Integrating Polygenic Scores into Multifactorial Breast Cancer Risk Assessment: Insights from the First Year of Clinical Implementation in Western Austria

 , , , and
, , , and
Simple Summary
Abstract
1. Introduction
1.1. MFRA to Support Genetic Counseling
1.2. The Need for the MFRA Is Most Obvious in Patients with PVs in Breast Cancer Risk Susceptibility Genes of Moderate Penetrance
1.3. Implementation of Polygenic Scores and MFRA in Breast Cancer Prevention and the Clinical Management of Breast Cancer
1.4. Preemptive Considerations for Maximal Patient Safety and Acceptance
2. Methods
2.1. Polygenic Score Integration into Multifactorial Assessment
- Genotyping across a representative set of genome-wide variants using microarray;
- Performing rigorous quality control of the genotype data;
- Imputation of missing genotypes using the 1000 Genomes Reference Panel;
- Estimation of the ancestry of the patient with a principal component analysis (PCA);
- Computation of the raw PGS;
- Standardization of the score with a local reference population (z-score).
2.2. Genotyping, Quality Control, and Imputation
2.3. Ancestry Estimation
2.4. Polygenic Score Calculation
3. Results
3.1. Patients
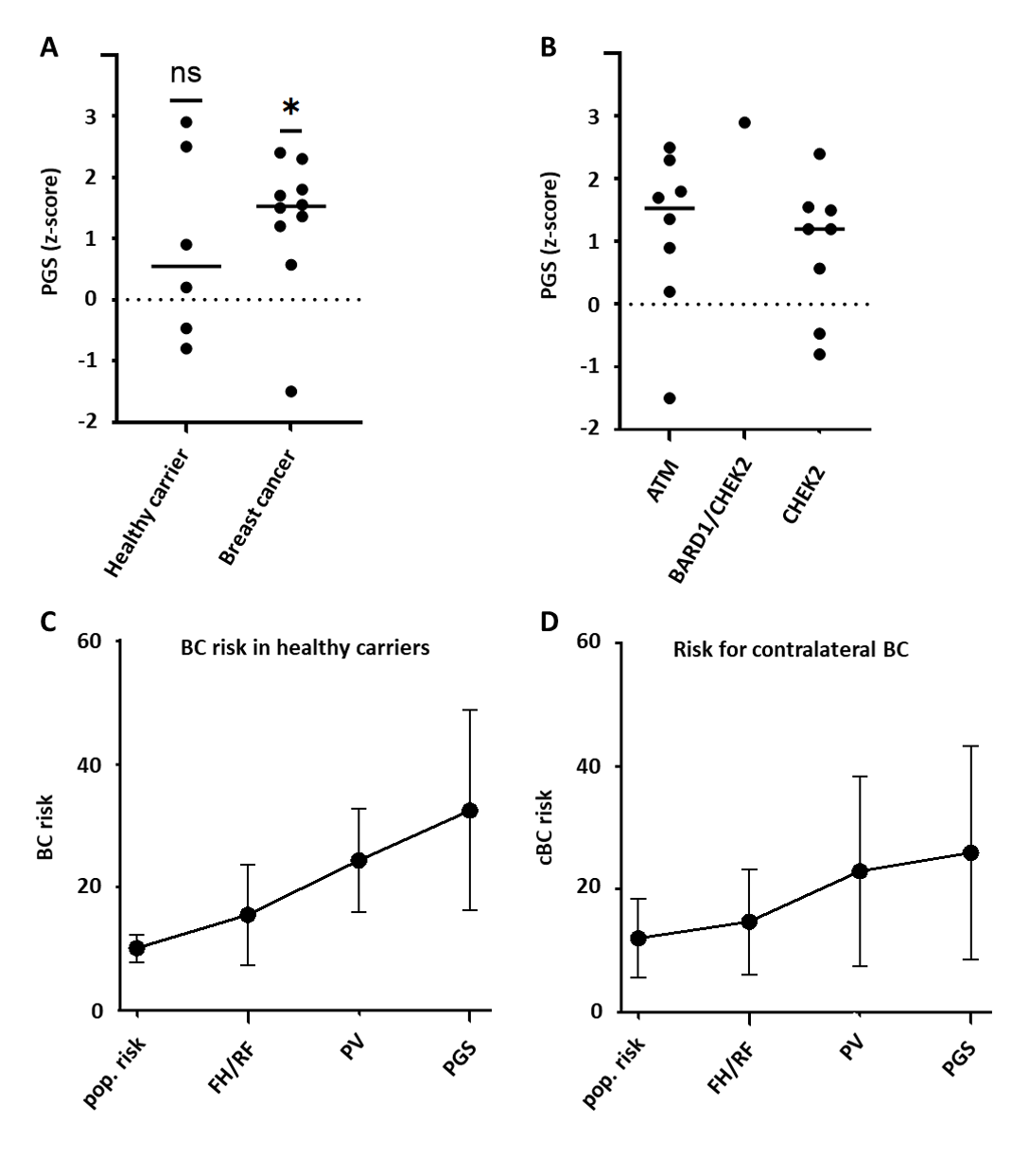
3.2. Cumulative Effect of MFRA and PGS Testing
3.3. Impact of Individualized Risk Prediction
- -
- For three breast cancer patients (aged 46–62 years), the MFRA had little or no impact. These patients already had a low population risk of 5.9–8.5% for contralateral breast cancer due to their comparably higher age. Following the MFRA, their individual risk increased from 8.2% to 12.2%.
- -
- Three breast cancer patients (aged 49–59 years) experienced a moderate increase in risk to 24.9–29.5% following the MFRA, compared to a population risk of 8.4% to 14.2%. One of these patients had an exceptionally low PGS that reduced the individual risk by more than 10%.
- -
- Two relatively young breast cancer patients (aged 37 and 41 years) experienced a marked increase in individual risk to 38.4% and 59.9% following the MFRA, compared to population risks of 19.7% and 23.3%, respectively.
- -
- Five of the patients with breast cancer decided to undergo bilateral mastectomies. However, the relative influence of the risk predicted by the MFRA, including PGS, compared to other clinical and non-clinical factors could not be quantified.
- -
- For two healthy heterozygotes in our cohort of patients (aged 41 and 61 years), the MFRA had a minor or moderate impact, with calculated risks of 17.1% and 20.7%, respectively (age-matched population risks of 7.3% and 11.4%, respectively), for breast cancer.
- -
