

Axl Regulation of NK Cell Activity Creates an Immunosuppressive Tumor Immune Microenvironment in Head and Neck Cancer

 , , , , ,
, , , , ,  and
and
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Genotype Analysis
2.3. Immunoblot Analysis
2.4. Cell Growth Assay
2.5. Cell Proliferation Assay
2.6. Flow Cytometry
2.6.1. Fluorescence Activated Cell Sorting (FACS)
2.6.2. Imaging Flow Cytometry
2.7. RNA Isolation, cDNA Synthesis, and Quantitative Polymerase Chain Reaction (qPCR)
2.8. In Vivo Mouse Experiments
2.8.1. Tumor Dissociation
2.8.2. NK Cell Depletion
2.9. Immunohistochemistry (IHC)
2.10. NK Cytotoxicity Assay
2.11. Murine Cytokine Array
2.12. Luminex
2.13. NanoString
2.14. Statistical Analysis
3. Results
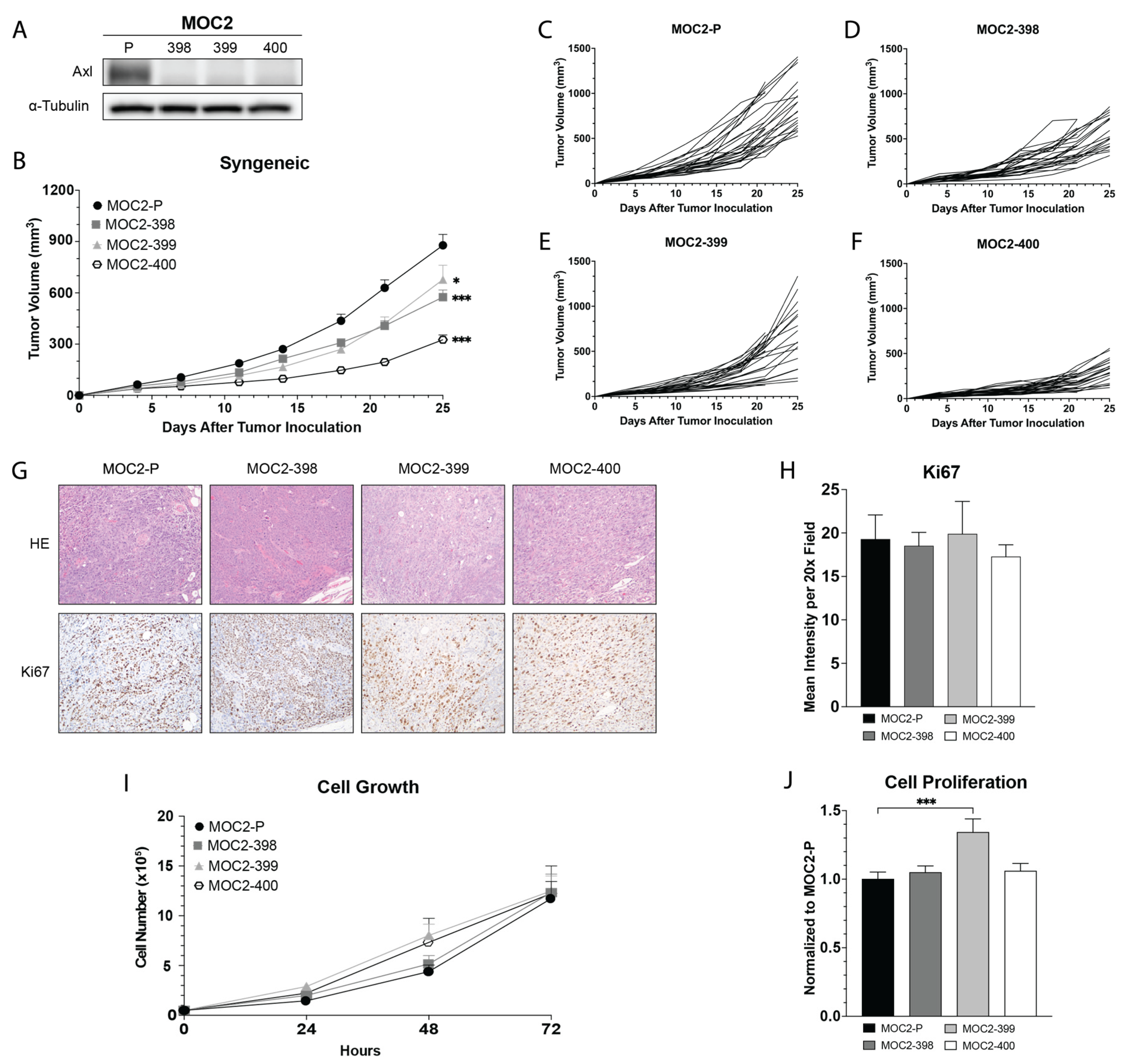
3.1. Development and In Vivo Growth of Axl KO MOC2 Cell Lines
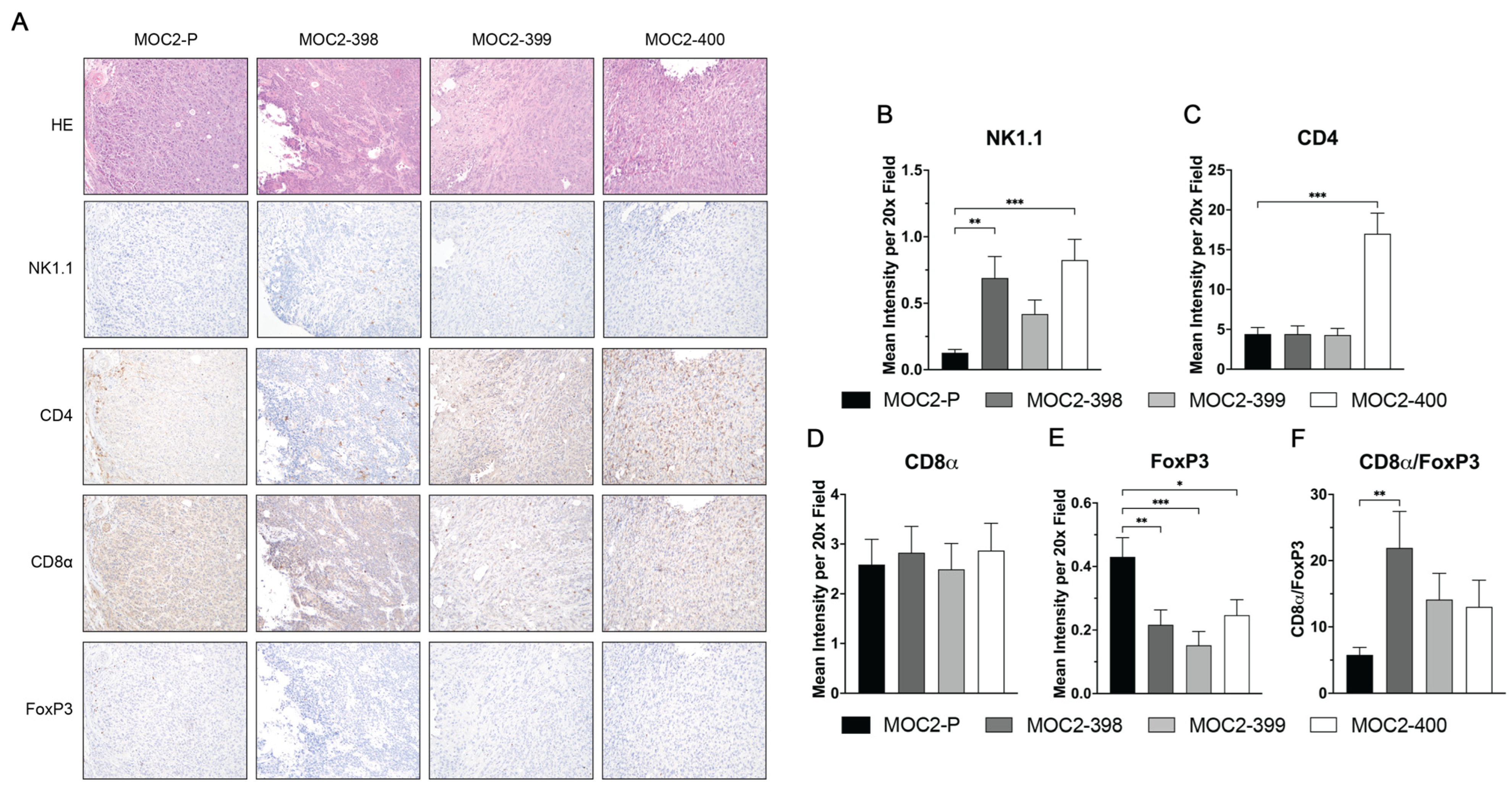
3.2. Axl KO Results in a Hotter TIME
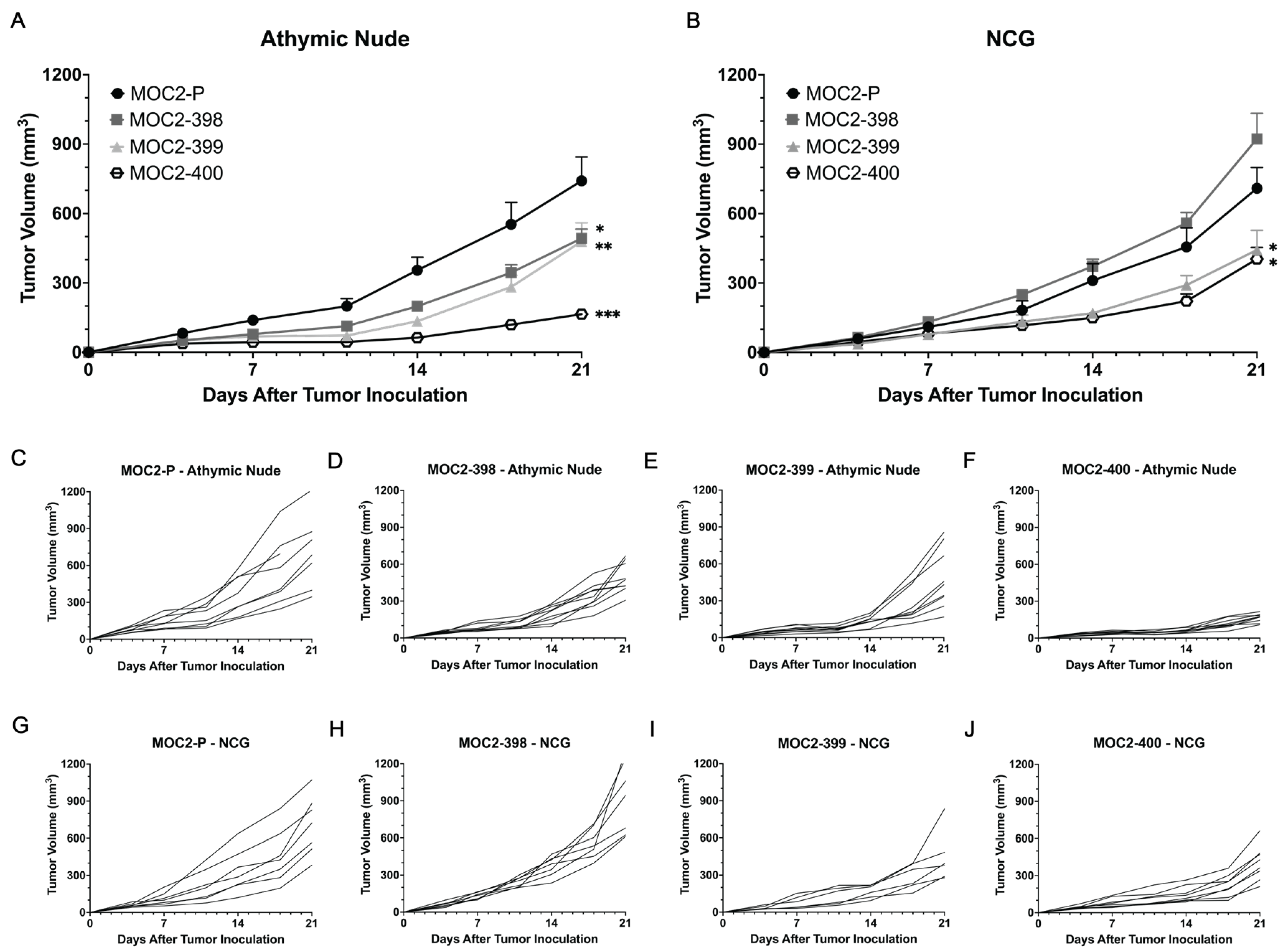
3.3. Tumor Growth Delay in Axl KO Cell Lines Is Mediated by Immune Cell Populations
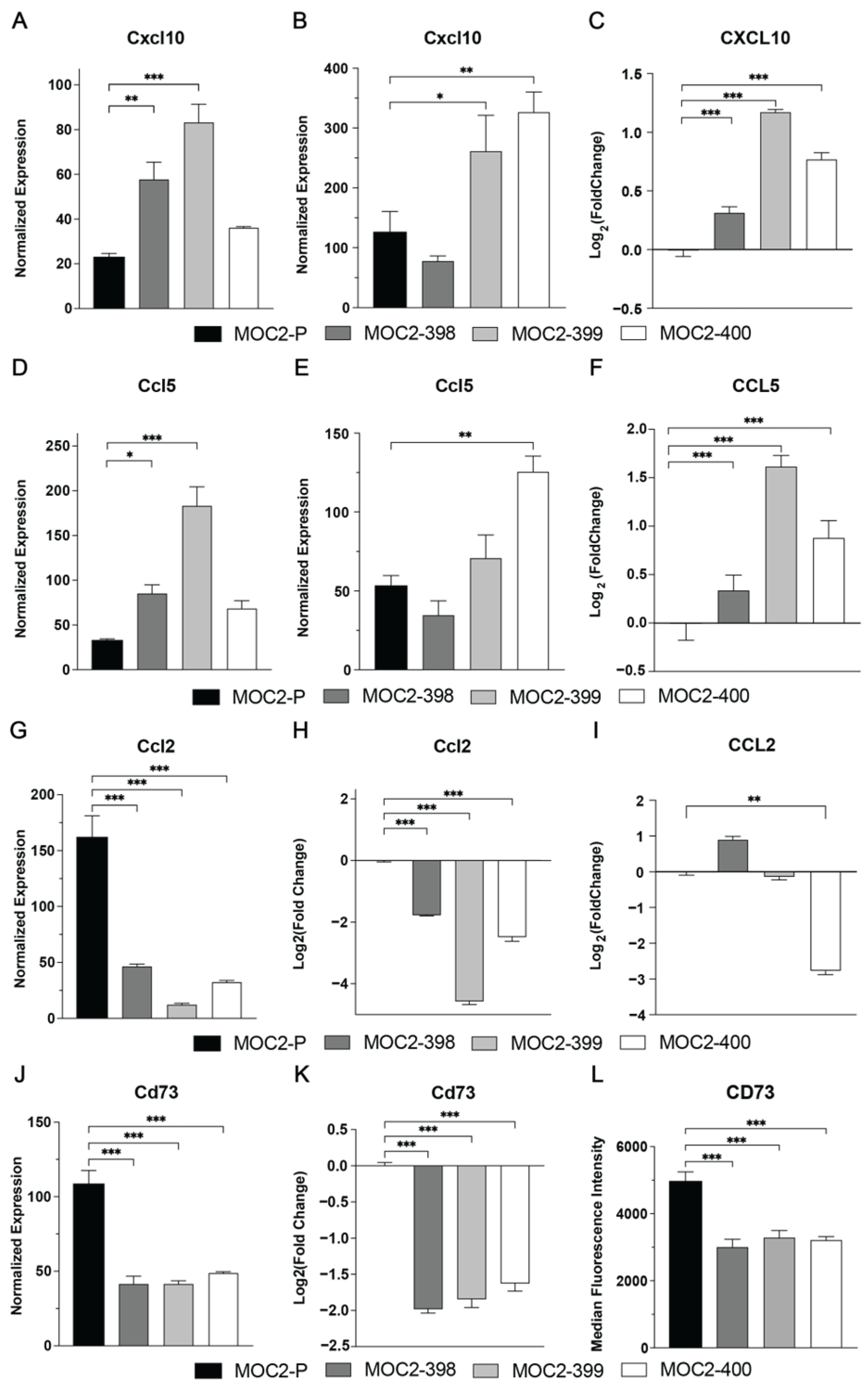
3.4. Axl KO Changes Expression of Immunomodulatory Cytokines and Chemokines
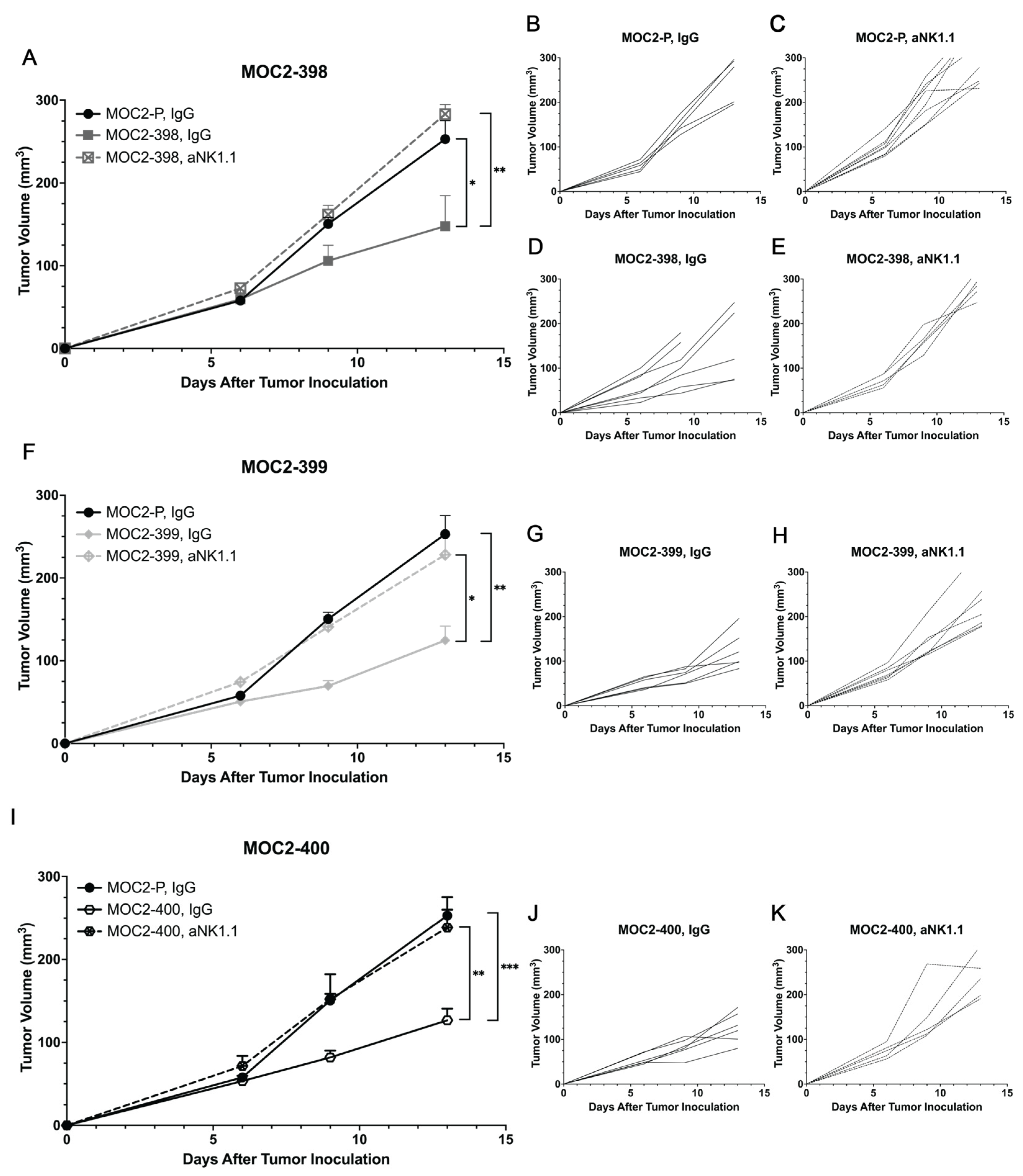
3.5. Axl KO Enhances NK Cell Cytotoxicity and Recruitment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Axl | Receptor tyrosine kinase UFO |
| HNC | Head and neck cancer |
| HPV | Human papillomavirus |
| TIME | Tumor immune microenvironment |
| RTK | Receptor tyrosine kinase |
| PDX | Patient-derived xenograft |
| NK | Natural killer cell |
| KO | Knockout |
| MOC | Mouse oral cancer |
| FBS | Fetal bovine serum |
| RNP | Ribonucleoprotein |
| gRNA | Guide RNA |
| PCR | Polymerase chain reaction |
| PMN-MDSCs | Polymorphonuclear myeloid-derived suppressor cells |
| IHC | Immunohistochemistry |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a Causal Association Between Human Papillomavirus and a Subset of Head and Neck Cancers. JNCI 2000, 92, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.; Gillison, M.L. Nivolumab for Squamous-Cell Cancer of Head and Neck. N. Engl. J. Med. 2017, 376, 596. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Gajewski, T.F. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Semin. Oncol. 2015, 42, 663–671. [Google Scholar] [CrossRef]
