

The Immune–Genomics of Cholangiocarcinoma: A Biological Footprint to Develop Novel Immunotherapies

 , ,
, ,  and
and 
Simple Summary
Abstract
1. Introduction
2. Biology of CCA
2.1. Tumor Phenotypes
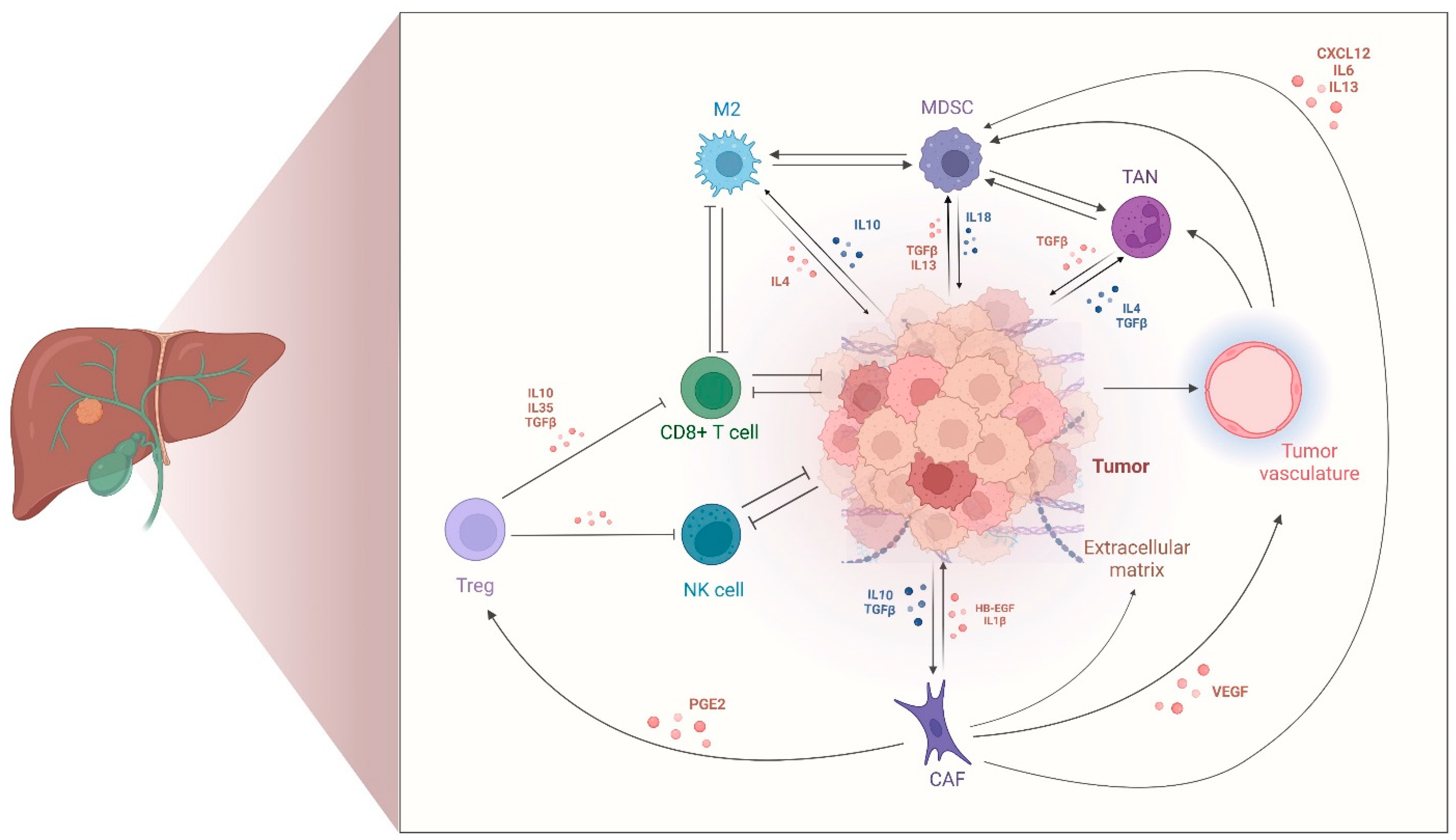
2.2. Tumor-Immune Microenvironment in CCA
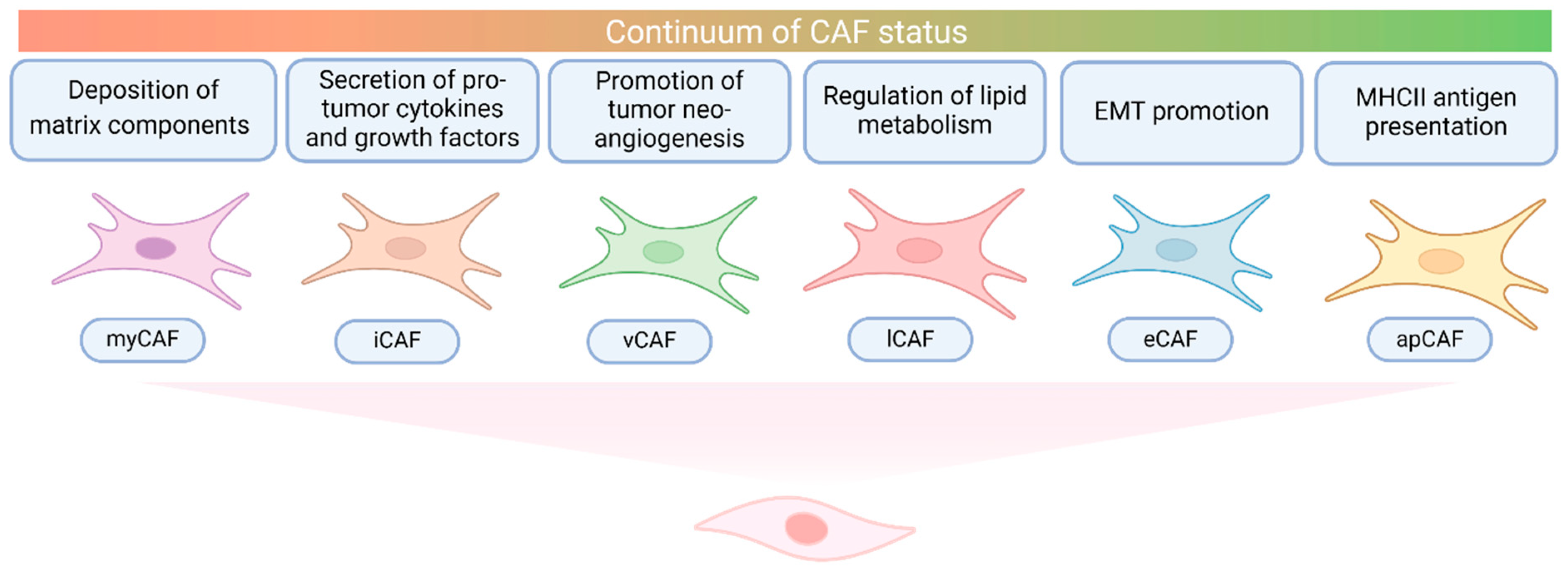
2.3. Cancer-Associated Fibroblasts in CCA
2.4. Immune-Based Classifications of CCA
3. Targeting the TIME of Clinics
4. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Valle, J.W.; Kelley, R.K.; Nervi, B.; Oh, D.-Y.; Zhu, A.X. Biliary tract cancer. Lancet 2021, 397, 428–444. [Google Scholar] [CrossRef]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic syndrome increases the risk of primary liver cancer in the United States: A study in the SEER-Medicare database. Hepatology 2011, 54, 463–471. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Greten, T.F.; Schwabe, R.; Bardeesy, N.; Ma, L.; Goyal, L.; Kelley, R.K.; Wang, X.W. Immunology and immunotherapy of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 349–365. [Google Scholar] [CrossRef]
- Loeuillard, E.; Yang, J.; Buckarma, E.; Wang, J.; Liu, Y.; Conboy, C.; Pavelko, K.D.; Li, Y.; O’Brien, D.; Wang, C.; et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J. Clin. Investig. 2020, 130, 5380–5396. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, Q.; Greten, T.F. MDSCs in liver cancer: A critical tumor-promoting player and a potential therapeutic target. Cell. Immunol. 2021, 361, 104295. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Zhang, Q.; Cao, Q.; Kong, R.; Xiang, X.; Liu, H.; Feng, M.; Wang, F.; Cheng, J.; Li, Z.; et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 2022, 612, 141–147. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, P.; Sun, R.; Li, J.; Hu, Z.; Xin, H.; Luo, C.; Zhou, J.; Fan, J.; Zhou, S. Tumor-associated neutrophils and macrophages interaction contributes to intrahepatic cholangiocarcinoma progression by activating STAT3. J. Immunother. Cancer 2021, 9, e001946. [Google Scholar] [CrossRef]
- Alvisi, G.; Termanini, A.; Soldani, C.; Portale, F.; Carriero, R.; Pilipow, K.; Costa, G.; Polidoro, M.; Franceschini, B.; Malenica, I.; et al. Multimodal single-cell profiling of intrahepatic cholangiocarcinoma defines hyperactivated Tregs as a potential therapeutic target. J. Hepatol. 2022, 77, 1359–1372. [Google Scholar] [CrossRef]
- Bao, X.; Li, Q.; Chen, J.; Chen, D.; Ye, C.; Dai, X.; Wang, Y.; Li, X.; Rong, X.; Cheng, F.; et al. Molecular Subgroups of Intrahepatic Cholangiocarcinoma Discovered by Single-Cell RNA Sequencing-Assisted Multiomics Analysis. Cancer Immunol. Res. 2022, 10, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef]
- Affo, S.; Nair, A.; Brundu, F.; Ravichandra, A.; Bhattacharjee, S.; Matsuda, M.; Chin, L.; Filliol, A.; Wen, W.; Song, X.; et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 2021, 39, 866–882.e11. [Google Scholar] [CrossRef]
- Sulpice, L.; Rayar, M.; Desille, M.; Turlin, B.; Fautrel, A.; Boucher, E.; Llamas-Gutierrez, F.; Meunier, B.; Boudjema, K.; Clément, B.; et al. Molecular profiling of stroma identifies osteopontin as an independent predictor of poor prognosis in intrahepatic cholangiocarcinoma. Hepatology 2013, 58, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-Y.; He, A.R.; Qin, S.; Chen, L.T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Ah Lee, M.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef]
- Kelley, R.K.; Ueno, M.; Finn, R.S.; Furuse, J.; Ren, Z.; Yau, T.; Klümpen, H.J.; Chan, S.L.; Ozaka, M.; Verslype, C.; et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2023, 401, 1853–1865. [Google Scholar] [CrossRef]
