
Targeting Kinesins for Therapeutic Exploitation of Chromosomal Instability in Lung Cancer


Simple Summary
Abstract
1. Introduction
1.1. Lung Cancer Background
1.2. Chromosomal Instability in Cancer
2. Exploiting Chromosomal Instability (CIN) for Cancer Therapy
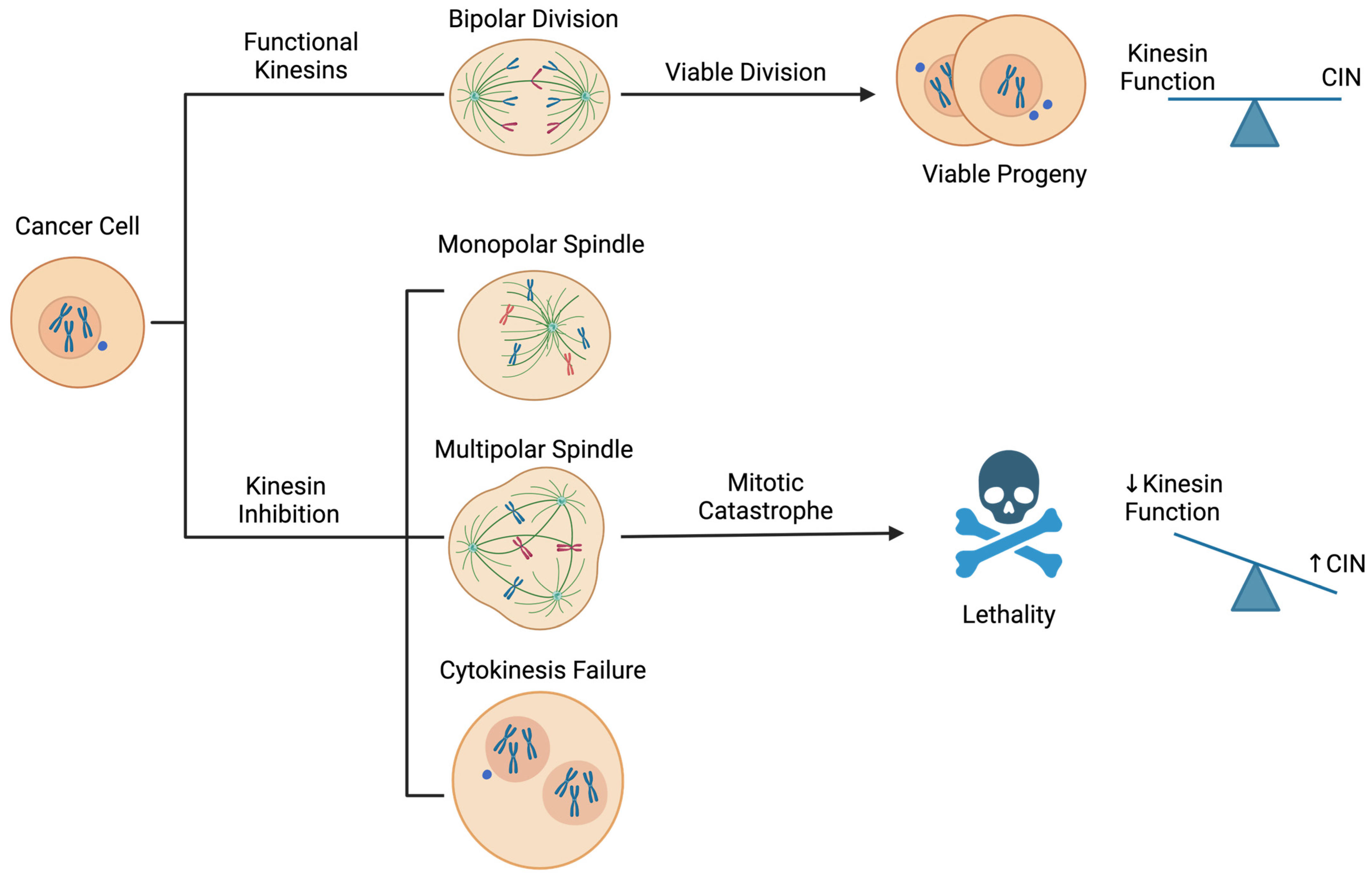
2.1. CIN: A Vulnerability in Cancer Cells
2.2. Strategies for Targeting CIN in Cancer
3. Kinesins: Classification, Structure, and General Functions
4. Kinesins in Lung Cancer
4.1. Kinesin-3 Family: KIF14
4.2. Kinesin-4 Family: KIF4A
4.3. Kinesin-5 Family: KIF11 (Eg5)
4.4. Kinesin-6 Family: KIF20A (MKLP2), KIF20B (MPP1), and KIF23 (MKLP1)
4.5. Kinesin-7 Family: KIF10 (CENPE)
4.6. Kinesin-8 Family: KIF18A, KIF18B
4.7. Kinesin-10 Family: KIF22 (KID)
4.8. Kinesin-12 Family: KIF15
4.9. Kinesin-13 Family: KIF2A, KIF2B, KIF2C, and KIF24
4.10. Kinesin-14A Family: KIFC1
4.11. Kinesin-14B Family: KIFC3
5. Non-Mitotic Kinesins Implicated in Lung Cancer
6. Outlook and Perspective
6.1. Potency and Selectivity of Kinesin Inhibitors
6.2. Therapeutic Index
6.3. Effects of CIN in Non-Malignant Cells
6.4. Enhancing Efficacy with Combination Strategies
6.5. Biomarkers to Inform Patient Selection
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Sun, S.; Lam, S.; Lockwood, W.W. New Insights into the Biology and Development of Lung Cancer in Never Smokers-Implications for Early Detection and Treatment. J. Transl. Med. 2023, 21, 585. [Google Scholar] [CrossRef]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-Small-Cell Lung Cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef]
- Politi, K.; Herbst, R.S. Lung Cancer in the Era of Precision Medicine. Clin. Cancer Res. 2015, 21, 2213–2220. [Google Scholar] [CrossRef] [PubMed]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non–small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef]
- Pikor, L.; Thu, K.; Vucic, E.; Lam, W. The Detection and Implication of Genome Instability in Cancer. Cancer Metastasis Rev. 2013, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and Clinical Implications of Chromosomal Instability in Cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef]
- Cunningham, C.E.; MacAuley, M.J.; Yadav, G.; Vizeacoumar, F.S.; Freywald, A.; Vizeacoumar, F.J. Targeting the CINful Genome: Strategies to Overcome Tumor Heterogeneity. Prog. Biophys. Mol. Biol. 2019, 147, 77–91. [Google Scholar] [CrossRef]
- Hosea, R.; Hillary, S.; Naqvi, S.; Wu, S.; Kasim, V. The Two Sides of Chromosomal Instability: Drivers and Brakes in Cancer. Signal Transduct. Target. Ther. 2024, 9, 75. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Jeusset, L.M.-P.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarró, L.; Couturier, D.-L.; Liu, L.; Schneider, M.; et al. A Pan-Cancer Compendium of Chromosomal Instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.-Z.; Wala, J.; Mermel, C.H.; et al. Pan-Cancer Patterns of Somatic Copy Number Alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.D.; Abbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar, A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of Copy Number Alterations in Human Cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef]
- Thu, K.L.; Soria-Bretones, I.; Mak, T.W.; Cescon, D.W. Targeting the Cell Cycle in Breast Cancer: Towards the next Phase. Cell Cycle 2018, 17, 1871–1885. [Google Scholar] [CrossRef] [PubMed]
- Slade, D. PARP and PARG Inhibitors in Cancer Treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef]
- Rath, O.; Kozielski, F. Kinesins and Cancer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin Superfamily Motor Proteins and Intracellular Transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- Lucanus, A.J.; Yip, G.W. Kinesin Superfamily: Roles in Breast Cancer, Patient Prognosis and Therapeutics. Oncogene 2018, 37, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Sabat-Pośpiech, D.; Fabian-Kolpanowicz, K.; Prior, I.A.; Coulson, J.M.; Fielding, A.B. Targeting Centrosome Amplification, an Achilles’ Heel of Cancer. Biochem. Soc. Trans. 2019, 47, 1209–1222. [Google Scholar] [CrossRef]
- Ling, B.; Liao, X.; Huang, Y.; Liang, L.; Jiang, Y.; Pang, Y.; Qi, G. Identification of Prognostic Markers of Lung Cancer through Bioinformatics Analysis and in Vitro Experiments. Int. J. Oncol. 2020, 56, 193–205. [Google Scholar] [CrossRef]
- Feng, Z.; Li, B.; Chen, Q.; Zhang, H.; Guo, Z.; Qin, J. Identification and Validation of a GPX4-Related Immune Prognostic Signature for Lung Adenocarcinoma. J. Oncol. 2022, 2022, 9054983. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Xu, H.; Liu, S.; Yang, B.; Li, L.; Zhao, H.; Jiang, C. Integrated Bioinformatics Analysis Identifies a New Stemness Index-Related Survival Model for Prognostic Prediction in Lung Adenocarcinoma. Front. Genet. 2022, 13, 860268. [Google Scholar] [CrossRef] [PubMed]
- Corson, T.W.; Huang, A.; Tsao, M.-S.; Gallie, B.L. KIF14 Is a Candidate Oncogene in the 1q Minimal Region of Genomic Gain in Multiple Cancers. Oncogene 2005, 24, 4741–4753. [Google Scholar] [CrossRef] [PubMed]
- Corson, T.W.; Zhu, C.Q.; Lau, S.K.; Shepherd, F.A.; Tsao, M.-S.; Gallie, B.L. KIF14 Messenger RNA Expression Is Independently Prognostic for Outcome in Lung Cancer. Clin. Cancer Res. 2007, 13, 3229–3234. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, G.; Wang, X.; Liao, X.; Huang, R.; Huang, C.; Huang, P.; Zhang, J.; Wang, P. Genome-wide Investigation of the Clinical Significance and Prospective Molecular Mechanisms of Kinesin Family Member Genes in Patients with Lung Adenocarcinoma. Oncol. Rep. 2019, 42, 1017–1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tang, F.; Tang, P.; Zhang, L.; Gan, Q.; Li, Y. Noncoding RNAs-Mediated Overexpression of KIF14 Is Associated with Tumor Immune Infiltration and Unfavorable Prognosis in Lung Adenocarcinoma. Aging 2022, 14, 8013–8031. [Google Scholar] [CrossRef] [PubMed]
- Hung, P.-F.; Hong, T.-M.; Hsu, Y.-C.; Chen, H.-Y.; Chang, Y.-L.; Wu, C.-T.; Chang, G.-C.; Jou, Y.-S.; Pan, S.-H.; Yang, P.-C. The Motor Protein KIF14 Inhibits Tumor Growth and Cancer Metastasis in Lung Adenocarcinoma. PLoS ONE 2013, 8, e61664. [Google Scholar] [CrossRef]
- Li, Y.; Hong, X.; Zhai, J.; Liu, Y.; Li, R.; Wang, X.; Zhang, Y.; Lv, Q. Novel Circular RNA Circ-0002727 Regulates miR-144-3p/KIF14 Pathway to Promote Lung Adenocarcinoma Progression. Front. Cell Dev. Biol. 2023, 11, 1249174. [Google Scholar] [CrossRef]
- Lao, Y.; Li, T.; Xie, X.; Chen, K.; Li, M.; Huang, L. MiR-195-3p Is a Novel Prognostic Biomarker Associated with Immune Infiltrates of Lung Adenocarcinoma. Int. J. Gen. Med. 2022, 15, 191–203. [Google Scholar] [CrossRef]
- Akinduro, O.O.; Suarez-Meade, P.; Roberts, M.; Tzeng, S.Y.; Sarabia-Estrada, R.; Schiapparelli, P.; Norton, E.S.; Gokaslan, Z.L.; Anastasiadis, P.Z.; Guerrero-Cázares, H.; et al. Verteporfin-Loaded Microparticles for Radiosensitization of Preclinical Lung and Breast Metastatic Spine Cancer. J. Neurosurg. Spine 2023, 38, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Dehan, E.; Ben-Dor, A.; Liao, W.; Lipson, D.; Frimer, H.; Rienstein, S.; Simansky, D.; Krupsky, M.; Yaron, P.; Friedman, E.; et al. Chromosomal Aberrations and Gene Expression Profiles in Non-Small Cell Lung Cancer. Lung Cancer 2007, 56, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Baudis, M.; Cleary, M.L. Progenetix.net: An Online Repository for Molecular Cytogenetic Aberration Data. Bioinformatics 2001, 17, 1228–1229. [Google Scholar] [CrossRef]
- Ma, J.; Gao, M.; Lu, Y.; Feng, X.; Zhang, J.; Lin, D.; Xiao, T.; Hu, Z.; Yuan, J.; Su, K.; et al. Gain of 1q25-32, 12q23-24.3, and 17q12-22 Facilitates Tumorigenesis and Progression of Human Squamous Cell Lung Cancer. J. Pathol. 2006, 210, 205–213. [Google Scholar] [CrossRef]
- Wu, G.; Zhou, L.; Khidr, L.; Guo, X.E.; Kim, W.; Lee, Y.M.; Krasieva, T.; Chen, P.-L. A Novel Role of the Chromokinesin Kif4A in DNA Damage Response. Cell Cycle 2008, 7, 2013–2020. [Google Scholar] [CrossRef]
- Barisic, M.; Aguiar, P.; Geley, S.; Maiato, H. Kinetochore Motors Drive Congression of Peripheral Polar Chromosomes by Overcoming Random Arm-Ejection Forces. Nat. Cell Biol. 2014, 16, 1249–1256. [Google Scholar] [CrossRef]
- Mazumdar, M.; Sundareshan, S.; Misteli, T. Human Chromokinesin KIF4A Functions in Chromosome Condensation and Segregation. J. Cell Biol. 2004, 166, 613–620. [Google Scholar] [CrossRef]