- Rausch, M.P.; Hastings, K.T. Chapter 9—Immune Checkpoint Inhibitors in the Treatment of Melanoma: From Basic Science to Clinical Application. In Cutaneous Melanoma: Etiology and Therapy; National Library of Medicine: Bethesda, MD, USA, 2017. [Google Scholar]
- Vareki, S.M. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. ImmunoTherapy Cancer 2018, 6, 157. [Google Scholar] [CrossRef]
- Xia, L.; Liu, Y.; Wang, Y. PD-1/PD-L1 Blockade Therapy in Advanced Non-Small-Cell Lung Cancer: Current Status and Future Directions. Oncologist 2019, 24, S31–S41. [Google Scholar] [CrossRef]
- Zemek, R.M.; De Jong, E.; Chin, W.L.; Schuster, I.S.; Fear, V.S.; Casey, T.H.; Forbes, C.; Dart, S.J.; Leslie, C.; Zaltouny, A.; et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci. Transl. Med. 2019, 11, eaav7816. [Google Scholar] [CrossRef]
- Ran, X.; Yang, K. Inhibitors of the PD-1/PD-L1 axis for the treatment of head and neck cancer: Current status and future perspectives. Drug Des. Devel Ther. 2017, 11, 2007–2014. [Google Scholar] [CrossRef] [PubMed]
- Kostecki, K.L.; Iida, M.; Crossman, B.E.; Salgia, R.; Harari, P.M.; Bruce, J.Y.; Wheeler, D.L. Immune Escape Strategies in Head and Neck Cancer: Evade, Resist, Inhibit, Recruit. Cancers 2024, 16, 312. [Google Scholar] [CrossRef] [PubMed]
- O’bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R.; Le Beau, M.M.; Earp, H.S.; Liu, E.T. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Pearson, H.E.; Bahrar, H.; Fowler, T.L.; Bednarz, B.P.; et al. AXL Is a Logical Molecular Target in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 2601–2612. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Luthar, N.; Toulany, M.; Gill, P.S.; Salgia, R.; Kimple, R.J.; et al. AXL mediates resistance to cetuximab therapy. Cancer Res. 2014, 74, 5152–5164. [Google Scholar] [CrossRef]
- McDaniel, N.K.; Iida, M.; Nickel, K.P.; Longhurst, C.A.; Fischbach, S.R.; Rodems, T.S.; Kranjac, C.A.; Bo, A.Y.; Luo, Q.; Gallagher, M.M.; et al. AXL Mediates Cetuximab and Radiation Resistance Through Tyrosine 821 and the c-ABL Kinase Pathway in Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 4349–4359. [Google Scholar] [CrossRef]
- Mints, M.; Landin, D.; Näsman, A.; Mirzaie, L.; Ursu, R.G.; Zupancic, M.; Marklund, L.; Dalianis, T.; Munck-Wikland, E.; Ramqvist, T. Tumour inflammation signature and expression of S100A12 and HLA class I improve survival in HPV-negative hypopharyngeal cancer. Sci. Rep. 2021, 11, 1782. [Google Scholar] [CrossRef]