- Verlingue, L.; Malka, D.; Allorant, A.; Massard, C.; Ferté, C.; Lacroix, L.; Rouleau, E.; Auger, N.; Ngo, M.; Nicotra, C.; et al. Precision medicine for patients with advanced biliary tract cancers: An effective strategy within the prospective MOSCATO-01 trial. Eur. J. Cancer 2017, 87, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Valle, J.W.; Morizane, C.; Karasic, T.B.; Abrams, T.A.; Furuse, J.; Kelley, R.K.; Cassier, P.A.; et al. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. N. Engl. J. Med. 2023, 388, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Borad, M.J.; Azad, N.S.; Kurzrock, R.; Abou-Alfa, G.K.; George, B.; Hainsworth, J.; Meric-Bernstam, F.; Swanton, C.; Sweeney, C.J.; et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): A multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021, 22, 1290–1300. [Google Scholar] [CrossRef]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): A phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 2020, 21, 1234–1243. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.-C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation profiling in cholangiocarcinoma: Prognostic and therapeutic implications. PLoS ONE. 2014, 9, e115383. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, Z.; Li, C.; Wang, C.; Jiang, W.; Chang, J.; Han, S.; Lu, Z.; Shao, Z.; Wang, Y.; et al. The genomic landscape of cholangiocarcinoma reveals the disruption of post-transcriptional modifiers. Nat. Commun. 2022, 13, 3061. [Google Scholar] [CrossRef] [PubMed]
- Wardell, C.P.; Fujita, M.; Yamada, T.; Simbolo, M.; Fassan, M.; Karlic, R.; Polak, P.; Kim, J.; Hatanaka, Y.; Maejima, K.; et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J. Hepatol. 2018, 68, 959–969. [Google Scholar] [CrossRef]
- Quinn, L.M.; Haldenby, S.; Antzcak, P.; Fowler, A.; Bullock, K.; Kenny, J.; Gilbert, T.; Andrews, T.; Diaz-Nieto, R.; Fenwick, S.; et al. Genomic profiling of idiopathic peri-hilar cholangiocarcinoma reveals new targets and mutational pathways. Sci. Rep. 2023, 13, 6681. [Google Scholar] [CrossRef]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.; Wong, B.H.; et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef]
- Zou, S.; Li, J.; Zhou, H.; Hou, Y.R.; Liu, K.X.; Sun, R.Q.; Wang, P.C.; Luo, C.B.; Li, J.; Zou, J.X.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012, 142, 1021–1031.e15. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef]
- Montal, R.; Sia, D.; Montironi, C.; Leow, W.Q.; Esteban-Fabró, R.; Pinyol, R.; Torres-Martin, M.; Bassaganyas, L.; Moeini, A.; Peix, J.; et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Silverman, I.M.; Murugesan, K.; Lihou, C.F.; Féliz, L.; Frampton, G.M.; Newton, R.C.; Tada, H.; Albacker, L.A.; Burn, T.C. Comprehensive genomic profiling in FIGHT-202 reveals the landscape of actionable alterations in advanced cholangiocarcinoma. J. Clin. Oncol. 2019, 37 (Suppl. S15), 4080. [Google Scholar] [CrossRef]
- Javle, M.; Bekaii-Saab, T.; Jain, A.; Wang, Y.; Kelley, R.K.; Wang, K.; Kang, H.C.; Catenacci, D.; Ali, S.; Krishnan, S.; et al. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer 2016, 122, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Bibeau, K.; Schultz, N.; Yaqubie, A.; Millang, B.; Ren, H.; Féliz, L. Effect of FGFR2 Alterations on Overall and Progression-Free Survival in Patients Receiving Systemic Therapy for Intrahepatic Cholangiocarcinoma. Target Oncol. 2022, 17, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Subimerb, C.; Pinlaor, S.; Khuntikeo, N.; Leelayuwat, C.; Morris, A.; McGrath, M.S.; Wongkham, S. Tissue invasive macrophage density is correlated with prognosis in cholangiocarcinoma. Mol. Med. Rep. 2010, 3, 597–605. [Google Scholar] [CrossRef]
- Charbel, A.; Tavernar, L.; Albrecht, T.; Brinkmann, F.; Verheij, J.; Roos, E.; Vogel, M.N.; Köhler, B.; Springfeld, C.; Brobeil, A.; et al. Spatiotemporal analysis of tumour-infiltrating immune cells in biliary carcinogenesis. Br. J. Cancer 2022, 127, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, T.; Dong, P.; Brinkmann, F.; Verheij, J.; Roos, E.; Vogel, M.N.; Köhler, B.; Springfeld, C.; Brobeil, A.; et al. M2-polarized tumor-associated macrophages promote epithelial-mesenchymal transition via activation of the AKT3/PRAS40 signaling pathway in intrahepatic cholangiocarcinoma. J. Cell Biochem. 2020, 121, 2828–2838. [Google Scholar] [CrossRef]
- Hasita, H.; Komohara, Y.; Okabe, H.; Masuda, T.; Ohnishi, K.; Lei, X.F.; Beppu, T.; Baba, H.; Takeya, M. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010, 101, 1913–1919. [Google Scholar] [CrossRef]
