

Neuroendocrine Transformation as a Mechanism of Resistance to Targeted Lung Cancer Therapies: Emerging Mechanisms and Their Therapeutic Implications


Simple Summary
Abstract
1. Introduction
2. The Context of SCLC Transformation
2.1. EGFR-Mutant NSCLC
2.2. Beyond EGFR-Mutant NSCLC
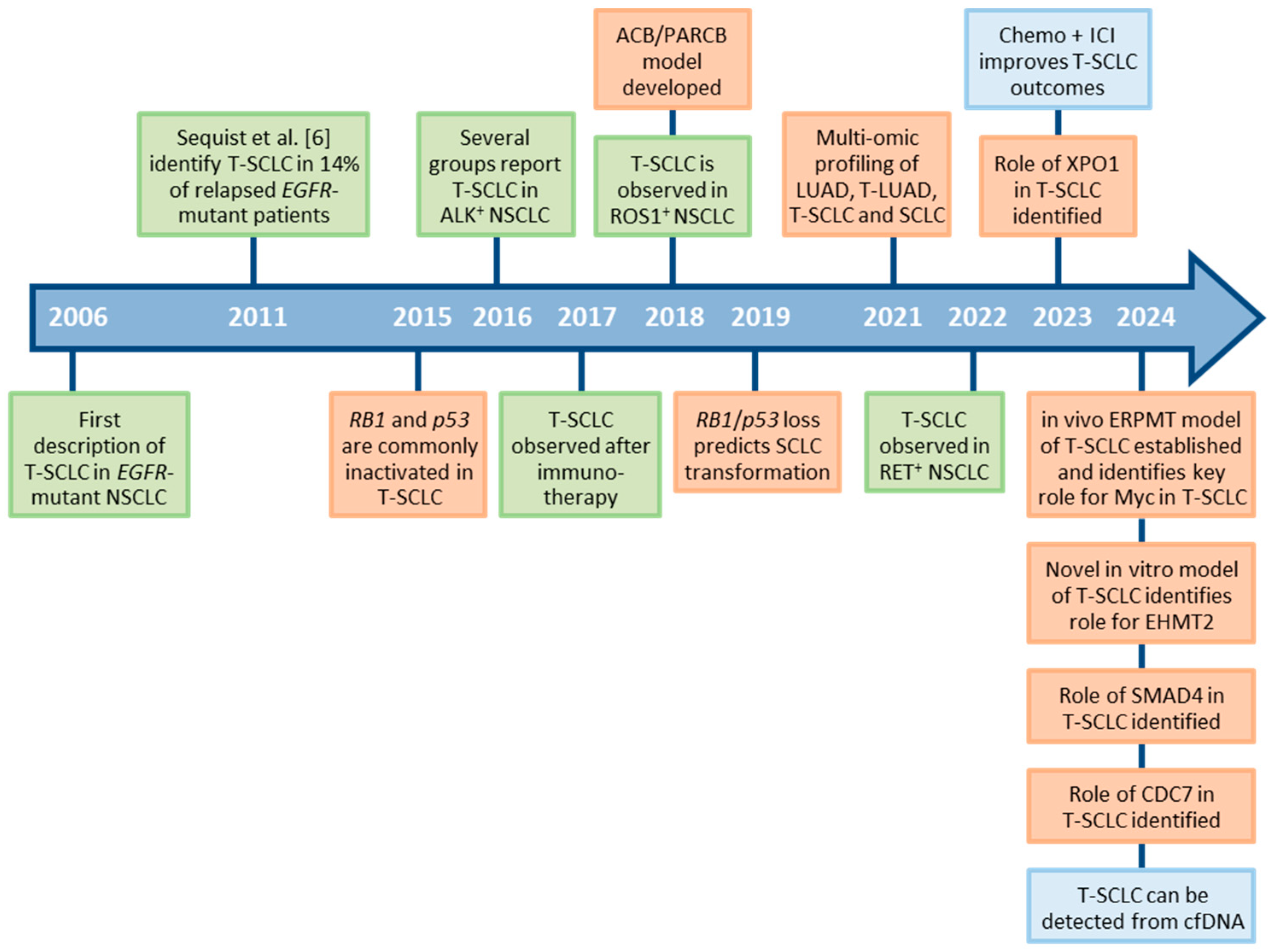
3. Clinical Course of SCLC Transformation
4. The Origins of T-SCLC
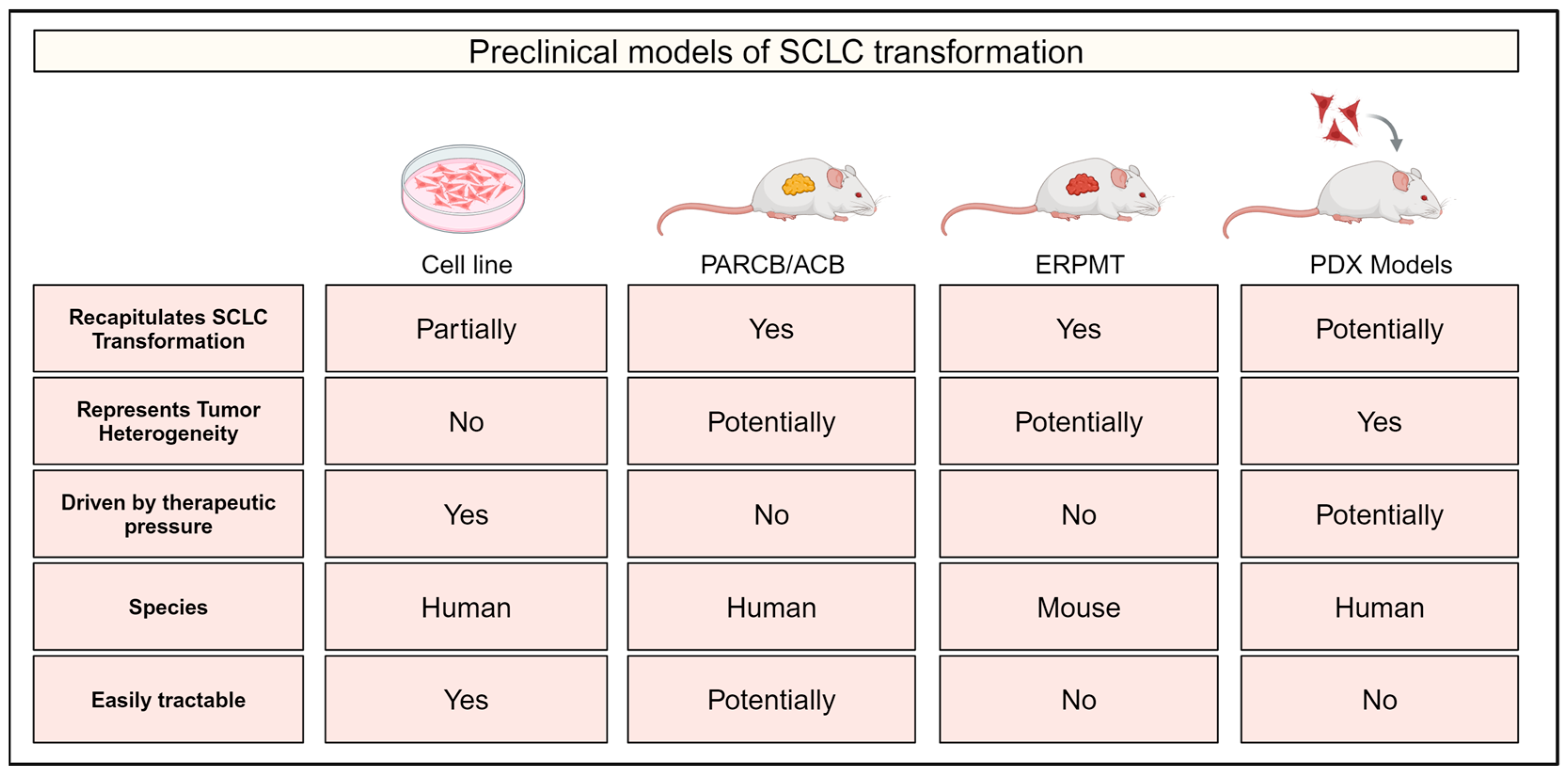
5. Preclinical Models of SCLC Transformation
5.1. Cell Line Models
5.2. The ACB/PARCB Model
5.3. The Genetically Engineered ERPMT Mouse Model
5.4. Patient-Derived Xenograft Models
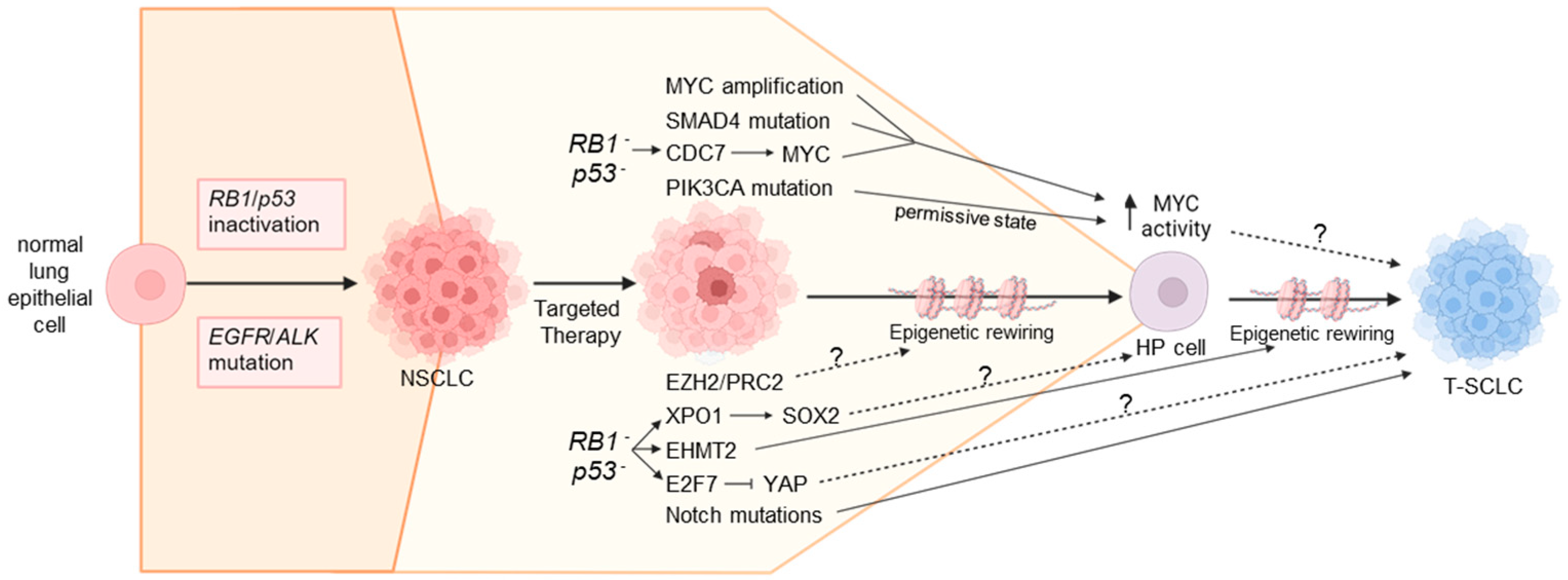
6. Mechanisms Underlying NSCLC-to-SCLC Transformation
6.1. Genomic Alterations Influencing SCLC Transformation
6.1.1. EGFR Exon19 Deletions
6.1.2. RB1 and p53 Inactivation
6.1.3. MYC Amplification
6.1.4. Notch Pathway Mutations
6.1.5. SMAD4 Mutations
6.2. Transcriptional Mechanisms Influencing SCLC Transformation
6.2.1. Exportin 1 Upregulation
6.2.2. CDC7 Upregulation
6.2.3. FGF9 Upregulation
6.2.4. YAP and TAZ Downregulation
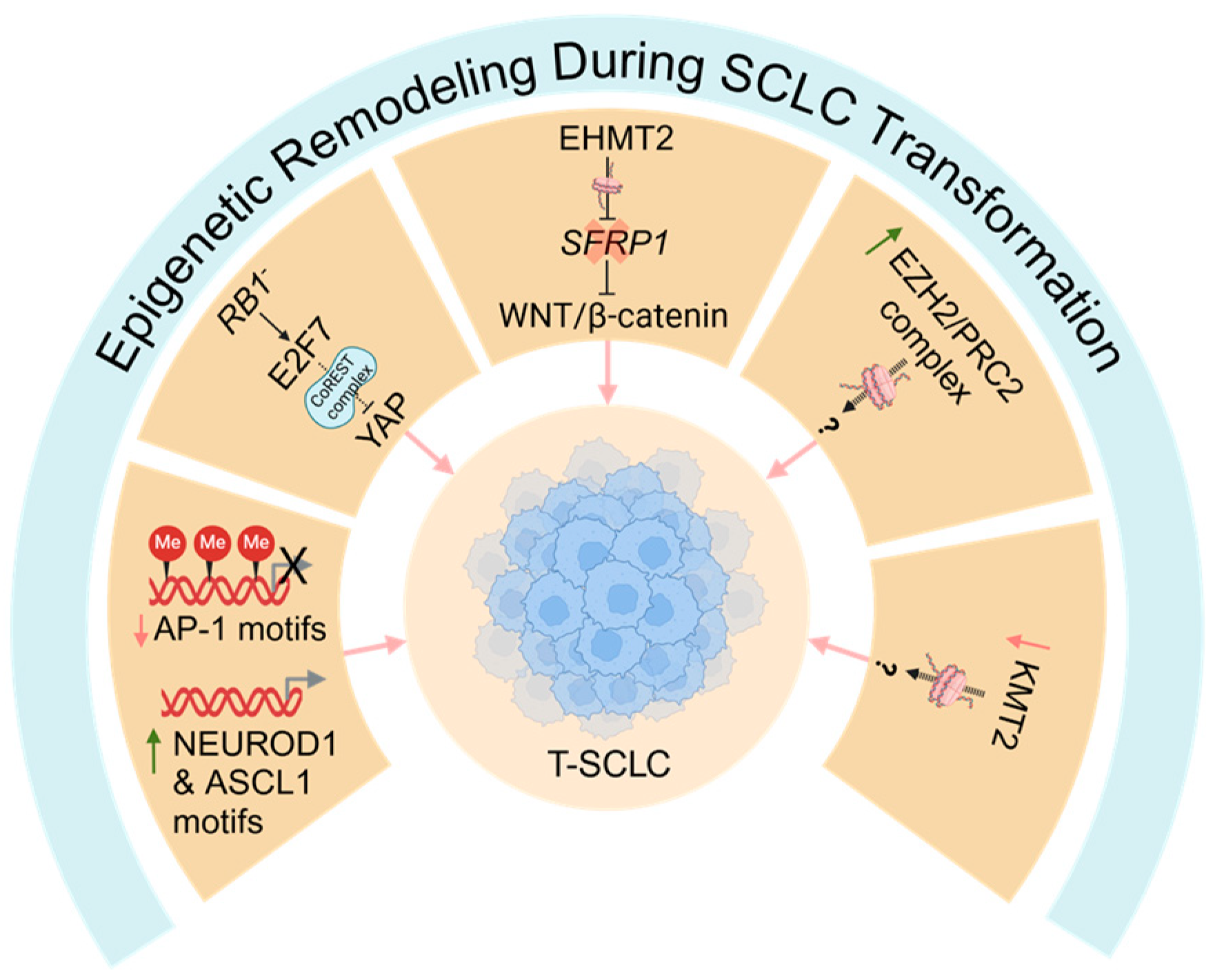
6.3. Epigenetic Remodeling During SCLC Transformation
Mediators of Epigenetic Remodeling During SCLC Transformation
7. Timing of Events That Drive SCLC Transformation
8. Prospects for T-SCLC Therapies
9. Future Perspectives
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Basumallik, N.; Agarwal, M. Small Cell Lung Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-Small Cell Lung Cancer: Current Treatment and Future Advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The Emerging Treatment Landscape of Targeted Therapy in Non-Small-Cell Lung Cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from Non-Small-Cell Lung Cancer to Small-Cell Lung Cancer: Molecular Drivers and Cells of Origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB Loss in Resistant EGFR Mutant Lung Adenocarcinomas That Transform to Small-Cell Lung Cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Ding, X.; Shi, M.; Liu, D.; Cao, J.; Zhang, K.; Zhang, R.; Zhang, L.; Ai, K.; Su, B.; Zhang, J. Transformation to Small Cell Lung Cancer Is Irrespective of EGFR and Accelerated by SMAD4-Mediated ASCL1 Transcription Independently of RB1 in Non-Small Cell Lung Cancer. Cell Commun. Signal. 2024, 22, 45. [Google Scholar] [CrossRef]
- Ferrer, L.; Giaj Levra, M.; Brevet, M.; Antoine, M.; Mazieres, J.; Rossi, G.; Chiari, R.; Westeel, V.; Poudenx, M.; Letreut, J.; et al. A Brief Report of Transformation From NSCLC to SCLC: Molecular and Therapeutic Characteristics. J. Thorac. Oncol. 2019, 14, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lewis, W.; Stewart, C.A.; Morris, B.B.; Solis, L.M.; Serrano, A.; Xi, Y.; Wang, Q.; Lopez, E.R.; Concannon, K.; et al. Brief Report: Comprehensive Clinicogenomic Profiling of Small Cell Transformation From EGFR-Mutant NSCLC Informs Potential Therapeutic Targets. JTO Clin. Res. Rep. 2024, 5, 100623. [Google Scholar] [CrossRef]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. 2019, 37, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at Risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 1784–1793. [Google Scholar] [CrossRef]
- Zakowski, M.F.; Ladanyi, M.; Kris, M.G. EGFR Mutations in Small-Cell Lung Cancers in Patients Who Have Never Smoked. N. Engl. J. Med. 2006, 355, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, R.; Okamoto, I.; Furuta, K.; Kawano, Y.; Sekijima, M.; Dote, K.; Satou, T.; Nishio, K.; Fukuoka, M.; Nakagawa, K. Sequential Occurrence of Non-Small Cell and Small Cell Lung Cancer with the Same EGFR Mutation. Lung Cancer 2007, 58, 411–413. [Google Scholar] [CrossRef]
- Okamoto, I.; Araki, J.; Suto, R.; Shimada, M.; Nakagawa, K.; Fukuoka, M. EGFR Mutation in Gefitinib-Responsive Small-Cell Lung Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2006, 17, 1028–1029. [Google Scholar] [CrossRef]
