.; Khan, N.; Vijayaraghavan, R.; Casimiro, S.; Fernandes, A.; Borralho, P.; et al. Genetic and Microenvironmental Intra-Tumor Heterogeneity Impacts Colorectal Cancer Evolution and Metastatic Development. Commun. Biol. 2022, 5, 937. [Google Scholar] [CrossRef]
- Chen, M.; Linstra, R.; van Vugt, M.A.T.M. Genomic Instability, Inflammatory Signaling and Response to Cancer Immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188661. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hubisz, M.J.; Earlie, E.M.; Duran, M.A.; Hong, C.; Varela, A.A.; Lettera, E.; Deyell, M.; Tavora, B.; Havel, J.J.; et al. Non-Cell-Autonomous Cancer Progression from Chromosomal Instability. Nature 2023, 620, 1080–1088. [Google Scholar] [CrossRef]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.-K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 3010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhuang, G.; Sun, X.; Shen, Y.; Wang, W.; Li, Q.; Di, W. TP53 Mutation-Mediated Genomic Instability Induces the Evolution of Chemoresistance and Recurrence in Epithelial Ovarian Cancer. Diagn. Pathol. 2017, 12, 16. [Google Scholar] [CrossRef]
- Xun, Z.; Wang, Y.; Long, J.; Li, Y.; Yang, X.; Sun, H.; Zhao, H. Development and Validation of a Genomic Instability-Related lncRNA Prognostic Model for Hepatocellular Carcinoma. Front. Genet. 2022, 13, 1034979. [Google Scholar] [CrossRef]
- Zhu, X.; Yu, R.; Peng, Y.; Miao, Y.; Jiang, K.; Li, Q. Identification of Genomic Instability Related lncRNA Signature with Prognostic Value and Its Role in Cancer Immunotherapy in Pancreatic Cancer. Front. Genet. 2022, 13, 990661. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, N.; He, Y.; Tao, C.; Liang, Z.; Xin, W.; Zhang, Q.; Wang, F. Bioinformatic Identification of Genomic Instability-Associated lncRNAs Signatures for Improving the Clinical Outcome of Cervical Cancer by a Prognostic Model. Sci. Rep. 2021, 11, 20929. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhao, S.; Song, X.; Zhang, M.; Zhu, X.; Li, C.; Chen, W.; Zhao, D. The Potential Use and Experimental Validation of Genomic Instability-Related lncRNA in Pancreatic Carcinoma. Medicine 2023, 102, e35300. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; He, X.; Yao, R.; Fan, L.; Zhao, L.; Lu, B.; Pang, Z. Genome Instability-Related LINC02577, LINC01133 and AC107464.2 Are lncRNA Prognostic Markers Correlated with Immune Microenvironment in Pancreatic Adenocarcinoma. BMC Cancer 2023, 23, 430. [Google Scholar] [CrossRef]
- Xue, C.; Lv, L.; Jiang, J.; Li, L. Promising Long Noncoding RNA DLX6-AS1 in Malignant Tumors. Am. J. Transl. Res. 2020, 12, 7682–7692. [Google Scholar]
- Ghafouri-Fard, S.; Najafi, S.; Hussen, B.M.; Ganjo, A.R.; Taheri, M.; Samadian, M. DLX6-AS1: A Long Non-Coding RNA With Oncogenic Features. Front. Cell Dev. Biol. 2022, 10, 746443. [Google Scholar] [CrossRef]