- McDaniel, N.K.; Cummings, C.T.; Iida, M.; Hulse, J.; Pearson, H.E.; Vasileiadi, E.; Parker, R.E.; Orbuch, R.A.; Ondracek, O.J.; Welke, N.B.; et al. MERTK Mediates Intrinsic and Adaptive Resistance to AXL-targeting Agents. Mol. Cancer Ther. 2018, 17, 2297–2308. [Google Scholar] [CrossRef]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef]
- Kostecki, K.L.; Iida, M.; Wiley, A.L.; Kimani, S.; Mehall, B.; Tetreault, K.; Alexandridis, R.; Yu, M.; Hong, S.; Salgia, R.; et al. Dual Axl/MerTK inhibitor INCB081776 creates a proinflammatory tumor immune microenvironment and enhances anti-PDL1 efficacy in head and neck cancer. Head Neck 2023, 45, 1255–1271. [Google Scholar] [CrossRef]
- Engelsen, A.S.T.; Lotsberg, M.L.; Abou Khouzam, R.; Thiery, J.P.; Lorens, J.B.; Chouaib, S.; Terry, S. Dissecting the Role of AXL in Cancer Immune Escape and Resistance to Immune Checkpoint Inhibition. Front. Immunol. 2022, 13, 869676. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, Y.; Zhang, D.; Ma, J. Axl inhibition induces the antitumor immune response which can be further potentiated by PD-1 blockade in the mouse cancer models. Oncotarget 2017, 8, 89761–89774. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.F.; Du, W.; Sorrelle, N.B.; Wnuk-Lipinska, K.; Topalovski, M.; Toombs, J.E.; Cruz, V.H.; Yabuuchi, S.; Rajeshkumar, N.; Maitra, A. Small-molecule inhibition of Axl targets tumor immune suppression and enhances chemotherapy in pancreatic cancer. Cancer Res. 2018, 78, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Goyette, M.A.; Côté, J.F. AXL Receptor Tyrosine Kinase as a Promising Therapeutic Target Directing Multiple Aspects of Cancer Progression and Metastasis. Cancers 2022, 14, 466. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Favata, M.; Lasky, K.; Feldman, P.; Lo, Y.; Yang, G.; Stevens, C.; Wen, X.; Sehra, S.; Katiyar, K.; et al. A Potent and Selective Dual Inhibitor of AXL and MERTK Possesses Both Immunomodulatory and Tumor-Targeted Activity. Front. Oncol. 2020, 10, 598477. [Google Scholar] [CrossRef]
- Iida, M.; Brand, T.M.; Starr, M.M.; Huppert, E.J.; Luthar, N.; Bahrar, H.; Coan, J.P.; Pearson, H.E.; Salgia, R.; Wheeler, D.L. Overcoming acquired resistance to cetuximab by dual targeting HER family receptors with antibody-based therapy. Mol. Cancer 2014, 13, 242. [Google Scholar] [CrossRef]
- Crowe, A.R.; Yue, W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio Protoc. 2019, 9, e3465. [Google Scholar] [CrossRef]