- Three healthy heterozygotes (aged 27–56 years) experienced a moderate to strong increase in risk to 30.3–58.2% following the MFRA, with a population risk between 8.2 and 12%.
4. Discussion
5. Limitations of This Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turnbull, C.; Sud, A.; Houlston, R.S. Cancer genetics, precision prevention and a call to action. Nat. Genet. 2018, 50, 1212–1218. [Google Scholar] [PubMed]
- Lee, A.; Mavaddat, N.; Wilcox, A.N.; Cunningham, A.P.; Carver, T.; Hartley, S.; de Villiers, C.B.; Izquierdo, A.; Simard, S.; Schmidt, M.K.; et al. BOADICEA: A comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 2019, 21, 1708–1718. [Google Scholar]
- Carver, T.; Hartley, S.; Lee, A.; Cunningham, A.P.; Archer, S.; Babb de Villiers, C.; Roberts, J.; Ruston, R.; Walter, F.M.; Tischkowitz, M.; et al. CanRisk Tool-A Web Interface for the Prediction of Breast and Ovarian Cancer Risk and the Likelihood of Carrying Genetic Pathogenic Variants. Cancer Epidemiol. Biomark. Prev. 2021, 30, 469–473. [Google Scholar]
- Archer, S.; Babb de Villiers, C.; Scheibl, F.; Carver, T.; Hartley, S.; Lee, A.; Cunningham, A.P.; Easton, D.F.; McIntosh, J.G.; Emery, J.; et al. Evaluating clinician acceptability of the prototype CanRisk tool for predicting risk of breast and ovarian cancer: A multi-methods study. PLoS ONE 2020, 15, e0229999. [Google Scholar]
- Lakeman, I.M.M.; Rodriguez-Girondo, M.D.M.; Lee, A.; Celosse, N.; Braspenning, M.E.; van Engelen, K.; van de Beek, I.; van der Hout, A.H.; Gómez García, E.B.; Mensenkamp, A.R.; et al. Clinical applicability of the Polygenic Risk Score for breast cancer risk prediction in familial cases. J. Med. Genet. 2023, 60, 327–336. [Google Scholar] [PubMed]
- Mars, N.; Widen, E.; Kerminen, S.; Meretoja, T.; Pirinen, M.; Della Briotta Parolo, P.; Palta, P.; Gen, F.; Palotie, A.; Kaprio, J.; et al. The role of polygenic risk and susceptibility genes in breast cancer over the course of life. Nat. Commun. 2020, 11, 6383. [Google Scholar]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.-H.; Wang, Q.; Bolla, M.K.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [PubMed]
- Tyrer, J.; Duffy, S.W.; Cuzick, J. A breast cancer prediction model incorporating familial and personal risk factors. Stat. Med. 2004, 23, 1111–1130. [Google Scholar]
- Pal Choudhury, P.; Brook, M.N.; Hurson, A.N.; Lee, A.; Mulder, C.V.; Coulson, P.; Schoemaker, M.J.; Jones, M.E.; Swerdlow, A.J.; Chatterjee, N.; et al. Comparative validation of the BOADICEA and Tyrer-Cuzick breast cancer risk models incorporating classical risk factors and polygenic risk in a population-based prospective cohort of women of European ancestry. Breast Cancer Res. 2021, 23, 22. [Google Scholar]