- Miura, T.; Yoshizawa, T.; Hirai, H.; Seino, H.; Morohashi, S.; Wu, Y.; Wakiya, T.; Kimura, N.; Kudo, D.; Ishido, K.; et al. Prognostic Impact of CD163+ Macrophages in Tumor Stroma and CD8+ T-Cells in Cancer Cell Nests in Invasive Extrahepatic Bile Duct Cancer. Anticancer Res. 2017, 37, 183–190. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, C.; Duan, Y.; Heinrich, B.; Rosato, U.; Diggs, L.P.; Ma, L.; Roy, S.; Fu, Q.; Brown, Z.J.; et al. Gut Microbiome Directs Hepatocytes to Recruit MDSCs and Promote Cholangiocarcinoma. Cancer Discov. 2021, 11, 1248–1267. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zeng, X.; Ren, X.; Zhang, Y.; Huang, M.; Tan, L.; Dai, Z.; Lai, J.; Xie, W.; Chen, Z.; et al. Targeting tumour-intrinsic N7-methylguanosine tRNA modification inhibits MDSC recruitment and improves anti-PD-1 efficacy. Gut 2023, 72, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Zhang, G.; Zheng, G.; Zhang, H.; Qiu, L. MUC1 induces the accumulation of Foxp3(+) Treg cells in the tumor microenvironment to promote the growth and metastasis of cholangiocarcinoma through the EGFR/PI3K/Akt signaling pathway. Int. Immunopharmacol. 2023, 118, 110091. [Google Scholar] [CrossRef] [PubMed]
- Jarman, E.J.; Horcas-Lopez, M.; Waddell, S.H.; MacMaster, S.; Gournopanos, K.; Soong, D.Y.H.; Musialik, K.I.; Tsokkou, P.; Ng, M.E.; Cambridge, W.A.; et al. DKK1 drives immune suppressive phenotypes in intrahepatic cholangiocarcinoma and can be targeted with anti-DKK1 therapeutic DKN-01. Liver Int. 2023, 43, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.J.; Koh, J.; Kim, K.; Chie, E.K.; Kim, B.; Lee, K.B.; Jang, J.Y.; Kim, S.W.; Oh, D.Y.; Bang, Y.J.; et al. High ratio of programmed cell death protein 1 (PD-1)(+)/CD8(+) tumor-infiltrating lymphocytes identifies a poor prognostic subset of extrahepatic bile duct cancer undergoing surgery plus adjuvant chemoradiotherapy. Radiother Oncol. 2015, 117, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Prithviraj, P.; Shrestha, R.; Sharma, R.; Anaka, M.; Bridle, K.R.; Kannourakis, G.; Crawford, D.H.G.; Jayachandran, A. Prognostic Role of Immune Checkpoint Regulators in Cholangiocarcinoma: A Pilot Study. J. Clin. Med. 2021, 10, 2191. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Wirta, E.V.; Szeto, S.; Koppatz, H.; Nordin, A.; Mäkisalo, H.; Arola, J.; Sirén, J.; Ahtiainen, M.; Böhm, J.; Mecklin, J.P.; et al. High immune cell infiltration predicts improved survival in cholangiocarcinoma. Front. Oncol. 2024, 14, 1333926. [Google Scholar] [CrossRef]
- Tian, L.; Ma, J.; Ma, L.; Zheng, B.; Liu, L.; Song, D.; Wang, Y.; Zhang, Z.; Gao, Q.; Song, K.; et al. PD-1/PD-L1 expression profiles within intrahepatic cholangiocarcinoma predict clinical outcome. World J. Surg. Oncol. 2020, 18, 303. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.; Kitano, Y.; Yamashita, Y.I.; Yamao, T.; Kajiyama, K.; Yoshizumi, T.; Fukuzawa, K.; Sugimachi, K.; Ikeda, Y.; Takamori, H.; et al. Cancer-associated fibroblast senescence and its relation with tumour-infiltrating lymphocytes and PD-L1 expressions in intrahepatic cholangiocarcinoma. Br. J. Cancer 2022, 126, 219–227. [Google Scholar] [CrossRef]
- Carapeto, F.; Bozorgui, B.; Shroff, R.T.; Chagani, S.; Solis Soto, L.; Foo, W.C.; Wistuba, I.; Meric-Bernstam, F.; Shalaby, A.; Javle, M.; et al. The immunogenomic landscape of resected intrahepatic cholangiocarcinoma. Hepatology 2022, 75, 297–308. [Google Scholar] [CrossRef]
- Vigano, L.; Soldani, C.; Franceschini, B.; Cimino, M.; Lleo, A.; Donadon, M.; Roncalli, M.; Aghemo, A.; Di Tommaso, L.; Torzilli, G. Tumor-Infiltrating Lymphocytes and Macrophages in Intrahepatic Cholangiocellular Carcinoma. Impact on Prognosis after Complete Surgery. J. Gastrointest Surg. 2019, 23, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.J.; Lu, J.C.; Zeng, H.Y.; Zhou, R.; Sun, Q.M.; Yang, G.H.; Pei, Y.Z.; Meng, X.L.; Shen, Y.H.; Zhang, P.F.; et al. CTLA-4 Synergizes With PD1/PD-L1 in the Inhibitory Tumor Microenvironment of Intrahepatic Cholangiocarcinoma. Front Immunol. 2021, 12, 705378. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Zhu, M.X.; Liu, W.F.; Rui, W.W.; Chen, Y.; Ding, X.Y.; Jiang, Y.S.; Wu, Z.Y.; Liu, B.B. Identification of clinically relevant subsets CD39(+)PD-1(+)CD8(+) T cells and CD39(+) regulatory T cells in intrahepatic cholangiocarcinoma using single-cell CyTOF. Transl. Oncol. 2024, 44, 101954. [Google Scholar] [CrossRef] [PubMed]