- Taniwaki, M.; Takano, A.; Ishikawa, N.; Yasui, W.; Inai, K.; Nishimura, H.; Tsuchiya, E.; Kohno, N.; Nakamura, Y.; Daigo, Y. Activation of KIF4A as a Prognostic Biomarker and Therapeutic Target for Lung Cancer. Clin. Cancer Res. 2007, 13, 6624–6631. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, G.; Singh, P.; Gurung, R.; Hakami, M.A.; Alkhorayef, N.; Alsaiari, A.A.; Alqahtani, L.S.; Hasan, M.R.; Rashid, S.; Kumar, A.; et al. Investigating the Role of Kinesin Family in Lung Adenocarcinoma via Integrated Bioinformatics Approach. Sci. Rep. 2023, 13, 9859. [Google Scholar] [CrossRef] [PubMed]
- Kahm, Y.-J.; Kim, I.-G.; Jung, U.; Lee, J.H.; Kim, R.-K. Impact of KIF4A on Cancer Stem Cells and EMT in Lung Cancer and Glioma. Cancers 2023, 15, 5523. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tang, W.; Li, H. Identification of KIF4A and Its Effect on the Progression of Lung Adenocarcinoma Based on the Bioinformatics Analysis. Biosci. Rep. 2021, 41, BSR20203973. [Google Scholar] [CrossRef] [PubMed]
- Singharajkomron, N.; Yodsurang, V.; Seephan, S.; Kungsukool, S.; Petchjorm, S.; Maneeganjanasing, N.; Promboon, W.; Dangwilailuck, W.; Pongrakhananon, V. Evaluating the Expression and Prognostic Value of Genes Encoding Microtubule-Associated Proteins in Lung Cancer. Int. J. Mol. Sci. 2022, 23, 14724. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wan, Y.; Deng, W.; Fei, F.; Wang, L.; Qi, F.; Zheng, Z. Promising Novel Biomarkers and Candidate Small-Molecule Drugs for Lung Adenocarcinoma: Evidence from Bioinformatics Analysis of High-Throughput Data. Open Med. 2022, 17, 96–112. [Google Scholar] [CrossRef]
- Zhang, L.; He, M.; Zhu, W.; Lv, X.; Zhao, Y.; Yan, Y.; Li, X.; Jiang, L.; Zhao, L.; Fan, Y.; et al. Identification of a Panel of Mitotic Spindle-Related Genes as a Signature Predicting Survival in Lung Adenocarcinoma. J. Cell. Physiol. 2020, 235, 4361–4375. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Yang, C.; Wei, J.; Li, W.; Qin, Y.; Liu, G. Identification and Verification of Microtubule Associated Genes in Lung Adenocarcinoma. Sci. Rep. 2023, 13, 16134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lu, W.; Samadi, N. KIF4A Knockdown Inhibits Tumor Progression and Promotes Chemo-Sensitivity via Induction of P21 in Lung Cancer Cells. Chem. Biol. Drug Des. 2023, 101, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Fu, C.; Lin, Y.; Dong, Z.; Pan, L.; Chen, W.; Zhao, J. Overexpression of Chromosome Kinesin Protein KIF4A Enhance Cisplatin Resistance in A549/DDP Cells. Int. J. Clin. Exp. Pathol. 2016, 9, 3331–3339. [Google Scholar]
- Pan, L.-N.; Zhang, Y.; Zhu, C.-J.; Dong, Z.-X. Kinesin KIF4A Is Associated with Chemotherapeutic Drug Resistance by Regulating Intracellular Trafficking of Lung Resistance-Related Protein. J. Zhejiang Univ. Sci. B 2017, 18, 1046–1054. [Google Scholar] [CrossRef]
- Wan, Q.; Shen, Y.; Zhao, H.; Wang, B.; Zhao, L.; Zhang, Y.; Bu, X.; Wan, M.; Shen, C. Impaired DNA Double-Strand Breaks Repair by Kinesin Family Member 4A Inhibition Renders Human H1299 Non-Small-Cell Lung Cancer Cells Sensitive to Cisplatin. J. Cell. Physiol. 2019, 234, 10360–10371. [Google Scholar] [CrossRef]
- Yan, T.; Jiang, Q.; Ni, G.; Ma, H.; Meng, Y.; Kang, G.; Xu, M.; Peng, F.; Li, H.; Chen, X.; et al. WZ-3146 Acts as a Novel Small Molecule Inhibitor of KIF4A to Inhibit Glioma Progression by Inducing Apoptosis. Cancer Cell Int. 2024, 24, 221. [Google Scholar] [CrossRef]
- Gao, W.; Lu, J.; Yang, Z.; Li, E.; Cao, Y.; Xie, L. Mitotic Functions and Characters of KIF11 in Cancers. Biomolecules 2024, 14, 386. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Tanaka, Y. Kinesin Superfamily Proteins (KIFs): Various Functions and Their Relevance for Important Phenomena in Life and Diseases. Exp. Cell Res. 2015, 334, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, X.; Lyu, Z. Analysis of Two-Gene Signatures and Related Drugs in Small-Cell Lung Cancer by Bioinformatics. Open Med. 2023, 18, 20230806. [Google Scholar] [CrossRef]
- Ling, J.; Wang, Y.; Ma, L.; Zheng, Y.; Tang, H.; Meng, L.; Zhang, L. KIF11, a plus End-Directed Kinesin, as a Key Gene in Benzo(a)pyrene-Induced Non-Small Cell Lung Cancer. Environ. Toxicol. Pharmacol. 2022, 89, 103775. [Google Scholar] [CrossRef]
- Schneider, M.A.; Christopoulos, P.; Muley, T.; Warth, A.; Klingmueller, U.; Thomas, M.; Herth, F.J.F.; Dienemann, H.; Mueller, N.S.; Theis, F.; et al. AURKA, DLGAP5, TPX2, KIF11 and CKAP5: Five Specific Mitosis-Associated Genes Correlate with Poor Prognosis for Non-Small Cell Lung Cancer Patients. Int. J. Oncol. 2017, 50, 365–372. [Google Scholar] [CrossRef]
- Li, Z.; Yu, B.; Qi, F.; Li, F. KIF11 Serves as an Independent Prognostic Factor and Therapeutic Target for Patients with Lung Adenocarcinoma. Front. Oncol. 2021, 11, 670218. [Google Scholar] [CrossRef]
- Maiti, P.; Sharma, P.; Nand, M.; Bhatt, I.D.; Ramakrishnan, M.A.; Mathpal, S.; Joshi, T.; Pant, R.; Mahmud, S.; Simal-Gandara, J.; et al. Integrated Machine Learning and Chemoinformatics-Based Screening of Mycotic Compounds against Kinesin Spindle ProteinEg5 for Lung Cancer Therapy. Molecules 2022, 27, 1639. [Google Scholar] [CrossRef]
- Sakuma, Y.; Hirai, S.; Yamaguchi, M.; Idogawa, M. Small Cell Lung Carcinoma Cells Depend on KIF11 for Survival. Int. J. Mol. Sci. 2024, 25, 7230. [Google Scholar] [CrossRef]
- Balasundaram, A. In Silico Analysis Revealed the Potential circRNA-miRNA-mRNA Regulative Network of Non-Small Cell Lung Cancer (NSCLC). Comput. Biol. Med. 2023, 152, 106315. [Google Scholar] [CrossRef]
- Mengyan, X.; Kun, D.; Xinming, J.; Yutian, W.; Yongqian, S. Identification and Verification of Hub Genes Associated with the Progression of Non-Small Cell Lung Cancer by Integrated Analysis. Front. Pharmacol. 2022, 13, 997842. [Google Scholar] [CrossRef]
- Liu, J.; Tian, Y.; Yi, L.; Gao, Z.; Lou, M.; Yuan, K. High KIF11 Expression Is Associated with Poor Outcome of NSCLC. Tumori J. 2022, 108, 40–46. [Google Scholar] [CrossRef]
- Fu, F.; Zhang, Y.; Gao, Z.; Zhao, Y.; Wen, Z.; Han, H.; Li, Y.; Chen, H. Development and Validation of a Five-Gene Model to Predict Postoperative Brain Metastasis in Operable Lung Adenocarcinoma. Int. J. Cancer 2020, 147, 584–592. [Google Scholar] [CrossRef]
- Saijo, T.; Ishii, G.; Ochiai, A.; Yoh, K.; Goto, K.; Nagai, K.; Kato, H.; Nishiwaki, Y.; Saijo, N. Eg5 Expression Is Closely Correlated with the Response of Advanced Non-Small Cell Lung Cancer to Antimitotic Agents Combined with Platinum Chemotherapy. Lung Cancer 2006, 54, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Sun, H.; Robichaux, J.; Pfeifer, M.; McDermott, U.; Travers, J.; Diao, L.; Xi, Y.; Tong, P.; Shen, L.; et al. A YAP/FOXM1 Axis Mediates EMT-Associated EGFR Inhibitor Resistance and Increased Expression of Spindle Assembly Checkpoint Components. Sci. Transl. Med. 2020, 12, eaaz4589. [Google Scholar] [CrossRef]
- Dong, A.; Wang, Z.-W.; Ni, N.; Li, L.; Kong, X.-Y. Similarity and Difference of Pathogenesis among Lung Cancer Subtypes Suggested by Expression Profile Data. Pathol. Res. Pract. 2021, 220, 153365. [Google Scholar] [CrossRef]
- Teicher, B.A.; Silvers, T.; Selby, M.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Parchment, R.; Krushkal, J.; Sonkin, D.; et al. Small Cell Lung Carcinoma Cell Line Screen of Etoposide/carboplatin plus a Third Agent. Cancer Med. 2017, 6, 1952–1964. [Google Scholar] [CrossRef]
- Kato, T.; Lee, D.; Huang, H.; Cruz, W.; Ujiie, H.; Fujino, K.; Wada, H.; Patel, P.; Hu, H.-P.; Hirohashi, K.; et al. Personalized siRNA-Nanoparticle Systemic Therapy Using Metastatic Lymph Node Specimens Obtained with EBUS-TBNA in Lung Cancer. Mol. Cancer Res. 2018, 16, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Good, J.A.D.; Wang, F.; Rath, O.; Kaan, H.Y.K.; Talapatra, S.K.; Podgórski, D.; MacKay, S.P.; Kozielski, F. Optimized S-Trityl-L-Cysteine-Based Inhibitors of Kinesin Spindle Protein with Potent in Vivo Antitumor Activity in Lung Cancer Xenograft Models. J. Med. Chem. 2013, 56, 1878–1893. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Fu, X.; Chang, M.; Wei, T.; Lin, R.; Tong, H.; Zhang, X.; Yuan, R.; Zhou, Z.; Huang, X.; et al. The Interaction of Calcium-Sensing Receptor with KIF11 Enhances Cisplatin Resistance in Lung Adenocarcinoma via BRCA1/cyclin B1 Pathway. Int. J. Biol. Sci. 2024, 20, 3892–3910. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, Y.; Hirai, S.; Sumi, T.; Niki, T.; Yamaguchi, M. Dual Inhibition of KIF11 and BCL2L1 Induces Apoptosis in Lung Adenocarcinoma Cells. Biochem. Biophys. Res. Commun. 2023, 678, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Tang, Y.; Shi, J.; Loy, C.T.; Amendt, C.; Wilm, C.; Zenke, F.T.; Mitchison, T.J. Quantitative Live Imaging of Cancer and Normal Cells Treated with Kinesin-5 Inhibitors Indicates Significant Differences in Phenotypic Responses and Cell Fate. Mol. Cancer Ther. 2008, 7, 3480–3489. [Google Scholar] [CrossRef] [PubMed]