- Das, S.; Forer, L.; Schonherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; Carthy, S.M.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Archer, S.; Donoso, F.S.; Carver, T.; Yue, A.; Cunningham, A.P.; Ficorella, L.; Tischkowitz, M.; Easton, D.F.; Antoniou, A.C.; Emery, J.; et al. Exploring the barriers to and facilitators of implementing CanRisk in primary care: A qualitative thematic framework analysis. Br. J. Gen. Pract. 2023, 73, e586–e596. [Google Scholar] [PubMed]
- Wand, H.; Kalia, S.S.; Helm, B.M.; Suckiel, S.A.; Brockman, D.; Vriesen, N.; Goudar, R.K.; Austin, J.; Yanes, T. Clinical genetic counseling and translation considerations for polygenic scores in personalized risk assessments: A Practice Resource from the National Society of Genetic Counselors. J. Genet. Couns. 2023, 32, 558–575. [Google Scholar] [PubMed]
- Tsoulaki, O.; Tischkowitz, M.; Antoniou, A.C.; Musgrave, H.; Rea, G.; Gandhi, A.; Cox, K.; Irvine, T.; Holcombe, S.; Eccles, D.; et al. Joint ABS-UKCGG-CanGene-CanVar consensus regarding the use of CanRisk in clinical practice. Br. J. Cancer 2024, 130, 2027–2036. [Google Scholar]
- Petrucelli, N.; Daly, M.B.; Pal, T. Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In GeneReviews((R)); University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Graffeo, R.; Rana, H.Q.; Conforti, F.; Bonanni, B.; Cardoso, M.J.; Paluch-Shimon, S.; Pagani, O.; Goldhirsch, A.; Partridge, A.H.; Lambertini, M.; et al. Moderate penetrance genes complicate genetic testing for breast cancer diagnosis: ATM, CHEK2, BARD1 and RAD51D. Breast 2022, 65, 32–40. [Google Scholar]
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar]
- Burton, H.; Chowdhury, S.; Dent, T.; Hall, A.; Pashayan, N.; Pharoah, P. Public health implications from COGS and potential for risk stratification and screening. Nat. Genet. 2013, 45, 349–351. [Google Scholar]
- Lambert, S.A.; Gil, L.; Jupp, S.; Ritchie, S.C.; Xu, Y.; Buniello, A.; McMahon, A.; Abraham, G.; Chapman, M.; Parkinson, M.; et al. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 2021, 53, 420–425. [Google Scholar]
- Witsch-Baumgartner, M.; Schwaninger, G.; Schnaiter, S.; Kollmann, F.; Burkhard, S.; Grobner, R.; Mühlegger, B.; Schamschula1, E.; Kirchmeier, P.; Zschocke, J. Array genotyping as diagnostic approach in medical genetics. Mol. Genet. Genom. Med. 2022, 10, e2016. [Google Scholar]
- Taliun, D.; Chothani, S.P.; Schonherr, S.; Forer, L.; Boehnke, M.; Abecasis, G.R.; Wang, C. LASER server: Ancestry tracing with genotypes or sequence reads. Bioinformatics 2017, 33, 2056–2058. [Google Scholar] [PubMed]