- Mocan, L.P.; Craciun, R.; Grapa, C.; Melincovici, C.S.; Rusu, I.; Al Hajjar, N.; Sparchez, Z.; Leucuta, D.; Ilies, M.; Sparchez, M.; et al. PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor for patients with intrahepatic cholangiocarcinoma. Cancer Immunol. Immunother. 2023, 72, 1003–1014. [Google Scholar] [CrossRef]
- Fontugne, J.; Augustin, J.; Pujals, A.; Compagnon, P.; Rousseau, B.; Luciani, A.; Tournigand, C.; Cherqui, D.; Azoulay, D.; Pawlotsky, J.M.; et al. PD-L1 expression in perihilar and intrahepatic cholangiocarcinoma. Oncotarget 2017, 8, 24644–24651. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Herrmann, E.; Schnitzbauer, A.A.; Zeuzem, S.; Hansmann, M.L.; Peveling-Oberhag, J.; Hartmann, S. PD-L1 expression in extrahepatic cholangiocarcinoma. Histopathology 2017, 71, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Lee, J.-C.; Shin, D.W.; Kim, J.; Hwang, J.-H. High PD-L1 expression is associated with therapeutic response to pembrolizumab in patients with advanced biliary tract cancer. Sci. Rep. 2020, 10, 12348. [Google Scholar] [CrossRef]
- Oh, D.-Y.; He, A.R.; Bouattour, M.; Okusaka, T.; Qin, S.; Chen, L.T.; Kitano, M.; Lee, C.K.; Kim, J.W.; Chen, M.H.; et al. Durvalumab or placebo plus gemcitabine and cisplatin in participants with advanced biliary tract cancer (TOPAZ-1): Updated overall survival from a randomised phase 3 study. Lancet Gastroenterol. Hepatol. 2024, 9, 694–704. [Google Scholar] [CrossRef]
- Filliol, A.; Saito, Y.; Nair, A.; Dapito, D.H.; Yu, L.X.; Ravichandra, A.; Bhattacharjee, S.; Affo, S.; Fujiwara, N.; Su, H.; et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature 2022, 610, 356–365. [Google Scholar] [CrossRef]
- Vaquero, J.; Aoudjehane, L.; Fouassier, L. Cancer-associated fibroblasts in cholangiocarcinoma. Curr. Opin. Gastroenterol. 2020, 36, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Cantallops Vilà, P.; Ravichandra, A.; Agirre Lizaso, A.; Perugorria, M.J.; Affò, S. Heterogeneity, crosstalk, and targeting of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2024, 79, 941–958. [Google Scholar] [CrossRef]
- Chuaysri, C.; Thuwajit, P.; Paupairoj, A.; Chau-In, S.; Suthiphongchai, T.; Thuwajit, C. Alpha-smooth muscle actin-positive fibroblasts promote biliary cell proliferation and correlate with poor survival in cholangiocarcinoma. Oncol. Rep. 2009, 21, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Sirica, A.E.; Dumur, C.I.; Campbell, D.J.; Almenara, J.A.; Ogunwobi, O.O.; Dewitt, J.L. Intrahepatic cholangiocarcinoma progression: Prognostic factors and basic mechanisms. Clin. Gastroenterol. Hepatol. 2009, 7 (Suppl. 11), S68–S78. [Google Scholar] [CrossRef]
- Loosen, S.H.; Roderburg, C.; Kauertz, K.L.; Pombeiro, I.; Leyh, C.; Benz, F.; Vucur, M.; Longerich, T.; Koch, A.; Braunschweig, T.; et al. Elevated levels of circulating osteopontin are associated with a poor survival after resection of cholangiocarcinoma. J. Hepatol. 2017, 67, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Clapéron, A.; Mergey, M.; Aoudjehane, L.; Ho-Bouldoires, T.H.; Wendum, D.; Prignon, A.; Merabtene, F.; Firrincieli, D.; Desbois-Mouthon, C.; Scatton, O.; et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology 2013, 58, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Clapéron, A.; Mergey, M.; Nguyen Ho-Bouldoires, T.H.; Vignjevic, D.; Wendum, D.; Chrétien, Y.; Merabtene, F.; Frazao, A.; Paradis, V.; Housset, C.; et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial-mesenchymal transition. J. Hepatol. 2014, 61, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Lai, G.H.; Radaeva, S.; Nakamura, T.; Sirica, A.E. Unique epithelial cell production of hepatocyte growth factor/scatter factor by putative precancerous intestinal metaplasias and associated “intestinal-type” biliary cancer chemically induced in rat liver. Hepatology 2000, 31, 1257–1265. [Google Scholar] [CrossRef]
- Fingas, C.D.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Guicciardi, M.E.; Cazanave, S.C.; Mertens, J.C.; Sirica, A.E.; Gores, G.J. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology 2011, 54, 2076–2088. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, A.; Rombouts, K.; Galastri, S.; Caligiuri, A.; Mingarelli, E.; Mello, T.; Marra, F.; Mantero, S.; Roncalli, M.; Invernizzi, P.; et al. Role of the stromal-derived factor-1 (SDF-1)-CXCR4 axis in the interaction between hepatic stellate cells and cholangiocarcinoma. J. Hepatol. 2012, 57, 813–820. [Google Scholar] [CrossRef]