- Rello-Varona, S.; Vitale, I.; Kepp, O.; Senovilla, L.; Jemaá, M.; Métivier, D.; Castedo, M.; Kroemer, G. Preferential Killing of Tetraploid Tumor Cells by Targeting the Mitotic Kinesin Eg5. Cell Cycle 2009, 8, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Saez, I.; Skoufias, D.A. Eg5 Targeting Agents: From New Anti-Mitotic Based Inhibitor Discovery to Cancer Therapy and Resistance. Biochem. Pharmacol. 2021, 184, 114364. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Kaufman, J.L.; Htut, M.; Agrawal, M.; Mazumder, A.; Cornell, R.F.; Zonder, J.A.; Fay, J.W.; Modiano, M.R.; Moshier, E.L.; et al. Filanesib plus Bortezomib and Dexamethasone in Relapsed/refractory t(11;14) and 1q21 Gain Multiple Myeloma. Cancer Med. 2022, 11, 358–370. [Google Scholar] [CrossRef]
- Hill, E.; Clarke, M.; Barr, F.A. The Rab6-Binding Kinesin, Rab6-KIFL, Is Required for Cytokinesis. EMBO J. 2000, 19, 5711–5719. [Google Scholar] [CrossRef]
- Jin, Z.; Peng, F.; Zhang, C.; Tao, S.; Xu, D.; Zhu, Z. Expression, Regulating Mechanism and Therapeutic Target of KIF20A in Multiple Cancer. Heliyon 2023, 9, e13195. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. Biomed Res. Int. 2016, 2016, 7969286. [Google Scholar] [CrossRef]
- Shi, C.; Huang, D.; Lu, N.; Chen, D.; Zhang, M.; Yan, Y.; Deng, L.; Lu, Q.; Lu, H.; Luo, S. Aberrantly Activated Gli2-KIF20A Axis Is Crucial for Growth of Hepatocellular Carcinoma and Predicts Poor Prognosis. Oncotarget 2016, 7, 26206–26219. [Google Scholar] [CrossRef]
- Xiu, G.; Sui, X.; Wang, Y.; Zhang, Z. FOXM1 Regulates Radiosensitivity of Lung Cancer Cell Partly by Upregulating KIF20A. Eur. J. Pharmacol. 2018, 833, 79–85. [Google Scholar] [CrossRef]
- Mushtaq, A.; Singh, P.; Tabassum, G.; Mohammad, T.; Hassan, M.I.; Syed, M.A.; Dohare, R. Unravelling Hub Genes as Potential Therapeutic Targets in Lung Cancer Using Integrated Transcriptomic Meta-Analysis and in Silico Approach. J. Biomol. Struct. Dyn. 2023, 41, 9089–9102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Mu, H.-J.; Shi, H.B.; Bi, H.X.; Jiang, Y.F.; Liu, G.H.; Zheng, H.Y.; Liu, B. Identification of Therapeutic Targets and Mechanisms of Tumorigenesis in Non-Small Cell Lung Cancer Using Multiple-Microarray Analysis. Medicine 2020, 99, e22815. [Google Scholar] [CrossRef]
- Cheng, Y.; Hou, K.; Wang, Y.; Chen, Y.; Zheng, X.; Qi, J.; Yang, B.; Tang, S.; Han, X.; Shi, D.; et al. Identification of Prognostic Signature and Gliclazide as Candidate Drugs in Lung Adenocarcinoma. Front. Oncol. 2021, 11, 665276. [Google Scholar] [CrossRef]
- Dai, B.; Ren, L.-Q.; Han, X.-Y.; Liu, D.-J. Bioinformatics Analysis Reveals 6 Key Biomarkers Associated with Non-Small-Cell Lung Cancer. J. Int. Med. Res. 2020, 48, 300060519887637. [Google Scholar] [CrossRef]
- Xing, B.; Shi, L.; Bao, Z.; Liang, Y.; Liu, B.; Liu, R. Molecular Clustering Based on Gene Set Expression and Its Relationship with Prognosis in Patients with Lung Adenocarcinoma. J. Thorac. Dis. 2022, 14, 1638–1650. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhou, L.-L.; Li, X.; Ni, J.; Chen, P.; Ma, R.; Wu, J.; Feng, J. Overexpression of KIF20A Confers Malignant Phenotype of Lung Adenocarcinoma by Promoting Cell Proliferation and Inhibiting Apoptosis. Cancer Med. 2018, 7, 4678–4689. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, W.; Xu, X.R.; Chen, S. Drug Resistance Related Genes in Lung Adenocarcinoma Predict Patient Prognosis and Influence the Tumor Microenvironment. Sci. Rep. 2023, 13, 9682. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Liu, X.; Wu, J.; Zhang, D.; Tian, J.; Wang, T.; Liu, S.; Meng, Z.; Wang, K.; Duan, X.; et al. Identification of Candidate Biomarkers Correlated with the Pathogenesis and Prognosis of Non-Small Cell Lung Cancer via Integrated Bioinformatics Analysis. Front. Genet. 2018, 9, 469. [Google Scholar] [CrossRef]
- Sun, D.; Zhang, H.; Zhang, C.; Wang, L. An Evaluation of KIF20A as a Prognostic Factor and Therapeutic Target for Lung Adenocarcinoma Using Integrated Bioinformatics Analysis. Front. Bioeng. Biotechnol. 2022, 10, 993820. [Google Scholar] [CrossRef]
- He, H.; Liang, L.; Huang, J.; Jiang, S.; Liu, Y.; Sun, X.; Li, Y.; Cong, L.; Jiang, Y. KIF20A Is Associated with Clinical Prognosis and Synergistic Effect of Gemcitabine Combined with Ferroptosis Inducer in Lung Adenocarcinoma. Front. Pharmacol. 2022, 13, 1007429. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O. Advancing Cancer Therapy: The Role of KIF20A as a Target for Inhibitor Development and Immunotherapy. Cancers 2024, 16, 2958. [Google Scholar] [CrossRef] [PubMed]
- Tcherniuk, S.; Skoufias, D.A.; Labriere, C.; Rath, O.; Gueritte, F.; Guillou, C.; Kozielski, F. Relocation of Aurora B and Survivin from Centromeres to the Central Spindle Impaired by a Kinesin-Specific MKLP-2 Inhibitor. Angew. Chem. Int. Ed. Engl. 2010, 49, 8228–8231. [Google Scholar] [CrossRef]
- Schrock, M.S.; Scarberry, L.; Stromberg, B.R.; Sears, C.; Torres, A.E.; Tallman, D.; Krupinski, L.; Chakravarti, A.; Summers, M.K. MKLP2 Functions in Early Mitosis to Ensure Proper Chromosome Congression. J. Cell Sci. 2022, 135, jcs259560. [Google Scholar] [CrossRef] [PubMed]
- Labrière, C.; Talapatra, S.K.; Thoret, S.; Bougeret, C.; Kozielski, F.; Guillou, C. New MKLP-2 Inhibitors in the Paprotrain Series: Design, Synthesis and Biological Evaluations. Bioorg. Med. Chem. 2016, 24, 721–734. [Google Scholar] [CrossRef]
- Ferrero, H.; Corachán, A.; Quiñonero, A.; Bougeret, C.; Pouletty, P.; Pellicer, A.; Domínguez, F. Inhibition of KIF20A by BKS0349 Reduces Endometriotic Lesions in a Xenograft Mouse Model. Mol. Hum. Reprod. 2019, 25, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, H.; Wu, Q.; Zhong, S. Berberine Targets KIF20A and CCNE2 to Inhibit the Progression of Nonsmall Cell Lung Cancer via the PI3K/AKT Pathway. Drug Dev. Res. 2023, 84, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Hirata, S.; Irie, A.; Senju, S.; Ikuta, Y.; Yokomine, K.; Harao, M.; Inoue, M.; Tomita, Y.; Tsunoda, T.; et al. Identification of HLA-A2-Restricted CTL Epitopes of a Novel Tumour-Associated Antigen, KIF20A, Overexpressed in Pancreatic Cancer. Br. J. Cancer 2011, 104, 300–307. [Google Scholar] [CrossRef]
- Abaza, A.; Soleilhac, J.-M.; Westendorf, J.; Piel, M.; Crevel, I.; Roux, A.; Pirollet, F. M Phase Phosphoprotein 1 Is a Human plus-End-Directed Kinesin-Related Protein Required for Cytokinesis. J. Biol. Chem. 2003, 278, 27844–27852. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, L.; Han, X. KIF20B Correlates with LUAD Progression and Is an Independent Risk Factor. Crit. Rev. Eukaryot. Gene Expr. 2024, 34, 49–59. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Rath, O.; Clayton, E.; Tomasi, S.; Kozielski, F. Depsidones from Lichens as Natural Product Inhibitors of M-Phase Phosphoprotein 1, a Human Kinesin Required for Cytokinesis. J. Nat. Prod. 2016, 79, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Brier, S.; Carletti, E.; DeBonis, S.; Hewat, E.; Lemaire, D.; Kozielski, F. The Marine Natural Product Adociasulfate-2 as a Tool to Identify the MT-Binding Region of Kinesins. Biochemistry 2006, 45, 15644–15653. [Google Scholar] [CrossRef] [PubMed]
- Vikberg, A.-L.; Vooder, T.; Lokk, K.; Annilo, T.; Golovleva, I. Mutation Analysis and Copy Number Alterations of KIF23 in Non-Small-Cell Lung Cancer Exhibiting KIF23 over-Expression. Onco Targets Ther. 2017, 10, 4969–4979. [Google Scholar] [CrossRef]
- Kato, T.; Wada, H.; Patel, P.; Hu, H.-P.; Lee, D.; Ujiie, H.; Hirohashi, K.; Nakajima, T.; Sato, M.; Kaji, M.; et al. Overexpression of KIF23 Predicts Clinical Outcome in Primary Lung Cancer Patients. Lung Cancer 2016, 92, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Li, H.; Zhang, F.; Lv, T.; Liu, H.; Song, Y. Expression of KIF23 and its prognostic role in non-small cell lung cancer: Analysis based on the data-mining of Oncomine. Zhongguo Fei Ai Za Zhi 2017, 20, 822–826. [Google Scholar] [PubMed]
- Yang, X.; Feng, Q.; Jing, J.; Yan, J.; Zeng, Z.; Zheng, H.; Cheng, X. Identification of Differentially Expressed Genes Associated with Lung Adenocarcinoma via Bioinformatics Analysis. Gen. Physiol. Biophys. 2021, 40, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Välk, K.; Vooder, T.; Kolde, R.; Reintam, M.-A.; Petzold, C.; Vilo, J.; Metspalu, A. Gene Expression Profiles of Non-Small Cell Lung Cancer: Survival Prediction and New Biomarkers. Oncology 2010, 79, 283–292. [Google Scholar] [CrossRef]