- Li, J.Z.; Absher, D.M.; Tang, H.; Southwick, A.M.; Casto, A.M.; Ramachandran, S.; Cann, H.M.; Barsh, G.S.; Feldman, M.; Cavalli-Sforza, L.L.; et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008, 319, 1100–1104. [Google Scholar]
- Morales, J.; Welter, D.; Bowler, E.H.; Cerezo, M.; Harris, L.W.; McMahon, A.C.; Hall, P.; Junkins, H.A.; Milano, A.; Hastings, E.; et al. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 2018, 19, 21. [Google Scholar]
- Forer, L.; Taliun, D.; LeFaive, J.; Smith, A.V.; Boughton, A.P.; Coassin, S.; Lamina, C.; Kronenberg, F.; Fuchsberger, C.; Schönherr, S. Imputation Server PGS: An automated approach to calculate polygenic risk scores on imputation servers. Nucleic Acids Res. 2024, 52, W70–W77. [Google Scholar]
- Ding, Y.; Hou, K.; Xu, Z.; Pimplaskar, A.; Petter, E.; Boulier, K.; Privé, F.; Vilhjálmsson, B.J.; Olde Loohuis, L.M.; Pasaniuc, B. Polygenic scoring accuracy varies across the genetic ancestry continuum. Nature 2023, 618, 774–781. [Google Scholar]
- Mavaddat, N.; Ficorella, L.; Carver, T.; Lee, A.; Cunningham, A.P.; Lush, M.; Dennis, J.; Tischkowitz, M.; Downes, K.; Hu, D.; et al. Incorporating Alternative Polygenic Risk Scores into the BOADICEA Breast Cancer Risk Prediction Model. Cancer Epidemiol. Biomark. Prev. 2023, 32, 422–427. [Google Scholar]
- Marmolejo, D.H.; Wong, M.Y.Z.; Bajalica-Lagercrantz, S.; Tischkowitz, M.; Balmana, J.; extended ERN-GENTURIS Thematic Group 3. Overview of hereditary breast and ovarian cancer (HBOC) guidelines across Europe. Eur. J. Med. Genet. 2021, 64, 104350. [Google Scholar]
- Liu, C.; Zeinomar, N.; Chung, W.K.; Kiryluk, K.; Gharavi, A.G.; Hripcsak, G.; Crew, K.D.; Shang, N.; Khan, A.; Fasel, D.; et al. Generalizability of Polygenic Risk Scores for Breast Cancer Among Women With European, African, and Latinx Ancestry. JAMA Netw. Open 2021, 4, e2119084. [Google Scholar] [PubMed]
- Stolarova, L.; Kleiblova, P.; Zemankova, P.; Stastna, B.; Janatova, M.; Soukupova, J.; Achatz, M.I.; Ambrosone, C.; Apostolou, P.; Arun, B.K.; et al. ENIGMA CHEK2gether Project: A Comprehensive Study Identifies Functionally Impaired CHEK2 Germline Missense Variants Associated with Increased Breast Cancer Risk. Clin. Cancer Res. 2023, 29, 3037–3050. [Google Scholar]
- Yiangou, K.; Mavaddat, N.; Dennis, J.; Zanti, M.; Wang, Q.; Bolla, M.K.; Abubakar, M.; Ahearn, T.U.; Andrulis, I.L.; Anton-Culver, H.; et al. Polygenic score distribution differences across European ancestry populations: Implications for breast cancer risk prediction. Breast Cancer Res. 2024, 26, 189. [Google Scholar]