- Okabe, H.; Beppu, T.; Ueda, M.; Hayashi, H.; Ishiko, T.; Masuda, T.; Otao, R.; Horlad, H.; Mima, K.; Miyake, K.; et al. Identification of CXCL5/ENA-78 as a factor involved in the interaction between cholangiocarcinoma cells and cancer-associated fibroblasts. Int. J. Cancer 2012, 131, 2234–2241. [Google Scholar] [CrossRef]
- Okamoto, K.; Tajima, H.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; Nakamura, K.; Oyama, K.; Nakagawara, H.; et al. Angiotensin II enhances epithelial-to-mesenchymal transition through the interaction between activated hepatic stellate cells and the stromal cell-derived factor-1/CXCR4 axis in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2012, 41, 573–582. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, J.; Qin, C. Blockade of CXCL12/CXCR4 signaling inhibits intrahepatic cholangiocarcinoma progression and metastasis via inactivation of canonical Wnt pathway. J. Exp. Clin. Cancer Res. 2014, 33, 103. [Google Scholar] [CrossRef]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Yang, K.; Wang, Q.; Zhu, Z.; Li, S.; Li, B.; Feng, B.; Tang, C. Prediction of CAF-related genes in immunotherapy and drug sensitivity in hepatocellular carcinoma: A multi-database analysis. Genes Immun. 2024, 25, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, M.A.; Kepecs, B.; Torres-Martin, M.; Bramel, E.R.; Haber, P.K.; Merritt, E.; Rialdi, A.; Param, N.J.; Maeda, M.; Lindblad, K.E.; et al. Novel microenvironment-based classification of intrahepatic cholangiocarcinoma with therapeutic implications. Gut 2023, 72, 736–748. [Google Scholar] [CrossRef]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, B.; Yang, X.; Cai, Q.; Liu, W.; Tian, M.; Luo, H.; Yin, W.; Song, Y.; Shi, Y.; et al. Fibroblastic FAP promotes intrahepatic cholangiocarcinoma growth via MDSCs recruitment. Neoplasia 2019, 21, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, A.; Pastore, M.; Marra, F.; Raggi, C. The Role of Stroma in Cholangiocarcinoma: The Intriguing Interplay between Fibroblastic Component, Immune Cell Subsets and Tumor Epithelium. Int. J. Mol. Sci. 2018, 19, 2885. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, S.; Tavormina, J.; Tampe, D.; Zeisberg, M.; Wang, H.; Mahadevan, K.K.; Wu, C.J.; Sugimoto, H.; Chang, C.C.; et al. Oncogenic collagen I homotrimers from cancer cells bind to α3β1 integrin and impact tumor microbiome and immunity to promote pancreatic cancer. Cancer Cell. 2022, 40, 818–834.e9. [Google Scholar] [CrossRef]
- Cadamuro, M.; Brivio, S.; Mertens, J.; Vismara, M.; Moncsek, A.; Milani, C.; Fingas, C.; Cristina Malerba, M.; Nardo, G.; Dall’Olmo, L.; et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J. Hepatol. 2019, 70, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Inoue, K.; Klein, S.; Halvorsen, S.; Chen, J.; Matsui, A.; Nikmaneshi, M.R.; Kitahara, S.; Hato, T.; Chen, X.; et al. Placental growth factor promotes tumour desmoplasia and treatment resistance in intrahepatic cholangiocarcinoma. Gut 2022, 71, 185–193. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018, 24, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Job, S.; Rapoud, D.; Dos Santos, A.; Gonzalez, P.; Desterke, C.; Pascal, G.; Elarouci, N.; Ayadi, M.; Adam, R.; Azoulay, D.; et al. Identification of Four Immune Subtypes Characterized by Distinct Composition and Functions of Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 72, 965–981. [Google Scholar] [CrossRef]
- Lin, Y.; Peng, L.; Dong, L.; Liu, D.; Ma, J.; Lin, J.; Chen, X.; Lin, P.; Song, G.; Zhang, M.; et al. Geospatial Immune Heterogeneity Reflects the Diverse Tumor–Immune Interactions in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2022, 12, 2350–2371. [Google Scholar] [CrossRef]
- Mody, K.; Jain, P.; El-Refai, S.M.; Azad, N.S.; Zabransky, D.J.; Baretti, M.; Shroff, R.T.; Kelley, R.K.; El-Khouiery, A.B.; Hockenberry, A.J.; et al. Clinical, Genomic, and Transcriptomic Data Profiling of Biliary Tract Cancer Reveals Subtype-Specific Immune Signatures. JCO Precis. Oncol. 2022, 6, e2100510. [Google Scholar] [CrossRef]