- Fukui, T.; Tsuta, K.; Furuta, K.; Watanabe, S.I.; Asamura, H.; Ohe, Y.; Maeshima, A.M.; Shibata, T.; Masuda, N.; Matsuno, Y. Epidermal Growth Factor Receptor Mutation Status and Clinicopathological Features of Combined Small Cell Carcinoma with Adenocarcinoma of the Lung. Cancer Sci. 2007, 98, 1714–1719. [Google Scholar] [CrossRef]
- Tatematsu, A.; Shimizu, J.; Murakami, Y.; Horio, Y.; Nakamura, S.; Hida, T.; Mitsudomi, T.; Yatabe, Y. Epidermal Growth Factor Receptor Mutations in Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 6092–6096. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Masago, K.; Katakami, N.; Yatabe, Y. Transformation to SCLC after Treatment with the ALK Inhibitor Alectinib. J. Thorac. Oncol. 2016, 11, e67–e72. [Google Scholar] [CrossRef] [PubMed]
- Levacq, D.; D’Haene, N.; de Wind, R.; Remmelink, M.; Berghmans, T. Histological Transformation of ALK Rearranged Adenocarcinoma into Small Cell Lung Cancer: A New Mechanism of Resistance to ALK Inhibitors. Lung Cancer 2016, 102, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Takegawa, N.; Hayashi, H.; Iizuka, N.; Takahama, T.; Ueda, H.; Tanaka, K.; Takeda, M.; Nakagawa, K. Transformation of ALK Rearrangement-Positive Adenocarcinoma to Small-Cell Lung Cancer in Association with Acquired Resistance to Alectinib. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 953–955. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Ikushima, S.; Ono, R.; Awano, N.; Kondo, K.; Furuhata, Y.; Fukumoto, K.; Kumasaka, T. Transformation to Small-Cell Lung Cancer as a Mechanism of Acquired Resistance to Crizotinib and Alectinib. Jpn. J. Clin. Oncol. 2016, 46, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, S.; Moore, J.A.; Montesion, M.; Sharaf, R.; Lin, D.I.; Colón, C.I.; Fleishmann, Z.; Ebot, E.M.; Newberg, J.Y.; Mills, J.M.; et al. Integrative Analysis of a Large Real-World Cohort of Small Cell Lung Cancer Identifies Distinct Genetic Subtypes and Insights into Histologic Transformation. Cancer Discov. 2023, 13, 1572–1591. [Google Scholar] [CrossRef]
- Cha, Y.J.; Cho, B.C.; Kim, H.R.; Lee, H.-J.; Shim, H.S. A Case of ALK-Rearranged Adenocarcinoma with Small Cell Carcinoma-Like Transformation and Resistance to Crizotinib. J. Thorac. Oncol. 2016, 11, e55–e58. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-C.; Liao, X.-H.; Wang, W.-X.; Xu, C.-W.; Zhuang, W.; Zhong, L.-H.; Du, K.-Q.; Chen, Y.-P.; Chen, G.; Fang, M.-Y. Patients Harboring ALK Rearrangement Adenocarcinoma after Acquired Resistance to Crizotinib and Transformation to Small-Cell Lung Cancer: A Case Report. Onco Targets Ther. 2017, 10, 3187–3192. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Langenbucher, A.; Gupta, P.; Yoda, S.; Fetter, I.J.; Rooney, M.; Do, A.; Kem, M.; Chang, K.P.; Oh, A.Y.; et al. Small Cell Transformation of ROS1 Fusion-Positive Lung Cancer Resistant to ROS1 Inhibition. NPJ Precis. Oncol. 2020, 4, 21. [Google Scholar] [CrossRef]
- Wu, C.; Su, P.; Hsu, C.; Chu, C.; Lin, C. Small Cell Transformation in Crizotinib-resistant ROS1-rearranged Non-small Cell Lung Cancer with Retention of ROS1 Fusion: A Case Report. Thorac. Cancer 2021, 12, 3068–3071. [Google Scholar] [CrossRef]
- Dimou, A.; Lo, Y.-C.; Halling, K.; Mansfield, A.S. EP08.02-037 Small Cell Transformation in a Patient with RET Fusion Positive Lung Adenocarcinoma on Pralsetinib. J. Thorac. Oncol. 2022, 17, S414. [Google Scholar] [CrossRef]
- Gazeu, A.; Aubert, M.; Pissaloux, D.; Lantuejoul, S.; Pérol, M.; Ikhlef, N.; Bouhamama, A.; Franceschi, T.; Swalduz, A. Small-Cell Lung Cancer Transformation as a Mechanism of Resistance to Pralsetinib in RET-Rearranged Lung Adenocarcinoma: A Case Report. Clin. Lung Cancer 2023, 24, 72–75. [Google Scholar] [CrossRef]
- Peng, Y.; Zheng, Z.; Zewen, W.; Yanan, L.; Mingyan, Z.; Meili, S. Whole-Exome Sequencing Explored Mechanism of Selpercatinib Resistance in RET-Rearranged Lung Adenocarcinoma Transformation into Small-Cell Lung Cancer: A Case Report. BMC Pulm. Med. 2023, 23, 492. [Google Scholar] [CrossRef] [PubMed]