- Wu, X.; Sui, Z.; Zhang, H.; Wang, Y.; Yu, Z. Integrated Analysis of lncRNA-Mediated ceRNA Network in Lung Adenocarcinoma. Front. Oncol. 2020, 10, 554759. [Google Scholar] [CrossRef]
- Guo, L.; Li, H.; Li, W.; Tang, J. Construction and Investigation of a Combined Hypoxia and Stemness Index lncRNA-Associated ceRNA Regulatory Network in Lung Adenocarcinoma. BMC Med. Genom. 2020, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yu, C. Identification of Immune Infiltration and Prognostic Biomarkers in Small Cell Lung Cancer Based on Bioinformatic Methods from 3 Studies. Comb. Chem. High Throughput Screen. 2023, 26, 507–516. [Google Scholar] [PubMed]
- Chow, S.-E.; Hsu, C.-C.; Yang, C.-T.; Meir, Y.-J.J. YAP Co-Localizes with the Mitotic Spindle and Midbody to Safeguard Mitotic Division in Lung-Cancer Cells. FEBS J. 2023, 290, 5704–5719. [Google Scholar] [CrossRef] [PubMed]
- Iltzsche, F.; Simon, K.; Stopp, S.; Pattschull, G.; Francke, S.; Wolter, P.; Hauser, S.; Murphy, D.J.; Garcia, P.; Rosenwald, A.; et al. An Important Role for Myb-MuvB and Its Target Gene KIF23 in a Mouse Model of Lung Adenocarcinoma. Oncogene 2017, 36, 110–121. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Wei, Y.-L.; She, Z.-Y. Kinesin-7 CENP-E in Tumorigenesis: Chromosome Instability, Spindle Assembly Checkpoint, and Applications. Front. Mol. Biosci. 2024, 11, 1366113. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.D.; Wood, K.W.; Cleveland, D.W. The Kinesin-like Protein CENP-E Is Kinetochore-Associated throughout Poleward Chromosome Segregation during Anaphase-A. J. Cell Sci. 1996, 109 Pt 5, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Putkey, F.R.; Cramer, T.; Morphew, M.K.; Silk, A.D.; Johnson, R.S.; McIntosh, J.R.; Cleveland, D.W. Unstable Kinetochore-Microtubule Capture and Chromosomal Instability Following Deletion of CENP-E. Dev. Cell 2002, 3, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Qu, T. Expression of CENPE and Its Prognostic Role in Non-Small Cell Lung Cancer. Open Med. 2019, 14, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Tian, C.; Liu, L.; Zeng, X.; Zhang, Y. Targeting CENP-E Augments Immunotherapy in Non-Small Cell Lung Cancer via Stabilizing PD-L1. Int. Immunopharmacol. 2024, 126, 111294. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Zhao, M.; Lu, Y.; Ning, H.; Yang, S.; Song, Y.; Chai, W.; Shi, X. CENPE Promotes Lung Adenocarcinoma Proliferation and Is Directly Regulated by FOXM1. Int. J. Oncol. 2019, 55, 257–266. [Google Scholar]
- Ma, Q.; Xu, Y.; Liao, H.; Cai, Y.; Xu, L.; Xiao, D.; Liu, C.; Pu, W.; Zhong, X.; Guo, X. Identification and Validation of Key Genes Associated with Non-Small-Cell Lung Cancer. J. Cell. Physiol. 2019, 234, 22742–22752. [Google Scholar] [CrossRef]
- Pinto, B.; Silva, J.P.N.; Silva, P.M.A.; Barbosa, D.J.; Sarmento, B.; Tavares, J.C.; Bousbaa, H. Maximizing Anticancer Response with MPS1 and CENPE Inhibition alongside Apoptosis Induction. Pharmaceutics 2023, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, Y.; Deng, Y.; Dong, Y.; Jiang, J.; Chen, S.; Kang, W.; Deng, J.; Sun, H. Survival-Based Bioinformatics Analysis to Identify Hub Genes and Key Pathways in Non-Small Cell Lung Cancer. Transl. Cancer Res. 2019, 8, 1188–1198. [Google Scholar] [CrossRef]
- Valter, A.; Luhari, L.; Pisarev, H.; Truumees, B.; Planken, A.; Smolander, O.P.; Oselin, K. Genomic Alterations as Independent Prognostic Factors to Predict the Type of Lung Cancer Recurrence. Gene 2023, 885, 147690. [Google Scholar] [CrossRef] [PubMed]
- Tomoshige, K.; Matsumoto, K.; Tsuchiya, T.; Oikawa, M.; Miyazaki, T.; Yamasaki, N.; Mishima, H.; Kinoshita, A.; Kubo, T.; Fukushima, K.; et al. Germline Mutations Causing Familial Lung Cancer. J. Hum. Genet. 2015, 60, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Fan, L.Y.-N.; Lam, E.W.-F. The FOXO3-FOXM1 Axis: A Key Cancer Drug Target and a Modulator of Cancer Drug Resistance. Semin. Cancer Biol. 2018, 50, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; Heath, E.I.; Schelman, W.R.; Johnson, B.M.; Kirby, L.C.; Lynch, K.M.; Botbyl, J.D.; Lampkin, T.A.; Holen, K.D. First-Time-in-Human Study of GSK923295, a Novel Antimitotic Inhibitor of Centromere-Associated Protein E (CENP-E), in Patients with Refractory Cancer. Cancer Chemother. Pharmacol. 2012, 69, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; McDonald, A.; Zhou, H.-J.; Adams, N.D.; Parrish, C.A.; Duffy, K.J.; Fitch, D.M.; Tedesco, R.; Ashcraft, L.W.; Yao, B.; et al. Discovery of the First Potent and Selective Inhibitor of Centromere-Associated Protein E: GSK923295. ACS Med. Chem. Lett. 2010, 1, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Balamuth, N.J.; Wood, A.; Wang, Q.; Jagannathan, J.; Mayes, P.; Zhang, Z.; Chen, Z.; Rappaport, E.; Courtright, J.; Pawel, B.; et al. Serial Transcriptome Analysis and Cross-Species Integration Identifies Centromere-Associated Protein E as a Novel Neuroblastoma Target. Cancer Res. 2010, 70, 2749–2758. [Google Scholar] [CrossRef]
- Wood, K.W.; Lad, L.; Luo, L.; Qian, X.; Knight, S.D.; Nevins, N.; Brejc, K.; Sutton, D.; Gilmartin, A.G.; Chua, P.R.; et al. Antitumor Activity of an Allosteric Inhibitor of Centromere-Associated Protein-E. Proc. Natl. Acad. Sci. USA 2010, 107, 5839–5844. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Duijf, P.H.G.; Khanna, K.K. Mitotic Slippage: An Old Tale with a New Twist. Cell Cycle 2019, 18, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Tipton, A.R.; Ji, W.; Sturt-Gillespie, B.; Bekier, M.E., 2nd; Wang, K.; Taylor, W.R.; Liu, S.-T. Monopolar Spindle 1 (MPS1) Kinase Promotes Production of Closed MAD2 (C-MAD2) Conformer and Assembly of the Mitotic Checkpoint Complex. J. Biol. Chem. 2013, 288, 35149–35158. [Google Scholar] [CrossRef]
- Mayes, P.A.; Degenhardt, Y.Y.; Wood, A.; Toporovskya, Y.; Diskin, S.J.; Haglund, E.; Moy, C.; Wooster, R.; Maris, J.M. Mitogen-Activated Protein Kinase (MEK/ERK) Inhibition Sensitizes Cancer Cells to Centromere-Associated Protein E Inhibition. Int. J. Cancer 2013, 132, E149–E157. [Google Scholar] [CrossRef]
- Li, J.; Li, H.; Zhang, C.; Zhang, C.; Jiang, L.; Wang, H.; Liu, H. Identification of a Gene Signature Closely Related to Immunosuppressive Tumour Microenvironment Predicting Prognosis of Patients in EGFR Mutant Lung Adenocarcinoma. Front. Oncol. 2021, 11, 732841. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.B.; Carlsen, C.L.; Scribano, C.M.; Pattaswamy, S.M.; Burkard, M.E.; Weaver, B.A. CENP-E Inhibition Induces Chromosomal Instability and Synergizes with Diverse Microtubule-Targeting Agents in Breast Cancer. Cancer Res. 2024, 84, 2674–2689. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, K.; Matsura, A.; Shimada, M.; Ishida-Ishihara, S.; Sato, F.; Yamamoto, T.; Yaguchi, K.; Kawamoto, E.; Kuroda, T.; Matsuo, K.; et al. Tetraploidy-Linked Sensitization to CENP-E Inhibition in Human Cells. Mol. Oncol. 2023, 17, 1148–1166. [Google Scholar] [CrossRef] [PubMed]
- Mohd Amin, A.S.; Eastwood, S.; Pilcher, C.; Truong, J.Q.; Foitzik, R.; Boag, J.; Gorringe, K.L.; Holien, J.K. KIF18A Inhibition: The next Big Player in the Search for Cancer Therapeutics. Cancer Metastasis Rev. 2024, 44, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, M.; Zhang, Z.; Zhang, L.; Liang, X.; Sun, L.; Zhong, D. High Kinesin Family Member 18A Expression Correlates with Poor Prognosis in Primary Lung Adenocarcinoma. Thorac. Cancer 2019, 10, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Jiang, L.; Lin, H.; Li, X.; Long, X.; Zhou, Y.; Li, B.; Li, Z. Overexpression of KIF18A Promotes Cell Proliferation, Inhibits Apoptosis, and Independently Predicts Unfavorable Prognosis in Lung Adenocarcinoma: Biological and Prognostic Role of KIF18A in LUAD. IUBMB Life 2019, 71, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zeng, H.; Zheng, J.; He, Y.; Zhuang, X.; Cai, J.; Huang, H.; Huang, H.; Xu, M. Preliminary Study on the Clinical Significance of Kinesin Kif18a in Nonsmall Cell Lung Cancer: An Analysis of 100 Cases: An Analysis of 100 Cases. Medicine 2020, 99, e19011. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yang, K.; Chen, J.; Qi, L.; Zhou, X.; Wang, P. Comprehensive Pan-Cancer Analysis of KIF18A as a Marker for Prognosis and Immunity. Biomolecules 2023, 13, 326. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.-T.; Zhong, F.-K. Kinesin Family Member 18A (KIF18A) Contributes to the Proliferation, Migration, and Invasion of Lung Adenocarcinoma Cells in Vitro and in Vivo. Dis. Markers 2019, 2019, 6383685. [Google Scholar] [CrossRef]