- Sridharan, V.; Neyaz, A.; Chogule, A.; Baiev, I.; Reyes, S.; Barr Fritcher, E.G.; Lennerz, J.K.; Sukov, W.; Kipp, B.; Ting, D.T.; et al. FGFR mRNA Expression in Cholangiocarcinoma and Its Correlation with FGFR2 Fusion Status and Immune Signatures. Clin. Cancer Res. 2022, 28, 5431–5439. [Google Scholar] [CrossRef]
- Ding, G.Y.; Ma, J.Q.; Yun, J.P.; Chen, X.; Ling, Y.; Zhang, S.; Shi, J.Y.; Chang, Y.Q.; Ji, Y.; Wang, X.Y.; et al. Distribution and density of tertiary lymphoid structures predict clinical outcome in intrahepatic cholangiocarcinoma. J. Hepatol. 2022, 76, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Oh, D.Y.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: Results from the KEYNOTE-158 and KEYNOTE-028 studies. Int. J. Cancer 2020, 147, 2190–2198. [Google Scholar] [CrossRef]
- de Biasi, A.R.; Villena-Vargas, J.; Adusumilli, P.S. Cisplatin-induced antitumor immunomodulation: A review of preclinical and clinical evidence. Clin. Cancer Res. 2014, 20, 5384–5391. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Ueno, M.; Ikeda, M.; Morizane, C.; Kobayashi, S.; Ohno, I.; Kondo, S.; Okano, N.; Kimura, K.; Asada, S.; Namba, Y.; et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: A non-randomised, multicentre, open-label, phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 611–621. [Google Scholar] [CrossRef]
- Oh, D.Y.; Lee, K.H.; Lee, D.W.; Yoon, J.; Kim, T.Y.; Bang, J.H.; Nam, A.R.; Oh, K.S.; Kim, J.M.; Lee, Y.; et al. Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: An open-label, single-centre, phase 2 study. Lancet Gastroenterol. Hepatol. 2022, 7, 522–532. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Zhao, Y.; Yang, Q.; Dong, L.; Liu, J.; Li, X.; Zhao, Z.; Mei, Q.; Han, W.; et al. Efficacy and biomarker analysis of nivolumab plus gemcitabine and cisplatin in patients with unresectable or metastatic biliary tract cancers: Results from a phase II study. J. Immunother. Cancer 2020, 8, e000367. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Ueno, M.; Yoo, C.; Ren, Z.; Furuse, J.; Kelley, R.K.; Chan, S.L.; Edeline, J.; Klümpen, H.-J.; Yau, T.; et al. Three-year follow-up data from KEYNOTE-966: Pembrolizumab (pembro) plus gemcitabine and cisplatin (gem/cis) compared with gem/cis alone for patients (pts) with advanced biliary tract cancer (BTC). J. Clin. Oncol. 2024, 42 (Suppl. S16), 4093. [Google Scholar] [CrossRef]
- Rimini, M.; Fornaro, L.; Rizzato, M.D.; Antonuzzo, L.; Rossari, F.; Satake, T.; Vandeputte, H.; Vivaldi, C.; Pressiani, T.; Lucchetti, J.; et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer: A large real-life worldwide population. Eur. J. Cancer 2024, 208, 114199. [Google Scholar] [CrossRef]
- Ghosh, M.; Tiwari, A.; Dubey, A.K.; Bhattacharjee, S.; Manjunath, Y.; Kashipathi, S.; Krishna, S.; Mandal, T.; Madhusudhan, M.S.; Rodrigues, G. Abstract 7535: First-in-human phase 1 clinical trial of ZM008, a monoclonal IgG1 targeting LLT1, monotherapy and in combination with pembrolizumab in advanced solid tumors. Cancer Res. 2024, 84 (Suppl. S6), 7535. [Google Scholar] [CrossRef]
- Sangsuwannukul, T.; Supimon, K.; Sujjitjoon, J.; Phanthaphol, N.; Chieochansin, T.; Poungvarin, N.; Wongkham, S.; Junking, M.; Yenchitsomanus, P.T. Anti-tumour effect of the fourth-generation chimeric antigen receptor T cells targeting CD133 against cholangiocarcinoma cells. Int. Immunopharmacol. 2020, 89, 107069. [Google Scholar] [CrossRef] [PubMed]
- Supimon, K.; Sangsuwannukul, T.; Sujjitjoon, J.; Phanthaphol, N.; Chieochansin, T.; Poungvarin, N.; Wongkham, S.; Junking, M.; Yenchitsomanus, P.T. Anti-mucin 1 chimeric antigen receptor T cells for adoptive T cell therapy of cholangiocarcinoma. Sci. Rep. 2021, 11, 6276. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, L.; Lwin, Z.; Chung, H.C.; Gomez-Roca, C.; Longo, F.; Yanez, E.; Senellart, H.; Doherty, H.; García-Corbacho, J.; Hendifar, A.E.; et al. Lenvatinib plus pembrolizumab for patients with previously treated biliary tract cancers in the multicohort phase II LEAP-005 study. J. Clin. Oncol. 2021, 39 (Suppl. S3), 321. [Google Scholar] [CrossRef]