- Wu, C.; Lin, W.; Fu, F. Long Non-Coding RNA DLX6-AS1 Knockdown Suppresses the Tumorigenesis and Progression of Non-Small Cell Lung Cancer through microRNA-16-5p/BMI1 Axis. Transl. Cancer Res. 2021, 10, 3772–3787. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ye, Z.; Mei, D.; Gu, H.; Zhang, J. Long Noncoding RNA DLX6-AS1 Promotes Tumorigenesis by Modulating miR-497-5p/FZD4/FZD6/Wnt/β-Catenin Pathway in Pancreatic Cancer. Cancer Manag. Res. 2019, 11, 4209–4221. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, H.; Bao, Y.; Yu, H.; Xie, D.; Wang, X. Exosomal Long Non-Coding RNA DLX6-AS1 as a Potential Diagnostic Biomarker for Non-Small Cell Lung Cancer. Oncol. Lett. 2019, 18, 5197–5204. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Zhao, D.; Yao, S. Identification of a Genome Instability-Associated LncRNA Signature for Prognosis Prediction in Colon Cancer. Front. Genet. 2021, 12, 679150. [Google Scholar] [CrossRef]
- Sebastian-delaCruz, M.; Gonzalez-Moro, I.; Olazagoitia-Garmendia, A.; Castellanos-Rubio, A.; Santin, I. The Role of lncRNAs in Gene Expression Regulation through mRNA Stabilization. Non-Coding RNA 2021, 7, 3. [Google Scholar] [CrossRef]
- Segal, D.; Dostie, J. The Talented LncRNAs: Meshing into Transcriptional Regulatory Networks in Cancer. Cancers 2023, 15, 3433. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Li, Y.; Li, J.; Gao, Z.; Yang, Z.; Li, Y.; Liu, H.; Fan, T. Long Non-Coding RNAs: The Regulatory Mechanisms, Research Strategies, and Future Directions in Cancers. Front. Oncol. 2020, 10, 598817. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Abdelmohsen, K.; Gorospe, M. Functional Interactions among microRNAs and Long Noncoding RNAs. Semin. Cell Dev. Biol. 2014, 34, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Ikeya, Y.; Okumura, T.; Kimura, T. Post-Transcriptional Inducible Gene Regulation by Natural Antisense RNA. Front. Biosci. Landmark Ed. 2015, 20, 1–36. [Google Scholar] [CrossRef]
- Tang, S.-S.; Zheng, B.-Y.; Xiong, X.-D. LincRNA-P21: Implications in Human Diseases. Int. J. Mol. Sci. 2015, 16, 18732–18740. [Google Scholar] [CrossRef]
- Chen, J.; Sun, M.; Kent, W.J.; Huang, X.; Xie, H.; Wang, W.; Zhou, G.; Shi, R.Z.; Rowley, J.D. Over 20% of Human Transcripts Might Form Sense-Antisense Pairs. Nucleic Acids Res. 2004, 32, 4812–4820. [Google Scholar] [CrossRef]
- He, Y.; Vogelstein, B.; Velculescu, V.E.; Papadopoulos, N.; Kinzler, K.W. The Antisense Transcriptomes of Human Cells. Science 2008, 322, 1855–1857. [Google Scholar] [CrossRef]
- Balbin, O.A.; Malik, R.; Dhanasekaran, S.M.; Prensner, J.R.; Cao, X.; Wu, Y.-M.; Robinson, D.; Wang, R.; Chen, G.; Beer, D.G.; et al. The Landscape of Antisense Gene Expression in Human Cancers. Genome Res. 2015, 25, 1068–1079. [Google Scholar] [CrossRef]
- Pelechano, V.; Steinmetz, L.M. Gene Regulation by Antisense Transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef]