- Tooker, B.C.; Newman, L.S.; Bowler, R.P.; Karjalainen, A.; Oksa, P.; Vainio, H.; Pukkala, E.; Brandt-Rauf, P.W. Proteomic Detection of Cancer in Asbestosis Patients Using SELDI-TOF Discovered Serum Protein Biomarkers. Biomarkers 2011, 16, 181–191. [Google Scholar] [CrossRef]
- Marquis, C.; Fonseca, C.L.; Queen, K.A.; Wood, L.; Vandal, S.E.; Malaby, H.L.H.; Clayton, J.E.; Stumpff, J. Chromosomally Unstable Tumor Cells Specifically Require KIF18A for Proliferation. Nat. Commun. 2021, 12, 1213. [Google Scholar] [CrossRef]
- Cohen-Sharir, Y.; McFarland, J.M.; Abdusamad, M.; Marquis, C.; Bernhard, S.V.; Kazachkova, M.; Tang, H.; Ippolito, M.R.; Laue, K.; Zerbib, J.; et al. Aneuploidy Renders Cancer Cells Vulnerable to Mitotic Checkpoint Inhibition. Nature 2021, 590, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Quinton, R.J.; DiDomizio, A.; Vittoria, M.A.; Kotýnková, K.; Ticas, C.J.; Patel, S.; Koga, Y.; Vakhshoorzadeh, J.; Hermance, N.; Kuroda, T.S.; et al. Whole-Genome Doubling Confers Unique Genetic Vulnerabilities on Tumour Cells. Nature 2021, 590, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Payton, M.; Belmontes, B.; Hanestad, K.; Moriguchi, J.; Chen, K.; McCarter, J.D.; Chung, G.; Ninniri, M.S.; Sun, J.; Manoukian, R.; et al. Small-Molecule Inhibition of Kinesin KIF18A Reveals a Mitotic Vulnerability Enriched in Chromosomally Unstable Cancers. Nat. Cancer 2024, 5, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Gliech, C.R.; Yeow, Z.Y.; Tapias-Gomez, D.; Yang, Y.; Huang, Z.; Tijhuis, A.E.; Spierings, D.C.; Foijer, F.; Chung, G.; Tamayo, N.; et al. Weakened APC/C Activity at Mitotic Exit Drives Cancer Vulnerability to KIF18A Inhibition. EMBO J. 2024, 43, 666–694. [Google Scholar] [CrossRef]
- Chen, Q.; Le, X.; Li, Q.; Liu, S.; Chen, Z. Exploration of Inhibitors Targeting KIF18A with Ploidy-Specific Lethality. Drug Discov. Today 2024, 29, 104142. [Google Scholar] [CrossRef]
- Tu, P.; Zhang, C.; Lu, B.; Xia, Y.; Yang, F. Abstract 664: GSC000190, a Highly Potent Inhibitor of KIF18A, for Tumors with Chromosome Instability. Cancer Res. 2024, 84, 664. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Macurek, L.; van der Vaart, B.; Galli, M.; Akhmanova, A.; Medema, R.H. A Complex of Kif18b and MCAK Promotes Microtubule Depolymerization and Is Negatively Regulated by Aurora Kinases. Curr. Biol. 2011, 21, 1356–1365. [Google Scholar] [CrossRef]
- Zhu, Y.; Cao, F.; Liu, F.; Liu, S.; Meng, L.; Gu, L.; Zhao, H.; Sang, M.; Shan, B. Identification of Potential Circular RNA Biomarkers in Lung Adenocarcinoma: A Bioinformatics Analysis and Retrospective Clinical Study. Oncol. Lett. 2022, 23, 144. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Pan, X.; Shang, Y.; Ni, D.-T.; Wu, F.-L. KIF18B as a Regulator in Microtubule Movement Accelerates Tumor Progression and Triggers Poor Outcome in Lung Adenocarcinoma. Tissue Cell 2019, 61, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Jiang, L.; Long, X.; Zhou, Y.; Deng, S.; Lin, H.; Li, X. Clinical Significance And Integrative Analysis Of Kinesin Family Member 18B In Lung Adenocarcinoma. Onco Targets Ther. 2019, 12, 9249–9264. [Google Scholar] [CrossRef] [PubMed]
- Ohsugi, M.; Adachi, K.; Horai, R.; Kakuta, S.; Sudo, K.; Kotaki, H.; Tokai-Nishizumi, N.; Sagara, H.; Iwakura, Y.; Yamamoto, T. Kid-Mediated Chromosome Compaction Ensures Proper Nuclear Envelope Formation. Cell 2008, 132, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Pike, R.; Ortiz-Zapater, E.; Lumicisi, B.; Santis, G.; Parsons, M. KIF22 Coordinates CAR and EGFR Dynamics to Promote Cancer Cell Proliferation. Sci. Signal. 2018, 11, eaaq1060. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Macůrek, L.; Janssen, A.; Geers, E.F.; Alvarez-Fernández, M.; Medema, R.H. Kif15 Cooperates with eg5 to Promote Bipolar Spindle Assembly. Curr. Biol. 2009, 19, 1703–1711. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, B.; Xu, L.; Li, M.; Wu, J.; Zhou, Y.; Li, Y. Downregulation of KIF15 Inhibits the Tumorigenesis of Non-Small-Cell Lung Cancer via Inactivating Raf/MEK/ERK Signaling. Histol. Histopathol. 2022, 37, 269–285. [Google Scholar] [PubMed]
- Qiao, Y.; Chen, J.; Ma, C.; Liu, Y.; Li, P.; Wang, Y.; Hou, L.; Liu, Z. Increased KIF15 Expression Predicts a Poor Prognosis in Patients with Lung Adenocarcinoma. Cell. Physiol. Biochem. 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Qin, S.; Long, X.; Zhao, Q.; Zhao, W. Co-Expression Network Analysis Identified Genes Associated with Cancer Stem Cell Characteristics in Lung Squamous Cell Carcinoma. Cancer Investig. 2020, 38, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; An, J.; Jia, R.; Liu, C.; Zhang, Y. Identification and Verification of Metabolism-Related Immunotherapy Features and Prognosis in Lung Adenocarcinoma. Curr. Med. Chem. 2024, 31, 1423–1441. [Google Scholar] [CrossRef]
- Ma, H.T.; Erdal, S.; Huang, S.; Poon, R.Y.C. Synergism between Inhibitors of Aurora A and KIF11 Overcomes KIF15-Dependent Drug Resistance. Mol. Oncol. 2014, 8, 1404–1418. [Google Scholar] [CrossRef]
- Sturgill, E.G.; Norris, S.R.; Guo, Y.; Ohi, R. Kinesin-5 Inhibitor Resistance Is Driven by Kinesin-12. J. Cell Biol. 2016, 213, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Milic, B.; Chakraborty, A.; Han, K.; Bassik, M.C.; Block, S.M. KIF15 Nanomechanics and Kinesin Inhibitors, with Implications for Cancer Chemotherapeutics. Proc. Natl. Acad. Sci. USA 2018, 115, E4613–E4622. [Google Scholar] [CrossRef] [PubMed]
- Solon, A.L.; Zaniewski, T.M.; O’Brien, P.; Clasby, M.; Hancock, W.O.; Ohi, R. Synergy between Inhibitors of Two Mitotic Spindle Assembly Motors Undermines an Adaptive Response. Mol. Biol. Cell 2022, 33, ar132. [Google Scholar] [CrossRef]
- Sebastian, J. Dihydropyrazole and Dihydropyrrole Structures Based Design of Kif15 Inhibitors as Novel Therapeutic Agents for Cancer. Comput. Biol. Chem. 2017, 68, 164–174. [Google Scholar] [CrossRef]
- Dumas, M.E.; Chen, G.-Y.; Kendrick, N.D.; Xu, G.; Larsen, S.D.; Jana, S.; Waterson, A.G.; Bauer, J.A.; Hancock, W.; Sulikowski, G.A.; et al. Dual Inhibition of Kif15 by Oxindole and Quinazolinedione Chemical Probes. Bioorg. Med. Chem. Lett. 2019, 29, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wen, J.; Liu, D.; Xu, X.; Fan, M.; Zhang, Z. The Molecular Mechanism of Kinesin Family Member 2A (KIF2A) Underlying Non-Small Cell Lung Cancer: The Effect of Its Knockdown on Malignant Behaviors, Stemness, Chemosensitivity, and Potential Regulated Signaling Pathways. Am. J. Transl. Res. 2022, 14, 68–85. [Google Scholar]
- Xu, L.; Zhang, X.; Wang, Z.; Zhao, X.; Zhao, L.; Hu, Y. Kinesin Family Member 2A Promotes Cancer Cell Viability, Mobility, Stemness, and Chemoresistance to Cisplatin by Activating the PI3K/AKT/VEGF Signaling Pathway in Non-Small Cell Lung Cancer. Am. J. Transl. Res. 2021, 13, 2060–2076. [Google Scholar]
- Xie, T.; Li, X.; Ye, F.; Lu, C.; Huang, H.; Wang, F.; Cao, X.; Zhong, C. High KIF2A Expression Promotes Proliferation, Migration and Predicts Poor Prognosis in Lung Adenocarcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 65–72. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Z.; Yu, H. Kinesin Family Member 2A High Expression Correlates with Advanced Tumor Stages and Worse Prognosis in Non-Small Cell Lung Cancer Patients. J. Clin. Lab. Anal. 2020, 34, e23135. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wang, L.; Ge, L.; Xu, D.; Zhang, R. IGF2BP1 Facilitates Non-Small Cell Lung Cancer Progression by Regulating the KIF2A-Mediated Wnt/β-Catenin Pathway. Funct. Integr. Genom. 2023, 24, 4. [Google Scholar] [CrossRef]
- Zheng, P.; Jiang, J.; Li, L.; Wei, L.; Li, J.; Jin, L. Circ_SATB2 Knockdown Inhibits the Tumorigenesis of Non-Small Cell Lung Cancer via miR-760/KIF2A Axis. Histol. Histopathol. 2023, 38, 431–441. [Google Scholar] [PubMed]
- Uchida, A.; Seki, N.; Mizuno, K.; Yamada, Y.; Misono, S.; Sanada, H.; Kikkawa, N.; Kumamoto, T.; Suetsugu, T.; Inoue, H. Regulation of KIF2A by Antitumor miR-451a Inhibits Cancer Cell Aggressiveness Features in Lung Squamous Cell Carcinoma. Cancers 2019, 11, 258. [Google Scholar] [CrossRef]