- Imakita, T.; Fujita, K.; Kanai, O.; Terashima, T.; Mio, T. Small Cell Lung Cancer Transformation during Immunotherapy with Nivolumab: A Case Report. Respir. Med. Case Rep. 2017, 21, 52–55. [Google Scholar] [CrossRef]
- Imakita, T.; Fujita, K.; Kanai, O.; Okamura, M.; Hashimoto, M.; Nakatani, K.; Sawai, S.; Mio, T. Small Cell Transformation of Non-Small Cell Lung Cancer under Immunotherapy: Case Series and Literature Review. Thorac. Cancer 2021, 12, 3062–3067. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, N.; Nagasaka, M.; Abdulfatah, E.; Shi, D.; Wozniak, A.J.; Sukari, A. Non-Small Cell to Small Cell Lung Cancer on PD-1 Inhibitors: Two Cases on Potential Histologic Transformation. Lung Cancer Targets Ther. 2018, 9, 85. [Google Scholar] [CrossRef]
- Okeya, K.; Kawagishi, Y.; Muranaka, E.; Izumida, T.; Tsuji, H.; Takeda, S. Hyperprogressive Disease in Lung Cancer with Transformation of Adenocarcinoma to Small-Cell Carcinoma during Pembrolizumab Therapy. Intern. Med. 2019, 58, 3295. [Google Scholar] [CrossRef] [PubMed]
- Bar, J.; Ofek, E.; Barshack, I.; Gottfried, T.; Zadok, O.; Kamer, I.; Urban, D.; Perelman, M.; Onn, A. Transformation to Small Cell Lung Cancer as a Mechanism of Resistance to Immunotherapy in Non-Small Cell Lung Cancer. Lung Cancer 2019, 138, 109–115. [Google Scholar] [CrossRef]
- Iams, W.T.; Beckermann, K.E.; Almodovar, K.; Hernandez, J.; Vnencak-Jones, C.; Lim, L.P.; Raymond, C.K.; Horn, L.; Lovly, C.M. Small Cell Lung Cancer Transformation as a Mechanism of Resistance to PD-1 Therapy in KRAS Mutant Lung Adenocarcinoma: A Report of Two Cases. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 14, e45. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, K.; Varkaris, A.; Viray, H.; VanderLaan, P.A.; Rangachari, D.; Costa, D.B. Small Cell Transformation of Non-Small Cell Lung Cancer on Immune Checkpoint Inhibitors: Uncommon or under-Recognized? J. Immunother. Cancer 2020, 8, e000697. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Qu, J.; Sheng, L.; Gao, Q.; Zhou, J. Case Report: Transformation From Non-Small Cell Lung Cancer to Small Cell Lung Cancer During Anti-PD-1 Therapy: A Report of Two Cases. Front. Oncol. 2021, 11, 619371. [Google Scholar] [CrossRef]
- Wang, D.; Ye, W.; Chen, D.; Shi, Q.; Ma, D. Transformation of Lung Squamous Cell Carcinoma to Small Cell Lung Cancer After Immunotherapy Resistance: A Case Report. Cancer Manag. Res. 2023, 15, 803. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, H.; Fan, Y. Shaping the Tumor Immune Microenvironment of SCLC: Mechanisms, and Opportunities for Immunotherapy. Cancer Treat. Rev. 2023, 120, 102606. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Xu, Y.; Li, Y.; Zhou, Y.; Chu, L.; Yang, X.; Ni, J.; Li, Y.; Guo, T.; Zheng, Z.; et al. Neuroendocrine Transformation from EGFR/ALK-Wild Type or TKI-Naïve Non-Small Cell Lung Cancer: An under-Recognized Phenomenon. Lung Cancer 2022, 169, 22–30. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Sun, H.; Su, J.-W.; Chen, Y.-Q.; Zhang, S.-L.; Zheng, M.-Y.; Li, Y.-F.; Huang, J.; Zhang, C.; Tai, Z.-X.; et al. A Potential Treatment Option for Transformed Small-Cell Lung Cancer on PD-L1 Inhibitor-Based Combination Therapy Improved Survival. Lung Cancer 2023, 175, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-K.; Lee, J.; Kim, S.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.-W.; Heo, D.S.; et al. Clonal History and Genetic Predictors of Transformation into Small-Cell Carcinomas from Lung Adenocarcinomas. J. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Koba, H.; Kimura, H.; Yoneda, T.; Ogawa, N.; Tanimura, K.; Tambo, Y.; Sone, T.; Hosomichi, K.; Tajima, A.; Kasahara, K. NOTCH Alteration in EGFR-Mutated Lung Adenocarcinoma Leads to Histological Small-Cell Carcinoma Transformation under EGFR-TKI Treatment. Transl. Lung Cancer Res. 2021, 10, 4161–4173. [Google Scholar] [CrossRef]