- Judd, N.P.; Winkler, A.E.; Murillo-Sauca, O.; Brotman, J.J.; Law, J.H.; James, S.; Lewis, J.; Dunn, G.P.; Bui, J.D.; Sunwoo, J.B.; et al. ERK1/2 Regulation of CD44 Modulates Oral Cancer Aggressiveness. Cancer Res. 2011, 72, 365–374. [Google Scholar] [CrossRef]
- Cash, H.; Shah, S.; Moore, E.; Caruso, A.; Uppaluri, R.; Waes, C.V.; Allen, C. mTOR and MEK1/2 inhibition differentially modulate tumor growth and the immune microenvironment in syngeneic models of oral cavity cancer. Oncotarget 2015, 6, 36400–36417. [Google Scholar] [CrossRef]
- Ran, G.H.; Lin, Y.Q.; Tian, L.; Zhang, T.; Yan, D.M.; Yu, J.H.; Deng, Y.C. Natural killer cell homing and trafficking in tissues and tumors: From biology to application. Signal Transduct. Target. Ther. 2022, 7, 205. [Google Scholar] [CrossRef]
- Saudemont, A.; Jouy, N.; Hetuin, D.; Quesnel, B. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood 2005, 105, 2428–2435. [Google Scholar] [CrossRef] [PubMed]
- Mgrditchian, T.; Arakelian, T.; Paggetti, J.; Noman, M.Z.; Viry, E.; Moussay, E.; Van Moer, K.; Kreis, S.; Guerin, C.; Buart, S.; et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, E9271–E9279. [Google Scholar] [CrossRef] [PubMed]
- Bhat, H.; Zaun, G.; Hamdan, T.A.; Lang, J.; Adomati, T.; Schmitz, R.; Friedrich, S.K.; Bergerhausen, M.; Cham, L.B.; Li, F.; et al. Arenavirus Induced CCL5 Expression Causes NK Cell-Mediated Melanoma Regression. Front. Immunol. 2020, 11, 1849. [Google Scholar] [CrossRef]
- Allard, B.; Beavis, P.A.; Darcy, P.K.; Stagg, J. Immunosuppressive activities of adenosine in cancer. Curr. Opin. Pharmacol. 2016, 29, 7–16. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef]
- Oyer, J.L.; Gitto, S.B.; Altomare, D.A.; Copik, A.J. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology 2018, 7, e1509819. [Google Scholar] [CrossRef]
- Lee, C.-C.; Ho, K.-H.; Huang, T.-W.; Shih, C.-M.; Hsu, S.-Y.; Liu, A.-J.; Chen, K.-C. A regulatory loop among CD276, miR-29c-3p, and Myc exists in cancer cells against natural killer cell cytotoxicity. Life Sci. 2021, 277, 119438. [Google Scholar] [CrossRef]
- Karlhofer, F.M.; Yokoyama, W.M. Stimulation of murine natural killer (NK) cells by a monoclonal antibody specific for the NK1.1 antigen. IL-2-activated NK cells possess additional specific stimulation pathways. J. Immunol. 1991, 146, 3662–3673. [Google Scholar] [CrossRef]