- Zhu, Y.; Ma, C.; Lv, A.; Kou, C. Circular RNA circ_0010235 Sponges miR-338-3p to Play Oncogenic Role in Proliferation, Migration and Invasion of Non-Small-Cell Lung Cancer Cells through Modulating KIF2A. Ann. Med. 2021, 53, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Kreis, N.-N.; Moon, H.H.; Wordeman, L.; Louwen, F.; Solbach, C.; Yuan, J.; Ritter, A. KIF2C/MCAK a Prognostic Biomarker and Its Oncogenic Potential in Malignant Progression, and Prognosis of Cancer Patients: A Systematic Review and Meta-Analysis as Biomarker. Crit. Rev. Clin. Lab. Sci. 2024, 61, 404–434. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, W.; Sun, L.; Yu, H.; Wang, Y.; Feng, L.; Yang, H. KIF2C Accelerates the Development of Non-Small Cell Lung Cancer and Is Suppressed by miR-186-3p via the AKT-GSK3β-β-Catenin Pathway. Sci. Rep. 2023, 13, 7288. [Google Scholar] [CrossRef]
- Gan, H.; Lin, L.; Hu, N.; Yang, Y.; Gao, Y.; Pei, Y.; Chen, K.; Sun, B. KIF2C Exerts an Oncogenic Role in Nonsmall Cell Lung Cancer and Is Negatively Regulated by miR-325-3p. Cell Biochem. Funct. 2019, 37, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Miao, R.; Han, T.; Liu, Y.; Zhou, J.; Guo, J.; Xing, Y.; Bai, Y.; Wu, J.; Hu, D. KIF2C as a Potential Therapeutic Target: Insights from Lung Adenocarcinoma Subtype Classification and Functional Experiments. Mol. Omics 2024, 20, 417–429. [Google Scholar] [CrossRef]
- Bai, Y.; Xiong, L.; Zhu, M.; Yang, Z.; Zhao, J.; Tang, H. Co-Expression Network Analysis Identified KIF2C in Association with Progression and Prognosis in Lung Adenocarcinoma. Cancer Biomark. 2019, 24, 371–382. [Google Scholar] [CrossRef]
- Li, Z.; Qi, F.; Li, F. Establishment of a Gene Signature to Predict Prognosis for Patients with Lung Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 8479. [Google Scholar] [CrossRef]
- Song, Y.-J.; Tan, J.; Gao, X.-H.; Wang, L.-X. Integrated Analysis Reveals Key Genes with Prognostic Value in Lung Adenocarcinoma. Cancer Manag. Res. 2018, 10, 6097–6108. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Husted, S.; Pilrose, J.; Ems-McClung, S.C.; Stout, J.R.; Carpenter, R.L.; Walczak, C.E. MCAK Inhibitors Induce Aneuploidy in Triple-Negative Breast Cancer Models. Cancers 2023, 15, 3309. [Google Scholar] [CrossRef] [PubMed]
- Laucius, C.D.; Orr, B.; Compton, D.A. Chromosomal Instability Suppresses the Growth of K-Ras-Induced Lung Adenomas. Cell Cycle 2019, 18, 1702–1713. [Google Scholar] [CrossRef]
- Manning, A.L.; Ganem, N.J.; Bakhoum, S.F.; Wagenbach, M.; Wordeman, L.; Compton, D.A. The Kinesin-13 Proteins Kif2a, Kif2b, and Kif2c/MCAK Have Distinct Roles during Mitosis in Human Cells. Mol. Biol. Cell 2007, 18, 2970–2979. [Google Scholar] [CrossRef]
- Kobayashi, T.; Tsang, W.Y.; Li, J.; Lane, W.; Dynlacht, B.D. Centriolar Kinesin Kif24 Interacts with CP110 to Remodel Microtubules and Regulate Ciliogenesis. Cell 2011, 145, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ni, H.; Yang, D.; Niu, Y.; Chen, K.; Xu, J.; Wang, F.; Tang, S.; Shi, Y.; Zhang, H.; et al. Driver and Novel Genes Correlated with Metastasis of Non-Small Cell Lung Cancer: A Comprehensive Analysis. Pathol. Res. Pract. 2021, 224, 153551. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.-X.; Yang, W.-X. KIFC1: A Promising Chemotherapy Target for Cancer Treatment? Oncotarget 2016, 7, 48656–48670. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Duan, Y.; Gong, S.; Zhu, Q.; Liu, X.; Liu, Z. An Integrative Pan-Cancer Analysis of Kinesin Family Member C1 (KIFC1) in Human Tumors. Biomedicines 2022, 10, 637. [Google Scholar] [CrossRef]
- Sharma, N.; Setiawan, D.; Hamelberg, D.; Narayan, R.; Aneja, R. Computational Benchmarking of Putative KIFC1 Inhibitors. Med. Res. Rev. 2023, 43, 293–318. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, B.Z.; Di Ciano-Oliveira, C.; Wu, Y.F.; Khavkine Binstock, S.S.; Soria-Bretones, I.; Pham, N.-A.; Elia, A.J.; Chari, R.; Lam, W.L.; et al. Identification of KIFC1 as a Putative Vulnerability in Lung Cancers with Centrosome Amplification. Cancer Gene Ther. 2024, 31, 1559–1570. [Google Scholar] [CrossRef]
- Celik, B.; Pasin, O.; Sen, S.; Tuncer, S.B.; Kayım, Z.Y.; Erciyas, S.K.; Erdogan, O.S.; Gultaslar, B.K.; Ghafour, A.A.; Yazıcı, H.; et al. DNA Methylation of KIFC1 Gene in Determination of Histological Diagnosis, Prognosis and Metastasis of Lung Cancer. Pathol. Res. Pract. 2023, 249, 154742. [Google Scholar] [CrossRef]
- Liu, Y.; Zhan, P.; Zhou, Z.; Xing, Z.; Zhu, S.; Ma, C.; Li, Q.; Zhu, Q.; Miao, Y.; Zhang, J.; et al. The Overexpression of KIFC1 Was Associated with the Proliferation and Prognosis of Non-Small Cell Lung Cancer. J. Thorac. Dis. 2016, 8, 2911–2923. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, W.; Miao, X.; Wang, X. KIFC1 Aggravates Non-Small-Cell Lung Cancer Cell Proliferation and Metastasis via Provoking TGF-β/SMAD Signal. Cell. Mol. Biol. 2023, 69, 293–299. [Google Scholar] [PubMed]
- Li, X.; Wang, S.; Ruan, P.; Bajinka, O.; Zhang, W. High Expression of KIFC1 Is a Poor Prognostic Biomarker and Correlates with TP53 Mutation in Lung Cancer. Medicine 2024, 103, e37286. [Google Scholar] [CrossRef]
- Grinberg-Rashi, H.; Ofek, E.; Perelman, M.; Skarda, J.; Yaron, P.; Hajdúch, M.; Jacob-Hirsch, J.; Amariglio, N.; Krupsky, M.; Simansky, D.A.; et al. The Expression of Three Genes in Primary Non-Small Cell Lung Cancer Is Associated with Metastatic Spread to the Brain. Clin. Cancer Res. 2009, 15, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Godinho, S.A.; Chandhok, N.S.; Ganem, N.J.; Azioune, A.; Thery, M.; Pellman, D. Mechanisms to Suppress Multipolar Divisions in Cancer Cells with Extra Centrosomes. Genes Dev. 2008, 22, 2189–2203. [Google Scholar] [CrossRef]
- Patel, N.; Weekes, D.; Drosopoulos, K.; Gazinska, P.; Noel, E.; Rashid, M.; Mirza, H.; Quist, J.; Brasó-Maristany, F.; Mathew, S.; et al. Integrated Genomics and Functional Validation Identifies Malignant Cell Specific Dependencies in Triple Negative Breast Cancer. Nat. Commun. 2018, 9, 1044. [Google Scholar] [CrossRef] [PubMed]
- Huo, W.; Zhu, X.-M.; Pan, X.-Y.; Du, M.; Sun, Z.; Li, Z.-M. MicroRNA-527 Inhibits TGF-β/SMAD Induced Epithelial-Mesenchymal Transition via Downregulating SULF2 Expression in Non-Small-Cell Lung Cancer. Math. Biosci. Eng. 2019, 16, 4607–4621. [Google Scholar] [CrossRef] [PubMed]
- Hata, S.; Pastor Peidro, A.; Panic, M.; Liu, P.; Atorino, E.; Funaya, C.; Jäkle, U.; Pereira, G.; Schiebel, E. The Balance between KIFC3 and EG5 Tetrameric Kinesins Controls the Onset of Mitotic Spindle Assembly. Nat. Cell Biol. 2019, 21, 1138–1151. [Google Scholar] [CrossRef]
- Nachbar, J.; Lázaro-Diéguez, F.; Prekeris, R.; Cohen, D.; Müsch, A. KIFC3 Promotes Mitotic Progression and Integrity of the Central Spindle in Cytokinesis. Cell Cycle 2014, 13, 426–433. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Y.; Jiang, X.; Guan, J.; Wang, H.; Zhang, J.; Tong, Y.; Qiu, X.; Zhou, R. KIFC3 Promotes the Proliferation, Migration and Invasion of Non-Small Cell Lung Cancer through the PI3K/AKT Signaling Pathway. Sci. Rep. 2024, 14, 20471. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Liu, H.; Luo, A.; Zhang, Q. KIFC3 Promotes the Progression of Non-Small Cell Lung Cancer Cells through the PI3K/Akt Pathway. Thorac. Cancer 2024, 15, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, Y.; Sakaguchi, M.; Terabayashi, T.; Inenaga, T.; Inoue, S.; Kobayashi, C.; Oshima, N.; Kiyonari, H.; Nakagata, N.; Sato, Y.; et al. Kif26b, a Kinesin Family Gene, Regulates Adhesion of the Embryonic Kidney Mesenchyme. Proc. Natl. Acad. Sci. USA 2010, 107, 9240–9245. [Google Scholar] [CrossRef] [PubMed]
- Guillabert-Gourgues, A.; Jaspard-Vinassa, B.; Bats, M.-L.; Sewduth, R.N.; Franzl, N.; Peghaire, C.; Jeanningros, S.; Moreau, C.; Roux, E.; Larrieu-Lahargue, F.; et al. Kif26b Controls Endothelial Cell Polarity through the Dishevelled/Daam1-Dependent Planar Cell Polarity-Signaling Pathway. Mol. Biol. Cell 2016, 27, 941–953. [Google Scholar] [CrossRef]
- Chen, N.; Wu, Q.; Zhang, G.; Fu, J.; Geng, Q.; Zhang, Y. Deactivation of AKT/GSK-3β-Mediated Wnt/β-Catenin Pathway by Silencing of KIF26B Weakens the Malignant Behaviors of Non-Small Cell Lung Cancer. Tissue Cell 2022, 76, 101750. [Google Scholar] [CrossRef]
- Cheung, H.O.-L.; Zhang, X.; Ribeiro, A.; Mo, R.; Makino, S.; Puviindran, V.; Law, K.K.L.; Briscoe, J.; Hui, C.-C. The Kinesin Protein Kif7 Is a Critical Regulator of Gli Transcription Factors in Mammalian Hedgehog Signaling. Sci. Signal. 2009, 2, ra29. [Google Scholar] [CrossRef] [PubMed]
- Klejnot, M.; Kozielski, F. Structural Insights into Human Kif7, a Kinesin Involved in Hedgehog Signalling. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 154–159. [Google Scholar] [CrossRef]
- Hu, Y.; Wu, M.-Z.; Gu, N.-J.; Xu, H.-T.; Li, Q.-C.; Wu, G.-P. Human Papillomavirus 16 (HPV 16) E6 but Not E7 Inhibits the Antitumor Activity of LKB1 in Lung Cancer Cells by Downregulating the Expression of KIF7. Thorac. Cancer 2020, 11, 3175–3180. [Google Scholar] [CrossRef]