- Ferone, G.; Lee, M.C.; Sage, J.; Berns, A. Cells of Origin of Lung Cancers: Lessons from Mouse Studies. Genes Dev. 2020, 34, 1017–1032. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.-Y.; Berns, A. Cell of Origin of Small Cell Lung Cancer: Inactivation of Trp53 and Rb1 in Distinct Cell Types of Adult Mouse Lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Nikolic, A.; Farnsworth, D.; Shi, R.; Johnson, F.D.; Liu, A.; Ladanyi, M.; Somwar, R.; Gallo, M.; Lockwood, W.W. Extracellular Signal-Regulated Kinase Mediates Chromatin Rewiring and Lineage Transformation in Lung Cancer. eLife 2021, 10, e66524. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ma, S.; Zhang, J.; Han, Y.; Wan, L.; Zhou, W.; Dong, X.; Yang, W.; Chen, Y.; Gao, L.; et al. EHMT2-Mediated Transcriptional Reprogramming Drives Neuroendocrine Transformation in Non-Small Cell Lung Cancer. Proc. Natl. Acad. Sci. USA 2024, 121, e2317790121. [Google Scholar] [CrossRef]
- Pearson, J.D.; Huang, K.; Pacal, M.; McCurdy, S.R.; Lu, S.; Aubry, A.; Yu, T.; Wadosky, K.M.; Zhang, L.; Wang, T.; et al. Binary Pan-Cancer Classes with Distinct Vulnerabilities Defined by pro- or Anti-Cancer YAP/TEAD Activity. Cancer Cell 2021, 39, 1115–1134.e12. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming Normal Human Epithelial Tissues to a Common, Lethal Neuroendocrine Cancer Lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Tran, W.; Song, K.; Sugimoto, T.; Obusan, M.B.; Wang, L.; Sheu, K.M.; Cheng, D.; Ta, L.; Varuzhanyan, G.; et al. Temporal Evolution Reveals Bifurcated Lineages in Aggressive Neuroendocrine Small Cell Prostate Cancer Trans-Differentiation. Cancer Cell 2023, 41, 2066–2082.e9. [Google Scholar] [CrossRef]
- Wang, L.; Smith, B.A.; Balanis, N.G.; Tsai, B.L.; Nguyen, K.; Cheng, M.W.; Obusan, M.B.; Esedebe, F.N.; Patel, S.J.; Zhang, H.; et al. A Genetically Defined Disease Model Reveals That Urothelial Cells Can Initiate Divergent Bladder Cancer Phenotypes. Proc. Natl. Acad. Sci. USA 2020, 117, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Earlie, E.M.; Li, K.; Thomas, J.; Hubisz, M.J.; Stein, B.D.; Zhang, C.; Cantley, L.C.; Laughney, A.M.; Varmus, H. Lineage-Specific Intolerance to Oncogenic Drivers Restricts Histological Transformation. Science 2024, 383, eadj1415. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of Small Cell Lung Cancer by Somatic Inactivation of Both Trp53 and Rb1 in a Conditional Mouse Model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, A.; Taniguchi, H.; Zhan, Y.A.; Hasan, M.M.; Chavan, S.S.; Meng, F.; Uddin, F.; Manoj, P.; Donoghue, M.T.A.; Won, H.H.; et al. Multi-Omic Analysis of Lung Tumors Defines Pathways Activated in Neuroendocrine Transformation. Cancer Discov. 2021, 11, 3028–3047. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, A.; Durani, V.; Sabet, A.; Redin, E.; Kawasaki, K.; Shafer, M.; Karthaus, W.R.; Zaidi, S.; Zhan, Y.A.; Manoj, P.; et al. Exportin 1 Inhibition Prevents Neuroendocrine Transformation through SOX2 Down-Regulation in Lung and Prostate Cancers. Sci. Transl. Med. 2023, 15, eadf7006. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, A.; Kawasaki, K.; Redin, E.; Uddin, F.; Rakhade, S.; Durani, V.; Sabet, A.; Shafer, M.; Karthaus, W.R.; Zaidi, S.; et al. CDC7 Inhibition Impairs Neuroendocrine Transformation in Lung and Prostate Tumors through MYC Degradation. Signal Transduct. Target. Ther. 2024, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Jing, M.; He, X.; Cai, C.Z.; Ma, Q.Z.; Li, K.; Zhang, B.X.; Yin, Y.; Shi, M.S.; Wang, Y.S. Epidermal Growth Factor Receptor Regulates Lineage Plasticity Driving Transformation to Small Cell Lung Cancer. Biochem. Biophys. Res. Commun. 2023, 681, 218–224. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive Genomic Profiles of Small Cell Lung Cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative Genome Analyses Identify Key Somatic Driver Mutations of Small-Cell Lung Cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Pietanza, M.C.; Johnson, M.L.; Riely, G.J.; Miller, V.A.; Sima, C.S.; Zakowski, M.F.; Rusch, V.W.; Ladanyi, M.; Kris, M.G. Incidence of EGFR Exon 19 Deletions and L858R in Tumor Specimens From Men and Cigarette Smokers With Lung Adenocarcinomas. J. Clin. Oncol. 2011, 29, 2066–2070. [Google Scholar] [CrossRef]