- Guttman, M.; Rinn, J.L. Modular Regulatory Principles of Large Non-Coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef]
- Li, J.; Li, P.; Zhao, W.; Yang, R.; Chen, S.; Bai, Y.; Dun, S.; Chen, X.; Du, Y.; Wang, Y.; et al. Expression of Long Non-Coding RNA DLX6-AS1 in Lung Adenocarcinoma. Cancer Cell Int. 2015, 15, 48. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The Current Landscape of Nucleic Acid Therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Yokota, T. Knocking Down Long Noncoding RNAs Using Antisense Oligonucleotide Gapmers. Methods Mol. Biol. 2020, 2176, 49–56. [Google Scholar] [CrossRef]
- Karn, T.; Jiang, T.; Hatzis, C.; Sänger, N.; El-Balat, A.; Rody, A.; Holtrich, U.; Becker, S.; Bianchini, G.; Pusztai, L. Association Between Genomic Metrics and Immune Infiltration in Triple-Negative Breast Cancer. JAMA Oncol. 2017, 3, 1707–1711. [Google Scholar] [CrossRef]
- Burr, K.L.; Robinson, J.I.; Rastogi, S.; Boylan, M.T.; Coates, P.J.; Lorimore, S.A.; Wright, E.G. Radiation-Induced Delayed Bystander-Type Effects Mediated by Hemopoietic Cells. Radiat. Res. 2010, 173, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; He, M.; Tu, W.; Konishi, T.; Liu, W.; Xie, Y.; Dang, B.; Li, W.; Uchihori, Y.; Hei, T.K.; et al. The Differential Role of Human Macrophage in Triggering Secondary Bystander Effects after Either Gamma-Ray or Carbon Beam Irradiation. Cancer Lett. 2015, 363, 92–100. [Google Scholar] [CrossRef]
- Lorimore, S.A.; Chrystal, J.A.; Robinson, J.I.; Coates, P.J.; Wright, E.G. Chromosomal Instability in Unirradiated Hemaopoietic Cells Induced by Macrophages Exposed in Vivo to Ionizing Radiation. Cancer Res. 2008, 68, 8122–8126. [Google Scholar] [CrossRef]
- Mothersill, C.; Seymour, C.B. Radiation-Induced Bystander Effects—Implications for Cancer. Nat. Rev. Cancer 2004, 4, 158–164. [Google Scholar] [CrossRef]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. Long Non-Coding RNA UCA1 Increases Chemoresistance of Bladder Cancer Cells by Regulating Wnt Signaling. FEBS J. 2014, 281, 1750–1758. [Google Scholar] [CrossRef]
- Pan, J.; Li, X.; Wu, W.; Xue, M.; Hou, H.; Zhai, W.; Chen, W. Long Non-Coding RNA UCA1 Promotes Cisplatin/Gemcitabine Resistance through CREB Modulating miR-196a-5p in Bladder Cancer Cells. Cancer Lett. 2016, 382, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guan, Z.; He, K.; Qian, J.; Cao, J.; Teng, L. LncRNA UCA1 in Anti-Cancer Drug Resistance. Oncotarget 2017, 8, 64638–64650. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Dragomir, M.P.; Yang, C.; Li, Q.; Horst, D.; Calin, G.A. Targeting Non-Coding RNAs to Overcome Cancer Therapy Resistance. Signal Transduct. Target. Ther. 2022, 7, 121. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | High HRD Scores (n = 120) | Low HRD Scores (n = 134) | p-Value * | |
|---|---|---|---|---|
| Age | ≤65 | 70 (58.33%) | 48 (35.82%) | 0.001 |
| >65 | 46 (38.33%) | 79 (59.03%) | ||
| Unknown | 4 (3.33%) | 7 (5.22%) | ||