- Coles, G.L.; Baglia, L.A.; Ackerman, K.G. KIF7 Controls the Proliferation of Cells of the Respiratory Airway through Distinct Microtubule Dependent Mechanisms. PLoS Genet. 2015, 11, e1005525. [Google Scholar] [CrossRef] [PubMed]
- Muhia, M.; Thies, E.; Labonté, D.; Ghiretti, A.E.; Gromova, K.V.; Xompero, F.; Lappe-Siefke, C.; Hermans-Borgmeyer, I.; Kuhl, D.; Schweizer, M.; et al. The Kinesin KIF21B Regulates Microtubule Dynamics and Is Essential for Neuronal Morphology, Synapse Function, and Learning and Memory. Cell Rep. 2016, 15, 968–977. [Google Scholar] [CrossRef] [PubMed]
- van Riel, W.E.; Rai, A.; Bianchi, S.; Katrukha, E.A.; Liu, Q.; Heck, A., Jr.; Hoogenraad, C.C.; Steinmetz, M.O.; Kapitein, L.C.; Akhmanova, A. Kinesin-4 KIF21B Is a Potent Microtubule Pausing Factor. eLife 2017, 6, e24746. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-G.; Pan, F.; Shao, J.-B.; Yan, Q.-Q.; Lu, L.; Zhang, N. Kinesin Superfamily Protein 21B Acts as an Oncogene in Non-Small Cell Lung Cancer. Cancer Cell Int. 2020, 20, 233. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Noda, Y. Intracellular Transport and Kinesin Superfamily Proteins, KIFs: Structure, Function, and Dynamics. Physiol. Rev. 2008, 88, 1089–1118. [Google Scholar] [CrossRef]
- Dong, L.; Feng, C.; Cheng, W.; Huang, A.; Ying, K. FOXP3 Targets KIF5A to Increase Lactate Production and Promote Docetaxel Resistance in Lung Adenocarcinoma. Acta Biochim. Biophys. Sin. 2024, 56, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, R.; Auger, N.; Auclin, E.; Besse, B. Clinical and Translational Implications of RET Rearrangements in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Choi, Y.L.; Togashi, Y.; Soda, M.; Hatano, S.; Inamura, K.; Takada, S.; Ueno, T.; Yamashita, Y.; Satoh, Y.; et al. KIF5B-ALK, a Novel Fusion Oncokinase Identified by an Immunohistochemistry-Based Diagnostic System for ALK-Positive Lung Cancer. Clin. Cancer Res. 2009, 15, 3143–3149. [Google Scholar] [CrossRef] [PubMed]
- Gow, C.-H.; Liu, Y.-N.; Li, H.-Y.; Hsieh, M.-S.; Chang, S.-H.; Luo, S.-C.; Tsai, T.-H.; Chen, P.-L.; Tsai, M.-F.; Shih, J.-Y. Oncogenic Function of a KIF5B-MET Fusion Variant in Non-Small Cell Lung Cancer. Neoplasia 2018, 20, 838–847. [Google Scholar] [CrossRef]
- Cho, J.H.; Ku, B.M.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K.; Ahn, M.-J. KIF5B-MET Gene Rearrangement with Robust Antitumor Activity in Response to Crizotinib in Lung Adenocarcinoma. J. Thorac. Oncol. 2018, 13, e29–e31. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K. Discovery Stories of RET Fusions in Lung Cancer: A Mini-Review. Front. Physiol. 2019, 10, 216. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Liu, C.; Wang, W.; Li, M.; Lv, D.; Sun, G.; Chen, H.; Dong, X.; Miao, Z.; et al. Identification of a Novel KIF13A-RET Fusion in Lung Adenocarcinoma by next-Generation Sequencing. Lung Cancer 2018, 118, 27–29. [Google Scholar] [CrossRef]
- Mo, Z.; Cai, C.; Yao, J.; Zhao, J.; Zhang, M.; Liu, H.; Mu, X. First Case Report of a Novel KIF13A-ALK Fusion in a Lung Adenocarcinoma Patient and Response to Alectinib with a 4-Year Follow-Up. Front. Genet. 2023, 14, 1289346. [Google Scholar] [CrossRef]
- Xia, D.; Le, L.P.; Iafrate, A.J.; Lennerz, J. KIF13B-NRG1 Gene Fusion and KRAS Amplification in a Case of Natural Progression of Lung Cancer. Int. J. Surg. Pathol. 2017, 25, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Thankachan, J.M.; Setty, S.R.G. KIF13A-A Key Regulator of Recycling Endosome Dynamics. Front. Cell Dev. Biol. 2022, 10, 877532. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Wang, D.; Hirokawa, N. KIF13B Enhances the Endocytosis of LRP1 by Recruiting LRP1 to Caveolae. J. Cell Biol. 2014, 204, 395–408. [Google Scholar] [CrossRef]
- Yamada, K.H.; Kang, H.; Malik, A.B. Antiangiogenic Therapeutic Potential of Peptides Derived from the Molecular Motor KIF13B That Transports VEGFR2 to Plasmalemma in Endothelial Cells. Am. J. Pathol. 2017, 187, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Tang, D.; Zhao, Y.C.; Liu, H.; Luo, S.; Stinchcombe, T.E.; Glass, C.; Su, L.; Shen, S.; Christiani, D.C.; et al. Novel Genetic Variants in KIF16B and NEDD4L in the Endosome-Related Genes Are Associated with Nonsmall Cell Lung Cancer Survival. Int. J. Cancer 2020, 147, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Hoepfner, S.; Severin, F.; Cabezas, A.; Habermann, B.; Runge, A.; Gillooly, D.; Stenmark, H.; Zerial, M. Modulation of Receptor Recycling and Degradation by the Endosomal Kinesin KIF16B. Cell 2005, 121, 437–450. [Google Scholar] [CrossRef]
- Singh, M.; Venugopal, C.; Tokar, T.; McFarlane, N.; Subapanditha, M.K.; Qazi, M.; Bakhshinyan, D.; Vora, P.; Murty, N.K.; Jurisica, I.; et al. Therapeutic Targeting of the Premetastatic Stage in Human Lung-to-Brain Metastasis. Cancer Res. 2018, 78, 5124–5134. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, R.P.; Moss, K.R.; Janusz-Kaminska, A.; Knudson, L.A.; Denes, L.T.; Saxena, T.; Boggupalli, D.P.; Li, Z.; Lin, K.; Bassell, G.J.; et al. Muscleblind-like Proteins Use Modular Domains to Localize RNAs by Riding Kinesins and Docking to Membranes. Nat. Commun. 2023, 14, 3427. [Google Scholar] [CrossRef] [PubMed]
- Dorner, C.; Ciossek, T.; Müller, S.; Møller, P.H.; Ullrich, A.; Lammers, R. Characterization of KIF1C, a New Kinesin-like Protein Involved in Vesicle Transport from the Golgi Apparatus to the Endoplasmic Reticulum. J. Biol. Chem. 1998, 273, 20267–20275. [Google Scholar] [CrossRef]
- Scholey, J.M. Kinesin-2: A Family of Heterotrimeric and Homodimeric Motors with Diverse Intracellular Transport Functions. Annu. Rev. Cell Dev. Biol. 2013, 29, 443–469. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Nakata, T.; Okada, Y.; Hirokawa, N. KIF3A/B: A Heterodimeric Kinesin Superfamily Protein That Works as a Microtubule plus End-Directed Motor for Membrane Organelle Transport. J. Cell Biol. 1995, 130, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog Signalling in the Mouse Requires Intraflagellar Transport Proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Li, R.; Zhang, M.; Wang, H.; Qu, Y. Kinesin Family Member 3A Inhibits the Carcinogenesis of Non-small Cell Lung Cancer and Prolongs Survival. Oncol. Lett. 2020, 20, 348. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Suh, Y.-A.; Oh, J.-H.; Lee, B.R.; Kim, J.; Jang, S.J. KIF3A Binds to β-Arrestin for Suppressing Wnt/β-Catenin Signalling Independently of Primary Cilia in Lung Cancer. Sci. Rep. 2016, 6, 32770. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Vaghjiani, V.; Szczepny, A.; Jayasekara, W.S.N.; Gonzalez-Rajal, A.; Kikuchi, K.; McCaughan, G.W.; Burgess, A.; Gough, D.J.; Watkins, D.N.; et al. Trp53 and Rb1 Regulate Autophagy and Ligand-Dependent Hedgehog Signaling. J. Clin. Investig. 2020, 130, 4006–4018. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, R.; Hao, M.; Zhao, X.; Li, C. Kinesin Family Member 3C (KIF3C) Is a Novel Non-Small Cell Lung Cancer (NSCLC) Oncogene Whose Expression Is Modulated by microRNA-150-5p (miR-150-5p) and microRNA-186-3p (miR-186-3p). Bioengineered 2021, 12, 3077–3088. [Google Scholar] [CrossRef]
- Gumy, L.F.; Chew, D.J.; Tortosa, E.; Katrukha, E.A.; Kapitein, L.C.; Tolkovsky, A.M.; Hoogenraad, C.C.; Fawcett, J.W. The Kinesin-2 Family Member KIF3C Regulates Microtubule Dynamics and Is Required for Axon Growth and Regeneration. J. Neurosci. 2013, 33, 11329–11345. [Google Scholar] [CrossRef] [PubMed]
- da Costa, R.; Passos, G.F.; Quintão, N.L.M.; Fernandes, E.S.; Maia, J.R.L.C.B.; Campos, M.M.; Calixto, J.B. Taxane-Induced Neurotoxicity: Pathophysiology and Therapeutic Perspectives. Br. J. Pharmacol. 2020, 177, 3127–3146. [Google Scholar] [CrossRef]
- Shahin, R.; Aljamal, S. Kinesin Spindle Protein Inhibitors in Cancer: From High Throughput Screening to Novel Therapeutic Strategies. Future Sci. OA 2022, 8, FSO778. [Google Scholar] [CrossRef]
- Lad, L.; Luo, L.; Carson, J.D.; Wood, K.W.; Hartman, J.J.; Copeland, R.A.; Sakowicz, R. Mechanism of Inhibition of Human KSP by Ispinesib. Biochemistry 2008, 47, 3576–3585. [Google Scholar] [CrossRef]
- Lemieux, C.; DeWolf, W.; Voegtli, W.; DeLisle, R.K.; Laird, E.; Wallace, E.; Woessner, R.; Corrette, C.; Allen, S.; Hans, J. ARRY-520, a Novel, Highly Selective KSP Inhibitor with Potent Anti-Proliferative Activity. Proc. Am. Assoc. Cancer Res. 2007, 48, 5590. [Google Scholar]