- Fogel, L.A.; Sun, M.M.; Geurs, T.L.; Carayannopoulos, L.N.; French, A.R. Markers of nonselective and specific NK cell activation. J. Immunol. 2013, 190, 6269–6276. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef]
- Kashii, Y.; Giorda, R.; Herberman, R.B.; Whiteside, T.L.; Vujanovic, N.L. Constitutive Expression and Role of the TNF Family Ligands in Apoptotic Killing of Tumor Cells by Human NK Cells1. J. Immunol. 1999, 163, 5358–5366. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Davra, V.; Kumar, S.; Geng, K.; Calianese, D.; Mehta, D.; Gadiyar, V.; Kasikara, C.; Lahey, K.C.; Chang, Y.J.; Wichroski, M.; et al. Axl and Mertk Receptors Cooperate to Promote Breast Cancer Progression by Combined Oncogenic Signaling and Evasion of Host Antitumor Immunity. Cancer Res. 2021, 81, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Lew, E.D.; Seelige, R.; Tindall, E.A.; Walsh, C.; Fagan, P.C.; Lee, J.Y.; Nevarez, R.; Oh, J.; Tucker, K.D.; et al. Immuno-oncological Efficacy of RXDX-106, a Novel TAM (TYRO3, AXL, MER) Family Small-Molecule Kinase Inhibitor. Cancer Res. 2019, 79, 1996–2008. [Google Scholar] [CrossRef]
- Rankin, E.B.; Fuh, K.C.; Taylor, T.E.; Krieg, A.J.; Musser, M.; Yuan, J.; Wei, K.; Kuo, C.J.; Longacre, T.A.; Giaccia, A.J. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010, 70, 7570–7579. [Google Scholar] [CrossRef]
- Du, W.; Huang, H.; Sorrelle, N.; Brekken, R.A. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight 2018, 3, e124184. [Google Scholar] [CrossRef]
- Yan, D.; Earp, H.S.; DeRyckere, D.; Graham, D.K. Targeting MERTK and AXL in EGFR Mutant Non-Small Cell Lung Cancer. Cancers 2021, 13, 5639. [Google Scholar] [CrossRef]
- Holtzhausen, A.; Harris, W.; Ubil, E.; Hunter, D.M.; Zhao, J.; Zhang, Y.; Zhang, D.; Liu, Q.; Wang, X.; Graham, D.K.; et al. TAM Family Receptor Kinase Inhibition Reverses MDSC-Mediated Suppression and Augments Anti–PD-1 Therapy in Melanoma. Cancer Immunol. Res. 2019, 7, 1672–1686. [Google Scholar] [CrossRef]
- Aguilera, T.A.; Giaccia, A.J. Molecular Pathways: Oncologic Pathways and Their Role in T-cell Exclusion and Immune Evasion-A New Role for the AXL Receptor Tyrosine Kinase. Clin. Cancer Res. 2017, 23, 2928–2933. [Google Scholar] [CrossRef] [PubMed]