- Ficorella, L.; Yang, X.; Mavaddat, N.; Carver, T.; Hassan, H.; Dennis, J.; Tyrer, J.; Ho, W.-K.; Teo, S.-H.; Hartman, M.; et al. Adapting the BOADICEA breast and ovarian cancer risk models for the ethnically diverse UK population. medRxiv 2025. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence. Familial Breast Cancer: Classification, Care and Managing Breast Cancer and Related Risks in People with a Family History of Breast Cancer; Guidelines; National Institute for Health and Care Excellence: London, UK, 2019. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Patient ID | Breast Cancer (Age of Diagnosis) | Age at Testing | Gene | Variant | ACMG Classification |
|---|---|---|---|---|---|
| 1 | BC (42), cBC (57) | 58 | ATM | c.3802del | C5 |
| 2 | BC (49), cBC (57) | 57 | CHEK2 | c.(908+1_909-1)_(1095+1_1096-1)del | C5 |
| 3 | BC (35) | 37 | ATM | c.(4236+1_4237-1)_(4436+1_4437-1)del | C5 |
| 4 | BC (41) | 41 | ATM | c.7674del | C5 |
| 5 | BC (46) | 62 | CHEK2 | c.792+2T>C | C4 |
| 6 | BC (49) | 49 | CHEK2 | c.470T>C | risk factor |
| 7 | BC (57) | 57 | ATM | c.3137T>C | C4 |
| 8 | BC (59) | 59 | ATM | c.5644C>T | C5 |
| 9 | BC (62) | 63 | CHEK2 | c.470T>C | risk factor |
| 10 | BC (60) | 62 | CHEK2 | c.470T>C | risk factor |
| 11 | p | 41 | CHEK2 | c.1100del | C5 |
| PMS2 | c.1831dup | C5 | |||
| 12 | p | 35 | BARD1 | c.1725_1903+1208del | C5 |
| CHEK2 | c.470T>C | risk factor | |||
| 13 | p | 56 | ATM | c.3802del | C5 |
| 14 | p | 27 | ATM | c.4148C>A | C5 |
| 15 | p | 60 | ATM | c.8584G>T | C5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forer, L.; Schwaninger, G.; Taxer, K.; Schnitzer, F.; Egle, D.; Zschocke, J.; Schnaiter, S. Integrating Polygenic Scores into Multifactorial Breast Cancer Risk Assessment: Insights from the First Year of Clinical Implementation in Western Austria. Cancers 2025, 17, 2472. https://doi.org/10.3390/cancers17152472
Forer L, Schwaninger G, Taxer K, Schnitzer F, Egle D, Zschocke J, Schnaiter S. Integrating Polygenic Scores into Multifactorial Breast Cancer Risk Assessment: Insights from the First Year of Clinical Implementation in Western Austria. Cancers. 2025; 17(15):2472. https://doi.org/10.3390/cancers17152472
Chicago/Turabian StyleForer, Lukas, Gunda Schwaninger, Kathrin Taxer, Florian Schnitzer, Daniel Egle, Johannes Zschocke, and Simon Schnaiter. 2025. "Integrating Polygenic Scores into Multifactorial Breast Cancer Risk Assessment: Insights from the First Year of Clinical Implementation in Western Austria" Cancers 17, no. 15: 2472. https://doi.org/10.3390/cancers17152472
APA StyleForer, L., Schwaninger, G., Taxer, K., Schnitzer, F., Egle, D., Zschocke, J., & Schnaiter, S. (2025). Integrating Polygenic Scores into Multifactorial Breast Cancer Risk Assessment: Insights from the First Year of Clinical Implementation in Western Austria. Cancers, 17(15), 2472. https://doi.org/10.3390/cancers17152472