- Batra, U.; Biswas, B.; Prabhash, K.; Krishna, M.V. Differential Clinicopathological Features, Treatments and Outcomes in Patients with Exon 19 Deletion and Exon 21 L858R EGFR Mutation-Positive Adenocarcinoma Non-Small-Cell Lung Cancer. BMJ Open Respir. Res. 2023, 10, e001492. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, M.; Yu, X. Differences among Lesions with Exon 19, Exon 21 EGFR Mutations and Wild Types in Surgically Resected Non-Small Cell Lung Cancer. Sci. Rep. 2016, 6, 31636. [Google Scholar] [CrossRef] [PubMed]
- Mambetsariev, I.; Arvanitis, L.; Fricke, J.; Pharaon, R.; Baroz, A.R.; Afkhami, M.; Koczywas, M.; Massarelli, E.; Salgia, R. Small Cell Lung Cancer Transformation Following Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. J. Clin. Med. 2022, 11, 1429. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive Genomic Analysis Identifies SOX2 as a Frequently Amplified Gene in Small-Cell Lung Cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Maas, L.; Abedpour, N.; Cartolano, M.; Kaiser, L.; Fischer, R.N.; Scheel, A.H.; Weber, J.-P.; Hellmich, M.; Bosco, G.; et al. Evolutionary Trajectories of Small Cell Lung Cancer under Therapy. Nature 2024, 627, 880–889. [Google Scholar] [CrossRef]
- Talluri, S.; Dick, F.A. Regulation of Transcription and Chromatin Structure by pRB. Cell Cycle 2012, 11, 3189–3198. [Google Scholar] [CrossRef]
- Serra, F.; Nieto-Aliseda, A.; Fanlo-Escudero, L.; Rovirosa, L.; Cabrera-Pasadas, M.; Lazarenkov, A.; Urmeneta, B.; Alcalde-Merino, A.; Nola, E.M.; Okorokov, A.L.; et al. P53 Rapidly Restructures 3D Chromatin Organization to Trigger a Transcriptional Response. Nat. Commun. 2024, 15, 2821. [Google Scholar] [CrossRef]
- Kim, Y.H.; Girard, L.; Giacomini, C.P.; Wang, P.; Hernandez-Boussard, T.; Tibshirani, R.; Minna, J.D.; Pollack, J.R. Combined Microarray Analysis of Small Cell Lung Cancer Reveals Altered Apoptotic Balance and Distinct Expression Signatures of MYC Family Gene Amplification. Oncogene 2006, 25, 130–138. [Google Scholar] [CrossRef]
- Shue, Y.T.; Drainas, A.P.; Li, N.Y.; Pearsall, S.M.; Morgan, D.; Sinnott-Armstrong, N.; Hipkins, S.Q.; Coles, G.L.; Lim, J.S.; Oro, A.E.; et al. A Conserved YAP/Notch/REST Network Controls the Neuroendocrine Cell Fate in the Lungs. Nat. Commun. 2022, 13, 2690. [Google Scholar] [CrossRef]
- Sriuranpong, V.; Borges, M.W.; Strock, C.L.; Nakakura, E.K.; Watkins, D.N.; Blaumueller, C.M.; Nelkin, B.D.; Ball, D.W. Notch Signaling Induces Rapid Degradation of Achaete-Scute Homolog 1. Mol. Cell. Biol. 2002, 22, 3129–3139. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural Heterogeneity Generated by Notch Signalling Promotes Small-Cell Lung Cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef]
- Ku, S.-Y.; Wang, Y.; Garcia, M.M.; Yamada, Y.; Mizuno, K.; Long, M.D.; Rosario, S.; Chinnam, M.; Assaad, M.A.; Puca, L.; et al. Notch Signaling Suppresses Neuroendocrine Differentiation and Alters the Immune Microenvironment in Advanced Prostate Cancer. J. Clin. Investig. 2024, 134, e175217. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, J.S.; Singleton, C.S.; Miele, L. Notch Signaling in Neuroendocrine Tumors. Front. Oncol. 2016, 6, 94. [Google Scholar] [CrossRef]