- Schmid, E.T.; Pang, I.K.; Carrera Silva, E.A.; Bosurgi, L.; Miner, J.J.; Diamond, M.S.; Iwasaki, A.; Rothlin, C.V. AXL receptor tyrosine kinase is required for T cell priming and antiviral immunity. eLife 2016, 5, e12414. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Abdou, A.; Engelsen, A.S.T.; Buart, S.; Dessen, P.; Corgnac, S.; Collares, D.; Meurice, G.; Gausdal, G.; Baud, V.; et al. AXL Targeting Overcomes Human Lung Cancer Cell Resistance to NK- and CTL-Mediated Cytotoxicity. Cancer Immunol. Res. 2019, 7, 1789–1802. [Google Scholar] [CrossRef]
- Tsukita, Y.; Fujino, N.; Miyauchi, E.; Saito, R.; Fujishima, F.; Itakura, K.; Kyogoku, Y.; Okutomo, K.; Yamada, M.; Okazaki, T.; et al. Axl kinase drives immune checkpoint and chemokine signalling pathways in lung adenocarcinomas. Mol. Cancer 2019, 18, 24. [Google Scholar] [CrossRef]
- Goyette, M.A.; Elkholi, I.E.; Apcher, C.; Kuasne, H.; Rothlin, C.V.; Muller, W.J.; Richard, D.E.; Park, M.; Gratton, J.P.; Cote, J.F. Targeting Axl favors an antitumorigenic microenvironment that enhances immunotherapy responses by decreasing Hif-1alpha levels. Proc. Natl. Acad. Sci. USA 2021, 118, e2023868118. [Google Scholar] [CrossRef]
- Li, Y.; Wu, T.; Gong, S.; Zhou, H.; Yu, L.; Liang, M.; Shi, R.; Wu, Z.; Zhang, J.; Li, S. Analysis of the Prognosis and Therapeutic Value of the CXC Chemokine Family in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 570736. [Google Scholar] [CrossRef]
- Han, Y.; Guo, Z.; Jiang, L.; Li, X.; Chen, J.; Ouyang, L.; Li, Y.; Wang, X. CXCL10 and CCL5 as feasible biomarkers for immunotherapy of homologous recombination deficient ovarian cancer. Am. J. Cancer Res. 2023, 13, 1904–1922. [Google Scholar]
- Robin, R.; Jovian, Y.; Blake, A.F.; Emily, F.H.; Ken, H.; Thomas, F.G. Immune cell and tumor cell-derived CXCL10 is indicative of immunotherapy response in metastatic melanoma. J. ImmunoTherapy Cancer 2021, 9, e003521. [Google Scholar] [CrossRef]
- Tanaka, M.; Siemann, D.W. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers 2020, 12, 1850. [Google Scholar] [CrossRef]
- Liss, C.; Fekete, M.J.; Hasina, R.; Lam, C.D.; Lingen, M.W. Paracrine angiogenic loop between head-and-neck squamous-cell carcinomas and macrophages. Int. J. Cancer 2001, 93, 781–785. [Google Scholar] [CrossRef]
- Liss, C.; Fekete, M.J.; Hasina, R.; Lingen, M.W. Retinoic acid modulates the ability of macrophages to participate in the induction of the angiogenic phenotype in head and neck squamous cell carcinoma. Int. J. Cancer 2002, 100, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Matosevic, S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J. Mol. Med. 2018, 96, 903–913. [Google Scholar] [CrossRef]