| Sex | Female | 57 (47.50%) | 73 (54.48%) | 0.287 |
| Male | 63 (52.50%) | 61 (45.52%) | ||
| Race | Caucasian/ White | 85 (70.8%) | 102 (76.1%) | 0.281 |
| Black/African American | 20 (16.7%) | 12 (9.0%) | ||
| Asian | 2 (1.7%) | 1 (0.7%) | ||
| Unknown | 13 (10.8%) | 19 (14.2%) | ||
| Stage | Stages I–II | 97 (80.83%) | 103 (76.87%) | 0.757 |
| Stages III–IV | 21 (17.50%) | 26 (19.40%) | ||
| Unknown | 2 (1.67%) | 5 (3.73%) | ||
| T | T1–2 | 106 (88.33%) | 113 (84.33%) | 0.519 |
| T3–4 | 14 (11.67%) | 21 (15.67%) | ||
| N | N0 | 79 (65.83%) | 87 (64.93%) | 0.893 |
| N1–3 | 41 (34.17%) | 41 (30.60%) | ||
| NX | 0 (0.00%) | 6 (4.48%) | ||
| M | M0 | 77 (64.17%) | 95 (70.90%) | 0.950 |
| M1 | 4 (3.33%) | 5 (3.73%) | ||
| MX | 38 (31.67%) | 32 (23.88%) | ||
| Unknown | 1 (0.83%) | 2 (1.49%) | ||
| Genes | High HRD Scores (n = 120) | Low HRD Scores (n = 134) | p-Value * | |
|---|---|---|---|---|
| TP53 | Mutant | 96 (80%) | 18 (13.43%) | 1 × 10−4 |
| Wildtype | 22 (18.33%) | 111 (82.09%) | ||
| Unknown | 2 (1.67%) | 5 (3.73%) | ||
| EGFR | Mutant | 10 (8.33%) | 19 (14.18%) | 0.184 |
| Wildtype | 108 (90%) | 110 (82.09%) | ||
| Unknown | 2 (1.67%) | 5 (3.73%) | ||
| KRAS | Mutant | 25 (20.83%) | 38 (28.36%) | 0.179 |
| Wildtype | 93 (77.5%) | 91 (67.91%) | ||
| Unknown | 2 (1.67%) | 5 (3.73%) | ||
| PIK3CA | Mutant | 5 (4.17%) | 6 (4.48%) | 1 |
| Wildtype | 113 (94.17%) | 123 (91.79%) | ||
| Unknown | 2 (1.67%) | 5 (3.73%) | ||
| CDKN2A | Mutant | 9 (7.50%) | 5 (3.73%) | 0.318 |
| Wildtype | 109 (90.83%) | 124 (92.54%) | ||
| Unknown | 2 (1.67%) | 5 (3.73%) | ||
| lncRNA | Function | Target | miRNA | Disease |
|---|---|---|---|---|
| DLX6-AS1 | ceRNA | RUNX2 | miR-505-3p | Breast cancer |
| ARPP19 | miR-16-5p | Cervical cancer | ||
| CDK4 | miR-124-3p | Ewing’s sarcoma | ||
| PDK1 | miR-4290 | Gastric cancer | ||
| POU2F1 | miR-204-5p | Gastric cancer | ||
| MMP2 | miR-203a | Hepatocellular carcinoma | ||
| WEE1 | miR-424-5p | Liver cancer | ||
| E2F1 | miR-197-5p | Malignant glioma | ||
| YAP1 | miR-497-5p | Neuroblastoma | ||
| PRR11 | miR-144 | NSCLC | ||
| DLK1 | miR-129-5p | Osteosarcoma | ||
| GADD45A | miR-376c | Pre-eclampsia | ||
| ERP44 | miR-149-5p | Pre-eclampsia | ||
| PTEN | miR-26a | Renal cell carcinoma | ||
| protein interaction | NOTCH1 | Ovarian cancer | ||
| UPF1 | Thyroid cancer | |||
| NA | PIK3CA | Colorectal cancer | ||
| AKT1 | Colorectal cancer | |||
| MTOR | Colorectal cancer | |||
| DLX6 | Lung adenocarcinoma | |||
| sponge | miR-199a | Cervical cancer | ||
| miR-497-5p | Neuroblastoma/PDAC | |||
| miR-223 | Urinary bladder cancer | |||
| transcript reg | CADM1 | Hepatocellular carcinoma | ||
| LINC01224 | ceRNA | MYO6 | miR-485-5p | Colorectal cancer |
| AKT3 | miR-485-5p | Endometrial cancer | ||
| sponge | MIR2467 | Colorectal cancer/NSCLC | ||
| transcript reg | YY1 | Colorectal cancer | ||
| ELFN1-AS1 | ceRNA | MTA1 | miR-1250 | Colorectal cancer |