- D’Haene, N.; Le Mercier, M.; Salmon, I.; Mekinda, Z.; Remmelink, M.; Berghmans, T. SMAD4 Mutation in Small Cell Transformation of Epidermal Growth Factor Receptor Mutated Lung Adenocarcinoma. Oncologist 2019, 24, 9–13. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular Subtypes of Small Cell Lung Cancer: A Synthesis of Human and Mouse Model Data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Hong, T.H.; Park, S.; Jung, H.-A.; Sun, J.-M.; Ahn, J.S.; Ahn, M.-J.; Park, K.; Choi, Y.-L.; Lee, S.-H. Molecular Subtypes of Small Cell Lung Cancer Transformed from Adenocarcinoma after EGFR Tyrosine Kinase Inhibitor Treatment. Transl. Lung Cancer Res. 2021, 10, 4209–4220. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The Nuclear Export Protein XPO1—From Biology to Targeted Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Quintanal-Villalonga, A.; Taniguchi, H.; Hao, Y.; Chow, A.; Zhan, Y.A.; Chavan, S.S.; Uddin, F.; Allaj, V.; Manoj, P.; Shah, N.S.; et al. Inhibition of XPO1 Sensitizes Small Cell Lung Cancer to First- and Second-Line Chemotherapy. Cancer Res. 2022, 82, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, T.; Meng, Z.; Wang, L.; Li, M.; Chen, J.; Qin, T.; Yu, J.; Zhang, M.; Bie, Z.; et al. XPO1 Inhibition Synergizes with PARP1 Inhibition in Small Cell Lung Cancer by Targeting Nuclear Transport of FOXO3a. Cancer Lett. 2021, 503, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 Promotes Lineage Plasticity and Antiandrogen Resistance in TP53- and RB1-Deficient Prostate Cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Voigt, E.; Wallenburg, M.; Wollenzien, H.; Thompson, E.; Kumar, K.; Feiner, J.; McNally, M.; Friesen, H.; Mukherjee, M.; Afeworki, Y.; et al. Sox2 Is an Oncogenic Driver of Small Cell Lung Cancer and Promotes the Classic Neuroendocrine Subtype. Mol. Cancer Res. 2021, 19, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Ishioka, K.; Yasuda, H.; Hamamoto, J.; Terai, H.; Emoto, K.; Kim, T.-J.; Hirose, S.; Kamatani, T.; Mimaki, S.; Arai, D.; et al. Upregulation of FGF9 in Lung Adenocarcinoma Transdifferentiation to Small Cell Lung Cancer. Cancer Res. 2021, 81, 3916–3929. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as Master Regulators in Cancer: Modulation, Function and Therapeutic Approaches. Nat. Cancer 2023, 4, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Franklin, J.M.; Wu, Z.; Guan, K.-L. Insights into Recent Findings and Clinical Application of YAP and TAZ in Cancer. Nat. Rev. Cancer 2023, 23, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Baine, M.K.; Hsieh, M.-S.; Lai, W.V.; Egger, J.V.; Jungbluth, A.A.; Daneshbod, Y.; Beras, A.; Spencer, R.; Lopardo, J.; Bodd, F.; et al. SCLC Subtypes Defined by ASCL1, NEUROD1, POU2F3, and YAP1: A Comprehensive Immunohistochemical and Histopathologic Characterization. J. Thorac. Oncol. 2020, 15, 1823–1835. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Dwivedi, B.; Chen, Z.; Zhang, C.; Barwick, B.; Ernani, V.; Zhang, G.; Gilbert-Ross, M.; Carlisle, J.; Khuri, F.R.; et al. YAP1 Expression in Small Cell Lung Cancer Defines a Distinct Subtype with T-Cell Inflamed Phenotype. J. Thorac. Oncol. 2021, 16, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Ohshima, M.; Suzuki, H.I.; Nagase, T. YAP and TAZ Modulate Cell Phenotype in a Subset of Small Cell Lung Cancer. Cancer Sci. 2016, 107, 1755–1766. [Google Scholar] [CrossRef]
- Pearsall, S.M.; Humphrey, S.; Revill, M.; Morgan, D.; Frese, K.K.; Galvin, M.; Kerr, A.; Carter, M.; Priest, L.; Blackhall, F.; et al. The Rare YAP1 Subtype of SCLC Revisited in a Biobank of 39 Circulating Tumor Cell Patient Derived Explant Models: A Brief Report. J. Thorac. Oncol. 2020, 15, 1836–1843. [Google Scholar] [CrossRef]