- Hakozaki, K.; Tanaka, N.; Takamatsu, K.; Takahashi, R.; Yasumizu, Y.; Mikami, S.; Shinojima, T.; Kakimi, K.; Kamatani, T.; Miya, F.; et al. Landscape of prognostic signatures and immunogenomics of the AXL/GAS6 axis in renal cell carcinoma. Br. J. Cancer 2021, 125, 1533–1543. [Google Scholar] [CrossRef]
- Reinhardt, J.; Landsberg, J.; Schmid-Burgk, J.L.; Ramis, B.B.; Bald, T.; Glodde, N.; Lopez-Ramos, D.; Young, A.; Ngiow, S.F.; Nettersheim, D.; et al. MAPK Signaling and Inflammation Link Melanoma Phenotype Switching to Induction of CD73 during Immunotherapy. Cancer Res. 2017, 77, 4697–4709. [Google Scholar] [CrossRef]
- Hernandez, K.C.; Shah, A.M.; Lopez, V.A.; Tagliabracci, V.S.; Chen, K.; Xu, L.; Bassel-Duby, R.; Olson, E.N.; Liu, N. CD73 contributes to the pathogenesis of fusion-negative rhabdomyosarcoma through the purinergic signaling pathway. Proc. Natl. Acad. Sci. USA 2024, 121, e2315925121. [Google Scholar] [CrossRef]
- Vazirinejad, R.; Ahmadi, Z.; Kazemi Arababadi, M.; Hassanshahi, G.; Kennedy, D. The Biological Functions, Structure and Sources of CXCL10 and Its Outstanding Part in the Pathophysiology of Multiple Sclerosis. Neuroimmunomodulation 2014, 21, 322–330. [Google Scholar] [CrossRef]
- Appay, V.; Rowland-Jones, S.L. RANTES: A versatile and controversial chemokine. Trends Immunol. 2001, 22, 83–87. [Google Scholar] [CrossRef]
- Salanga, C.M.; Salanga, M.C. Genotype to Phenotype: CRISPR Gene Editing Reveals Genetic Compensation as a Mechanism for Phenotypic Disjunction of Morphants and Mutants. Int. J. Mol. Sci. 2021, 22, 3472. [Google Scholar] [CrossRef]
- Dong, S.; Zhao, M.; Zhu, J.; Li, T.; Yan, M.; Xing, K.; Liu, P.; Yu, S.; Ma, J.; He, H. Natural killer cells: A future star for immunotherapy of head and neck squamous cell carcinoma. Front. Immunol. 2024, 15, 1442673. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.K.; Chu, T.-H.; Kim, S.-A.; Vo, M.-C.; Nguyen, V.-T.; Lee, K.-H.; Jung, S.-H.; Yoon, M.; Cho, D.; Lee, J.-J.; et al. Efficacy of natural killer cell therapy combined with chemoradiotherapy in murine models of head and neck squamous cell carcinoma. Cytotherapy 2024, 26, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Rebuffet, L.; Narni-Mancinelli, E.; Cornen, S.; Igarashi, R.Y.; Fantin, V.R. Natural killer cell therapies. Nature 2024, 626, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.