- Arkenau, H.T.; Martin-Liberal, J.; Calvo, E.; Penel, N.; Krebs, M.G.; Herbst, R.S.; Walgren, R.A.; Widau, R.C.; Mi, G.; Jin, J.; et al. Ramucirumab Plus Pembrolizumab in Patients with Previously Treated Advanced or Metastatic Biliary Tract Cancer: Nonrandomized, Open-Label, Phase I Trial (JVDF). Oncologist 2018, 23, 1407-e136. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Ren, Z.; Chon, H.J.; Park, J.O.; Kim, J.W.; Pressiani, T.; Li, D.; Zhukova, L.; Zhu, A.X.; Chen, M.-H.; et al. Atezolizumab plus chemotherapy with or without bevacizumab in advanced biliary tract cancer: Results from a randomized proof-of-concept phase II trial (IMbrave151). J. Clin. Oncol. 2024, 42 (Suppl. S3), 435. [Google Scholar] [CrossRef]
- Iyer, R.V.; Pokuri, V.K.; Groman, A.; Ma, W.W.; Malhotra, U.; Iancu, D.M.; Grande, C.; Saab, T.B. A Multicenter Phase II Study of Gemcitabine, Capecitabine, and Bevacizumab for Locally Advanced or Metastatic Biliary Tract Cancer. Am. J. Clin. Oncol. 2018, 41, 649–655. [Google Scholar] [CrossRef]
- Lee, S.; Shroff, R.T.; Makawita, S.; Xiao, L.; Danner De Armas, A.; Bhosale, P.; Reddy, K.; Shalaby, A.; Raghav, K.; Pant, S.; et al. Phase II Study of Ramucirumab in Advanced Biliary Tract Cancer Previously Treated By Gemcitabine-Based Chemotherapy. Clin. Cancer Res. 2022, 28, 2229–2236. [Google Scholar] [CrossRef]
- LoRusso, P.; Yamamoto, N.; Patel, M.R.; Laurie, S.A.; Bauer, T.M.; Geng, J.; Davenport, T.; Teufel, M.; Li, J.; Lahmar, M.; et al. The MDM2-p53 Antagonist Brigimadlin (BI 907828) in Patients with Advanced or Metastatic Solid Tumors: Results of a Phase Ia, First-in-Human, Dose-Escalation Study. Cancer Discov. 2023, 13, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Tolcher, A.; Hafez, N.; Lugowska, I.; Ramlau, R.; Macarulla, T.; Geng, J.; Li, J.; Teufel, M.; Märten, A.; et al. Efficacy and Safety of the MDM2-p53 Antagonist Brigimadlin (BI 907828) in Patients with Advanced Biliary Tract Cancer: A Case Series. Oncol. Targets Ther. 2024, 17, 267–280. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, N.; Zhao, Y.; Pan, Q.; Mao, A.; Zhu, W.; Lin, Z.; Wang, L.; Wang, Y.L.; Zhou, J.; et al. 283P Postoperative adjuvant tislelizumab plus lenvatinib and capecitabine for biliary tract cancer: Interim results of a prospective, single-arm, phase II study. Ann. Oncol. 2024, 35, S119. [Google Scholar] [CrossRef]
- Yoo, C.; Park, J.O.; Kim, K.P.; Hyung, J.; Ryoo, B.Y.; Hong, J.Y.; Shin, S.H.; Song, T.J.; Oh, D.; Lee, W.; et al. 97P Neoadjuvant durvalumab plus gemcitabine and cisplatin (D+GemCis) versus gemcis alone for localized biliary tract cancer (BTC): Results of a randomized, multicenter, open-label, phase II trial (DEBATE). Ann. Oncol. 2023, 34, S216. [Google Scholar] [CrossRef]
- Bang, Y.H.; Lee, C.-K.; Bang, K.; Kim, H.D.; Kim, K.P.; Jeong, J.H.; Park, I.; Ryoo, B.Y.; Lee, D.K.; Choi, H.J.; et al. Artificial Intelligence-Powered Spatial Analysis of Tumor-Infiltrating Lymphocytes as a Potential Biomarker for Immune Checkpoint Inhibitors in Patients with Biliary Tract Cancer. Clin. Cancer Res. 2024, 30, 4635–4643. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Bridgewater, J.; Edeline, J.; Kelley, R.K.; Klümpen, H.J.; Malka, D.; Primrose, J.N.; Rimassa, L.; Stenzinger, A.; Valle, J.W.; et al. Biliary tract cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Mahipal, A.; Clemens, K.M.; Liao, J.; Weipert, C.M.; Bucheit, L.; Khushman, M.D.M. Real-world testing, treatment patterns, and outcomes following liquid biopsy in advanced cholangiocarcinoma. J. Clin. Oncol. 2024, 42 (Suppl. S3), 455. [Google Scholar] [CrossRef]
- Lapitz, A.; Azkargorta, M.; Milkiewicz, P.; Olaizola, P.; Zhuravleva, E.; Grimsrud, M.M.; Schramm, C.; Arbelaiz, A.; O’Rourke, C.J.; La Casta, A.; et al. Liquid biopsy-based protein biomarkers for risk prediction, early diagnosis, and prognostication of cholangiocarcinoma. J. Hepatol. 2023, 79, 93–108. [Google Scholar] [CrossRef] [PubMed]
- González-Medina, A.; Vila-Casadesús, M.; Gomez-Rey, M.; Fabregat-Franco, C.; Sierra, A.; Tian, T.V.; Castet, F.; Castillo, G.; Matito, J.; Martinez, P.; et al. Clinical Value of Liquid Biopsy in Patients with FGFR2 Fusion–Positive Cholangiocarcinoma During Targeted Therapy. Clin. Cancer Res. 2024, 30, 4491–4504. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- European Network for the Study of Cholangiocarcinoma. Available online: https://eurocholangionet.eu/european-network-for-the-study-of-cholangiocarcinoma-2/ (accessed on 23 November 2024).
- Precision BTC Network. Available online: https://precision-btc.eu/ (accessed on 4 October 2024).