| TRIM44 | miR-4644 | Colorectal cancer | ||
| GFPT1 | miR-183-3p | Esophageal cancer | ||
| NA | Erk | Colon adenocarcinoma | ||
| IRS1 | Colon adenocarcinoma | |||
| vimentin | Colon adenocarcinoma | |||
| TRPM2-AS | ceRNA | PYCR1 | miR-140-3p | Breast cancer |
| HMGA1 | miR-195 | Gastric cancer | ||
| WEE1 | miR-497 | Retinoblastoma | ||
| protein interaction | TAF15 | Colorectal cancer | ||
| NA | SHC1 | NSCLC | ||
| sponge | miR-140-3p | Breast cancer | ||
| transcript reg | ELK1 | Gastric cancer | ||
| CALML3-AS1 | ceRNA | ZBTB2 | miR-4316 | Urinary bladder cancer |
| MIR2052HG | histone mod and transcript reg | LMTK3 | Breast cancer |
| Gene | HR (Hazard Ratio) | p-Value | LogFC | |
|---|---|---|---|---|
| High HRD Scores | AC046195.1 | 2.255680678 | 0.014686109 | −2.24 |
| LHFPL3-AS2 | 2.226216673 | 0.015577954 | −2.52 | |
| LINC02772 | 2.202470692 | 0.021184545 | −2.09 | |
| LINC01224 | 2.150362909 | 0.0234937 | 2.30 |
| GI-lncRNA | Drug | DrugBank Accession Number | Cancer Type | PMID | FDA |
|---|---|---|---|---|---|
| KCNMB2-AS1 | Panobinostat | DB06603 | Nephroblastoma | 26176219 | approved |
| MIR9-1HG | Panobinostat | DB06603 | Nephroblastoma | 26176219 | approved |
| MIR9-1HG | Tetraarsenic Oxide | NA | Breast Cancer | 33932728 | NA |
| ELFN1-AS1 | Dexamethasone | DB01234 | Hepatocellular Carcinoma | 29409992 | approved |
| DLX6-AS1 | Panobinostat | DB06603 | Nephroblastoma | 26176219 | approved |
| LHFPL3-AS2 | Oxaliplatin | DB00526 | Gastric Cancer | 29156779 | approved |
| LHFPL3-AS2 | Panobinostat | DB06603 | Nephroblastoma | 26176219 | approved |
| LINC01612 | Oxaliplatin | DB00526 | Gastric Cancer | 29156779 | approved |
| SFTA1P | Panobinostat | DB06603 | Nephroblastoma | 26176219 | approved |
| LINC02688 | Panobinostat | DB06603 | Nephroblastoma | 26176219 | approved |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souza, V.G.P.; Benard, K.H.; Stewart, G.L.; Enfield, K.S.S.; Lam, W.L. Identification of Genomic Instability-Associated LncRNAs as Potential Therapeutic Targets in Lung Adenocarcinoma. Cancers 2025, 17, 996. https://doi.org/10.3390/cancers17060996
Souza VGP, Benard KH, Stewart GL, Enfield KSS, Lam WL. Identification of Genomic Instability-Associated LncRNAs as Potential Therapeutic Targets in Lung Adenocarcinoma. Cancers. 2025; 17(6):996. https://doi.org/10.3390/cancers17060996
Chicago/Turabian StyleSouza, Vanessa G. P., Katya H. Benard, Greg L. Stewart, Katey S. S. Enfield, and Wan L. Lam. 2025. "Identification of Genomic Instability-Associated LncRNAs as Potential Therapeutic Targets in Lung Adenocarcinoma" Cancers 17, no. 6: 996. https://doi.org/10.3390/cancers17060996
APA StyleSouza, V. G. P., Benard, K. H., Stewart, G. L., Enfield, K. S. S., & Lam, W. L. (2025). Identification of Genomic Instability-Associated LncRNAs as Potential Therapeutic Targets in Lung Adenocarcinoma. Cancers, 17(6), 996. https://doi.org/10.3390/cancers17060996