- Wu, Z.; Su, J.; Li, F.; Chen, T.; Mayner, J.; Engler, A.; Ma, S.; Li, Q.; Guan, K.-L. YAP Silencing by RB1 Mutation Is Essential for Small-Cell Lung Cancer Metastasis. Nat. Commun. 2023, 14, 5916. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Durmaz, Y.T.; Li, Y.; Sabet, A.H.; Vajdi, A.; Denize, T.; Walton, E.; Laimon, Y.N.; Doench, J.G.; Mahadevan, N.R.; et al. Regulation of Neuroendocrine Plasticity by the RNA-Binding Protein ZFP36L1. Nat. Commun. 2022, 13, 4998. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Guo, J.; Liu, Y.; Zheng, Q.; Li, X.; Wu, C.; Fang, D.; Chen, X.; Ma, L.; Xu, P.; et al. YAP Drives Fate Conversion and Chemoresistance of Small Cell Lung Cancer. Sci. Adv. 2021, 7, eabg1850. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Nanayakkara, J.; Claypool, D.; Saghafinia, S.; Wong, J.J.M.; Xu, M.; Wang, X.; Nicol, C.J.B.; Michael, I.P.; Hafner, M.; et al. A miR-375/YAP Axis Regulates Neuroendocrine Differentiation and Tumorigenesis in Lung Carcinoid Cells. Sci. Rep. 2021, 11, 10455. [Google Scholar] [CrossRef] [PubMed]
- Frost, T.C.; Gartin, A.K.; Liu, M.; Cheng, J.; Dharaneeswaran, H.; Keskin, D.B.; Wu, C.J.; Giobbie-Hurder, A.; Thakuria, M.; DeCaprio, J.A. YAP1 and WWTR1 Expression Inversely Correlates with Neuroendocrine Markers in Merkel Cell Carcinoma. J. Clin. Investig. 2023, 133, e157171. [Google Scholar] [CrossRef]
- Pearson, J.D.; Huang, K.; Dela Pena, L.G.; Ducarouge, B.; Mehlen, P.; Bremner, R. Netrin-1 and UNC5B Cooperate with Integrins to Mediate YAP-Driven Cytostasis. Cancer Res. Commun. 2024, 4, 2374–2383. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-Wide Association between YAP/TAZ/TEAD and AP-1 at Enhancers Drives Oncogenic Growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, H.; Rajurkar, M.; Li, Q.; Cotton, J.L.; Ou, J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M.; Park, J.-S.; et al. Tead and AP1 Coordinate Transcription and Motility. Cell Rep. 2016, 14, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- El Zarif, T.; Meador, C.B.; Qiu, X.; Seo, J.-H.; Davidsohn, M.P.; Savignano, H.; Lakshminarayanan, G.; McClure, H.M.; Canniff, J.; Fortunato, B.; et al. Detecting Small Cell Transformation in Patients with Advanced EGFR Mutant Lung Adenocarcinoma through Epigenomic cfDNA Profiling. Clin. Cancer Res. 2024, 30, 3798–3811. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Zoubeidi, A.; Selth, L.A. The Epigenetic and Transcriptional Landscape of Neuroendocrine Prostate Cancer. Endocr.-Relat. Cancer 2020, 27, R35–R50. [Google Scholar] [CrossRef]
- Ladu, S.; Calvisi, D.F.; Conner, E.A.; Farina, M.; Factor, V.M.; Thorgeirsson, S.S. E2F1 Inhibits C-Myc-Driven Apoptosis via PIK3CA/Akt/mTOR and COX-2 in a Mouse Model of Human Liver Cancer. Gastroenterology 2008, 135, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Driskill, J.H.; Pan, D. Control of Stem Cell Renewal and Fate by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2023, 24, 895–911. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, X.H.; Cai, Y.; Yang, D.; Shi, J.; Xing, P.; Xu, T.; Wu, L.; Su, W.; Xu, R.; et al. Rationale and Design of a Phase II Trial of Combined Serplulimab and Chemotherapy in Patients with Histologically Transformed Small Cell Lung Cancer: A Prospective, Single-Arm and Multicentre Study. Clin. Oncol. 2024, 36, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Goldberg, S.B.; Le, X.; Piotrowska, Z.; Goldman, J.W.; De Langen, A.J.; Okamoto, I.; Cho, B.C.; Smith, P.; Mensi, I.; et al. Biomarker-Directed Phase II Platform Study in Patients with EGFR Sensitizing Mutation-Positive Advanced/Metastatic Non-Small Cell Lung Cancer Whose Disease Has Progressed on First-Line Osimertinib Therapy (ORCHARD). Clin. Lung Cancer 2021, 22, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Dziadziuszko, R.; Sugawara, S.; Kao, S.; Hochmair, M.; Huemer, F.; de Castro, G.; Havel, L.; Caro, R.B.; Losonczy, G.; et al. Five-Year Survival in Patients with Extensive-Stage Small Cell Lung Cancer Treated with Atezolizumab in the Phase III IMpower133 Study and the Phase III IMbrella a Extension Study. Lung Cancer 2024, 196, 107924. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.V.; Reck, M.; Mansfield, A.S.; Mok, T.; Scherpereel, A.; Reinmuth, N.; Garassino, M.C.; De Castro Carpeno, J.; Califano, R.; Nishio, M.; et al. Updated Overall Survival and PD-L1 Subgroup Analysis of Patients With Extensive-Stage Small-Cell Lung Cancer Treated With Atezolizumab, Carboplatin, and Etoposide (IMpower133). J. Clin. Oncol. 2021, 39, 619–630. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus Platinum-Etoposide versus Platinum-Etoposide in First-Line Treatment of Extensive-Stage Small-Cell Lung Cancer (CASPIAN): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of Transcription Factor Programs and Immune Pathway Activation Define Four Major Subtypes of SCLC with Distinct Therapeutic Vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.F.; Rakhade, S.; Quintanal-Villalonga, A.; Lee, J.; Forsythe, B.; Moses, K.A.; Ahn, L.S.H.; Pupo, A.; Falcon, C.J.; Rekhtman, N.; et al. Osimertinib, Platinum, Etoposide as Initial Treatment for Patients with EGFR Mutant Lung Cancers with TP53 and RB1 Alterations. J. Clin. Oncol. 2024, 42, 8565. [Google Scholar] [CrossRef]
- Steeghs, N.; Pruis, M.; van Herpen, C.; Lu, V.; Redman, J.; Zhou, X. A Phase 1 Open-Label Study to Assess the Relative Bioavailability of TAK-931 Tablets in Reference to Powder-in-Capsule in Patients with Advanced Solid Tumors. Investig. New Drugs 2023, 41, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, Y.; Shimizu, T.; Yonemori, K.; Kojima, T.; Kondo, S.; Koganemaru, S.; Iwasa, S.; Harano, K.; Koyama, T.; Lu, V.; et al. Safety, Tolerability, and Pharmacokinetics of TAK-931, a Cell Division Cycle 7 Inhibitor, in Patients with Advanced Solid Tumors: A Phase I First-in-Human Study. Cancer Res. Commun. 2022, 2, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Jan, S.; Dar, M.I.; Wani, R.; Sandey, J.; Mushtaq, I.; Lateef, S.; Syed, S.H. Targeting EHMT2/G9a for Cancer Therapy: Progress and Perspective. Eur. J. Pharmacol. 2021, 893, 173827. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Approach | Drugs | Stage of Development | Treatment Phase | Reference |
|---|---|---|---|---|
| Standard chemotherapy | Platinum/ etoposide | Approved | T-SCLC | |
| Chemotherapy + immunotherapy | Serplulimab + platinum/ etoposide | Ongoing Phase 2 trials | T-SCLC | NCT05957510, NCT03944772 |
| Immunotherapy + PARP inhibition | Olaparib + Durvalumab | Ongoing Phase 2 trial | T-SCLC | NCT04538378 |
| EGFR inhibitor + chemotherapy | Osimertinib + platinum/ etoposide | Ongoing Phase 1 trial | NSCLC with EGFR/RB/TP53 alterations | NCT03567642 |
| XPO1 inhibition | Selinexor | Preclinical evidence | Not applicable | [56] |
| CDC7 inhibition | Simurosertib/ TAK-931 | Preclinical evidence | Not applicable | [57] |
| EHMT2 inhibition | UNC0638, UNC0642 | Preclinical evidence | Not applicable | [48] |
| WNT/β-catenin inhibition | MSAB | Preclinical evidence | Not applicable | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, A.; Bhaskar, N.; Pearson, J.D. Neuroendocrine Transformation as a Mechanism of Resistance to Targeted Lung Cancer Therapies: Emerging Mechanisms and Their Therapeutic Implications. Cancers 2025, 17, 260. https://doi.org/10.3390/cancers17020260
Joshi A, Bhaskar N, Pearson JD. Neuroendocrine Transformation as a Mechanism of Resistance to Targeted Lung Cancer Therapies: Emerging Mechanisms and Their Therapeutic Implications. Cancers. 2025; 17(2):260. https://doi.org/10.3390/cancers17020260
Chicago/Turabian StyleJoshi, Asim, Nivitha Bhaskar, and Joel D. Pearson. 2025. "Neuroendocrine Transformation as a Mechanism of Resistance to Targeted Lung Cancer Therapies: Emerging Mechanisms and Their Therapeutic Implications" Cancers 17, no. 2: 260. https://doi.org/10.3390/cancers17020260
APA StyleJoshi, A., Bhaskar, N., & Pearson, J. D. (2025). Neuroendocrine Transformation as a Mechanism of Resistance to Targeted Lung Cancer Therapies: Emerging Mechanisms and Their Therapeutic Implications. Cancers, 17(2), 260. https://doi.org/10.3390/cancers17020260