{kind=link}
{kind=link}
| Study Name/ID | Phase | Status | Experimental Regimen | Target | Class | Setting |
|---|---|---|---|---|---|---|
| NCT06451497 | 1 | Recruiting | ZM008 as single agent followed by combination with pembrolizumab | LLT1 | Monoclonal antibody | Metastatic |
| NCT06256055 | 1 | Recruiting | UCMYM802 | Mesothelin | Cellular therapy (CAR-T) | Metastatic |
| NCT06043466 | 1 | Recruiting | C-13-60 | CEA | Cellular therapy (CAR-T) | Metastatic |
| NCT06239194 | 1/2 | Recruiting | MDX2001 | CD3/CD28 on T cells and TROP2/c-MET on tumor cells | Tetraspecific T cell engager antibody | Metastatic |
| NCT06548412 | 1/2 | Not yet recruiting | CTX-009 + cisplatin, gemcitabine, durvalumab | VEGF-A/DLL4 | Bispecific antibody | Metastatic |
| NCT06501625 | 1/2 | Not yet recruiting | Ivosidenib + cisplatin, gemcitabine, durvalumab | IDH1 | Small molecule inhibitor | Metastatic |
| NCT05921760 | 1/2 | Not yet recruiting | Ivosidenib + nivolumab + ipilimumab | IDH1 + PD-1 + CTLA-4 | Small molecule inhibitor + monoclonal antibodies | Metastatic |
| NCT06708663 | 1/2 | Not yet recruiting | HX009 + IN10018 | PD-1/CD47 + FAK | Bispecific antibody + small molecule inhibitor | Metastatic |
| NCT04492033 | 1b/2 | Active, not recruiting | CTX-009 + irinotecan or paclitaxel | VEGF-A/DLL4 | Bispecific antibody | Metastatic |
| BLUESTAR (NCT05123482) | 1/2a | Recruiting | Rilvegostomig + AZD8205 | PD-1/TIGIT + B7-H4 | Bispecific antibody + ADC | Metastatic |
| GEMINI-Hepatobiliary (NCT05775159) | 2 | Recruiting | Volrustomig or rilvegostomig + cisplatin and gemcitabine | PD-1/CTLA-4 or PD-1/TIGIT | Bispecific antibody | Metastatic |
| NCT06569225 | 2 | Not yet recruiting | Rilvegostomig + cisplatin, gemcitabine, nab-paclitaxel | PD-1/TIGIT | Bispecific antibody | Metastatic |
| SEVILLA (NCT06529718) | 2 | Not yet recruiting | Ivonescimab | PD-1/VEGF | Bispecific antibody | Metastatic |
| NCT06591520 | 2 | Not yet recruiting | Ivonescimab + cisplatin and gemcitabine | PD-1/VEGF | Bispecific antibody | Metastatic |
| NCT06530823 | 2 | Not yet recruiting | Pemigatinib + durvalumab | FGFR1-3 | Small molecule inhibitor + monoclonal antibody | Metastatic |
| Brightline-2 (NCT05512377) | 2 | Recruiting | Brigimadlin | MDM2–TP53 | Small molecule inhibitor | Metastatic |
| NCT06654947 | 2 | Not yet recruiting | Surufatinib + toripalimab + GEMOX | VEGFR1-3, FGFR1, CSF1R + PD-1 | Small molecule inhibitor + Monoclonal antibody | Metastatic |
| NCT05506943 | 2/3 | Active, not recruiting | CTX-009 + paclitaxel | VEGF-A/DLL4 | Bispecific antibody | Metastatic |
| NCT06591520 | 3 | Not yet recruiting | Gemcitabine, cisplatin + AK112 or durvalumab | PD-1/VEGF | Bispecific antibody | Metastatic |
| TOURMALINE (NCT05771480) | 3b | Recruiting | Durvalumab + gemcitabine-based chemotherapy | PD-L1 | Monoclonal antibody | Metastatic |
| TopDouble (NCT05924880) | 3b | Active, not recruiting | Durvalumab + gemcitabine-based chemotherapy | PD-L1 | Monoclonal antibody | Metastatic |
| DEBATE (NCT04308174) | 2 | Active, not recruiting | Cisplatin, gemcitabine with or without durvalumab | PD-L1 | Monoclonal antibody | Neoadjuvant |
| NCT05254847 | 2 | Recruiting | Tislelizumab + lenvatinib + capecitabine | PD-L1 + VEGFR1-3, FGFR1-4, PDGFRα, KIT, and RET | Monoclonal antibody + multi-TKI | Adjuvant |
| NCT05239169 | 2 | Active, not recruiting | Durvalumab + tremelimumab with or without capecitabine | PD-L1 + CTLA-4 | Monoclonal antibodies | Adjuvant |
| ARTEMIDE-Biliary01 (NCT06109779) | 3 | Recruiting | Rilvegostomig/placebo + investigator’s choice of chemotherapy | PD-1/TIGIT | Bispecific antibody | Adjuvant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cammarota, A.; Balsano, R.; Pressiani, T.; Bozzarelli, S.; Rimassa, L.; Lleo, A. The Immune–Genomics of Cholangiocarcinoma: A Biological Footprint to Develop Novel Immunotherapies. Cancers 2025, 17, 272. https://doi.org/10.3390/cancers17020272
Cammarota A, Balsano R, Pressiani T, Bozzarelli S, Rimassa L, Lleo A. The Immune–Genomics of Cholangiocarcinoma: A Biological Footprint to Develop Novel Immunotherapies. Cancers. 2025; 17(2):272. https://doi.org/10.3390/cancers17020272
Chicago/Turabian StyleCammarota, Antonella, Rita Balsano, Tiziana Pressiani, Silvia Bozzarelli, Lorenza Rimassa, and Ana Lleo. 2025. "The Immune–Genomics of Cholangiocarcinoma: A Biological Footprint to Develop Novel Immunotherapies" Cancers 17, no. 2: 272. https://doi.org/10.3390/cancers17020272
APA StyleCammarota, A., Balsano, R., Pressiani, T., Bozzarelli, S., Rimassa, L., & Lleo, A. (2025). The Immune–Genomics of Cholangiocarcinoma: A Biological Footprint to Develop Novel Immunotherapies. Cancers, 17(2), 272. https://doi.org/10.3390/cancers17020272