-J.; Hagel, C.; Becker, B.; Oetting, A.; Möckelmann, N.; Droste, C.; Möller-Koop, C.; Witt, M.; Blaurock, M.; Loges, S.; et al. Tissue Microarray Analyses Suggest Axl as a Predictive Biomarker in HPV-Negative Head and Neck Cancer. Cancers 2022, 14, 1829. [Google Scholar] [CrossRef]
- Nowak, J.; Bentele, M.; Kutle, I.; Zimmermann, K.; Lühmann, J.L.; Steinemann, D.; Kloess, S.; Koehl, U.; Roßberg, W.; Ahmed, A.; et al. CAR-NK Cells Targeting HER1 (EGFR) Show Efficient Anti-Tumor Activity against Head and Neck Squamous Cell Carcinoma (HNSCC). Cancers 2023, 15, 3169. [Google Scholar] [CrossRef]
- Ciulean, I.S.; Fischer, J.; Quaiser, A.; Bach, C.; Abken, H.; Tretbar, U.S.; Fricke, S.; Koehl, U.; Schmiedel, D.; Grunwald, T. CD44v6 specific CAR-NK cells for targeted immunotherapy of head and neck squamous cell carcinoma. Front. Immunol. 2023, 14, 1290488. [Google Scholar] [CrossRef]
- Zhang, M.; Lam, K.P.; Xu, S. Natural Killer Cell Engagers (NKCEs): A new frontier in cancer immunotherapy. Front. Immunol. 2023, 14, 1207276. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostecki, K.L.; Harmon, R.L.; Iida, M.; Harris, M.A.; Crossman, B.E.; Bruce, J.Y.; Salgia, R.; Wheeler, D.L. Axl Regulation of NK Cell Activity Creates an Immunosuppressive Tumor Immune Microenvironment in Head and Neck Cancer. Cancers 2025, 17, 994. https://doi.org/10.3390/cancers17060994
Kostecki KL, Harmon RL, Iida M, Harris MA, Crossman BE, Bruce JY, Salgia R, Wheeler DL. Axl Regulation of NK Cell Activity Creates an Immunosuppressive Tumor Immune Microenvironment in Head and Neck Cancer. Cancers. 2025; 17(6):994. https://doi.org/10.3390/cancers17060994
Chicago/Turabian StyleKostecki, Kourtney L., Regan L. Harmon, Mari Iida, Madelyn A. Harris, Bridget E. Crossman, Justine Yang Bruce, Ravi Salgia, and Deric L. Wheeler. 2025. "Axl Regulation of NK Cell Activity Creates an Immunosuppressive Tumor Immune Microenvironment in Head and Neck Cancer" Cancers 17, no. 6: 994. https://doi.org/10.3390/cancers17060994
APA StyleKostecki, K. L., Harmon, R. L., Iida, M., Harris, M. A., Crossman, B. E., Bruce, J. Y., Salgia, R., & Wheeler, D. L. (2025). Axl Regulation of NK Cell Activity Creates an Immunosuppressive Tumor Immune Microenvironment in Head and Neck Cancer. Cancers, 17(6), 994. https://doi.org/10.3390/cancers17060994