- Yukawa, M.; Yamauchi, T.; Kurisawa, N.; Ahmed, S.; Kimura, K.-I.; Toda, T. Fission Yeast Cells Overproducing HSET/KIFC1 Provides a Useful Tool for Identification and Evaluation of Human Kinesin-14 Inhibitors. Fungal Genet. Biol. 2018, 116, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Watts, C.A.; Richards, F.M.; Bender, A.; Bond, P.J.; Korb, O.; Kern, O.; Riddick, M.; Owen, P.; Myers, R.M.; Raff, J.; et al. Design, Synthesis, and Biological Evaluation of an Allosteric Inhibitor of HSET That Targets Cancer Cells with Supernumerary Centrosomes. Chem. Biol. 2013, 20, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhai, L.; Wang, Y.; Boohaker, R.J.; Lu, W.; Gupta, V.V.; Padmalayam, I.; Bostwick, R.J.; White, E.L.; Ross, L.J.; et al. Discovery of a Novel Inhibitor of Kinesin-like Protein KIFC1. Biochem. J. 2016, 473, 1027–1035. [Google Scholar] [CrossRef]
- Punekar, S.R.; Velcheti, V.; Neel, B.G.; Wong, K.-K. The Current State of the Art and Future Trends in RAS-Targeted Cancer Therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Demoor-Goldschmidt, C.; de Vathaire, F. Review of Risk Factors of Secondary Cancers among Cancer Survivors. Br. J. Radiol. 2019, 92, 20180390. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Cao, Y.; Cao, M.; Wang, Y.; Cao, Y.; Gong, T. Nanomedicine in Cancer Therapy. Signal Transduct. Target. Ther. 2023, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Mahato, R.; Tai, W.; Cheng, K. Prodrugs for Improving Tumor Targetability and Efficiency. Adv. Drug Deliv. Rev. 2011, 63, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, R.; Gorospe, K.A.; Labouta, H.I.; Azad, T.; Lee, W.L.; Thu, K.L. Alternative Strategies for Delivering Immunotherapeutics Targeting the PD-1/PD-L1 Immune Checkpoint in Cancer. Pharmaceutics 2024, 16, 1181. [Google Scholar] [CrossRef] [PubMed]
- Komlodi-Pasztor, E.; Sackett, D.L.; Fojo, A.T. Inhibitors Targeting Mitosis: Tales of How Great Drugs against a Promising Target Were Brought down by a Flawed Rationale. Clin. Cancer Res. 2012, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.M.; Collins, I. Recent Findings and Future Directions for Interpolar Mitotic Kinesin Inhibitors in Cancer Therapy. Future Med. Chem. 2016, 8, 463–489. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. Cancer Cells Display Profound Intra- and Interline Variation Following Prolonged Exposure to Antimitotic Drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gong, H.; Huang, K. Oncogenic Role of Kinesin Proteins and Targeting Kinesin Therapy. Cancer Sci. 2013, 104, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Doménech, E.; Malumbres, M. Mitosis-Targeting Therapies: A Troubleshooting Guide. Curr. Opin. Pharmacol. 2013, 13, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.C.; Ribeiro, D.; Pedrosa, J.; Sarmento, B.; Silva, P.M.A.; Bousbaa, H. Mitosis Inhibitors in Anticancer Therapy: When Blocking the Exit Becomes a Solution. Cancer Lett. 2019, 440–441, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Tunquist, B.J.; Woessner, R.D.; Walker, D.H. Mcl-1 Stability Determines Mitotic Cell Fate of Human Multiple Myeloma Tumor Cells Treated with the Kinesin Spindle Protein Inhibitor ARRY-520. Mol. Cancer Ther. 2010, 9, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.C.; Morden, C.R.; Palmer, M.C.L.; Nachtigal, M.W.; McManus, K.J. Detecting Chromosome Instability in Cancer: Approaches to Resolve Cell-to-Cell Heterogeneity. Cancers 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A Signature of Chromosomal Instability Inferred from Gene Expression Profiles Predicts Clinical Outcome in Multiple Human Cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Greene, S.B.; Dago, A.E.; Leitz, L.J.; Wang, Y.; Lee, J.; Werner, S.L.; Gendreau, S.; Patel, P.; Jia, S.; Zhang, L.; et al. Chromosomal Instability Estimation Based on next Generation Sequencing and Single Cell Genome Wide Copy Number Variation Analysis. PLoS ONE 2016, 11, e0165089. [Google Scholar] [CrossRef]
- Mitchison, T.J. The Proliferation Rate Paradox in Antimitotic Chemotherapy. Mol. Biol. Cell 2012, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kriegsmann, M.; Warth, A. What Is Better/reliable, Mitosis Counting or Ki67/MIB1 Staining? Transl. Lung Cancer Res. 2016, 5, 543–546. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Family | Gene (Protein) Name | Genomic Location | Class | Type (N, M, C) | General Functions & |
|---|---|---|---|---|---|
| Kinesin-1 | KIF5A | 12q13.3 | Non-Mitotic | N, plus-end | axonal transport of vesicles, organelles, and macromolecules in neurons |
| KIF5B | 10p11.22 | Non-Mitotic | N, plus-end | transport of organelles like mitochondria and lysosomes | |
| Kinesin-2 | KIF3A | 5q31.1 | Non-Mitotic | N, plus-end | cilia formation and function, intraflagellar transport, organelle transport |
| KIF3C | 2p23.3 | Non-Mitotic | N, plus-end | microtubule dynamics, axonal growth, and regeneration | |
| Kinesin-3 | KIF1C | 17p13.2 | Non-Mitotic | N, plus-end | transports Golgi-derived vesicles and mRNA |
| KIF13A | 6p22.3 | Non-Mitotic | N, plus-end | recycling endosome dynamics, intracellular cargo transport | |
| KIF13B | 8p12 | Non-Mitotic | N, plus-end | endocytosis and angiogenesis via transport of VEGFR2 | |
| KIF14 | 1q32.1 | Mitotic | N, plus-end | cytokinesis, chromosome congression, and alignment | |
| KIF16B | 20p12.1 | Non-Mitotic | N, plus-end | endosome transport, recycling, and degradation of receptors | |
| Kinesin-4 | KIF4A | Xq13.1 | Mitotic | N, plus-end | chromosome condensation and segregation, and cytokinesis |
| KIF7 | 15q26.1 | Non-Mitotic | N, non-motile | microtubule dynamics, cilia formation, cell cycle, and Hedgehog signalling | |
| KIF21B | 1q32.1 | Non-Mitotic | N, plus-end | microtubule dynamics, cytoskeletal transport, and neuronal morphology | |
| Kinesin-5 | KIF11 (Eg5, KSP) | 10q23.33 | Mitotic | N, plus-end | bipolar spindle formation via centrosome separation |
| Kinesin-6 | KIF20A (MKLP2) | 5q31.2 | Mitotic | N, plus-end | cytokinesis, Golgi positioning, and vesicle secretion |
| KIF20B (MPP1) | 10q23.31 | Mitotic | N, plus-end | abscission and cytokinesis | |
| KIF23 (MKLP1) | 15q23 | Mitotic | N, plus-end | centralspindlin complex component and cytokinesis | |
| Kinesin-7 | KIF10 (CENPE) | 4q24 | Mitotic | N, plus-end | kinetochore-MT attachment, chromosome alignment, and spindle assembly checkpoint |
| Kinesin-8 | KIF18A | 11p14.1 | Mitotic | N, plus-end | chromosome congression and alignment, and microtubule dynamics |
| KIF18B | 17q21.31 | Mitotic | N, plus-end | chromosome alignment and microtubule dynamics | |
| Kinesin-10 | KIF22 (KID) | 16p11.2 | Mitotic | N, plus-end | chromosome congression, alignment, and compaction during anaphase |
| Kinesin-11 | KIF26B | 1q44 | Non-Mitotic | N, non-motile | cell adhesion, migration, and polarity |
| Kinesin-12 | KIF15 | 3p21.31 | Mitotic | N, plus-end | spindle elongation and spindle bipolarity via centrosome separation |
| Kinesin-13 | KIF2A | 5q12.1 | Mitotic | M, non-motile | spindle formation and bipolarity, chromosome alignment, and microtubule dynamics |
| KIF2B | 17q22 | Mitotic | M, non-motile | kinetochore-MT attachment and microtubule dynamics | |
| KIF2C (MCAK) | 1p34.1 | Mitotic | M, non-motile | kinetochore-MT attachment, chromosome congression and alignment, and microtubule dynamics | |
| KIF24 | 9p13.3 | Non-Mitotic | M, non-motile | microtubule dynamics and cilia formation | |
| Kinesin-14A | KIFC1 (HSET) | 6p21.32 | Mitotic | C, minus-end | spindle assembly and bipolarity, and intracellular cargo transport |
| Kinesin-14B | KIFC3 | 16q21 | Mitotic | C, minus-end | centrosome cohesion and spindle assembly, cytokinesis, and intracellular cargo transport |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Wu, B.Z.; Thu, K.L. Targeting Kinesins for Therapeutic Exploitation of Chromosomal Instability in Lung Cancer. Cancers 2025, 17, 685. https://doi.org/10.3390/cancers17040685
Zhang C, Wu BZ, Thu KL. Targeting Kinesins for Therapeutic Exploitation of Chromosomal Instability in Lung Cancer. Cancers. 2025; 17(4):685. https://doi.org/10.3390/cancers17040685
Chicago/Turabian StyleZhang, Christopher, Benson Z. Wu, and Kelsie L. Thu. 2025. "Targeting Kinesins for Therapeutic Exploitation of Chromosomal Instability in Lung Cancer" Cancers 17, no. 4: 685. https://doi.org/10.3390/cancers17040685
APA StyleZhang, C., Wu, B. Z., & Thu, K. L. (2025). Targeting Kinesins for Therapeutic Exploitation of Chromosomal Instability in Lung Cancer. Cancers, 17(4), 685. https://doi.org/10.3390/cancers17040685