
Treatment of KRAS-Mutated Pancreatic Cancer: New Hope for the Patients?

 , , , and
, , , and
Simple Summary
Abstract
1. Introduction
2. PDAC Molecular Subtypes
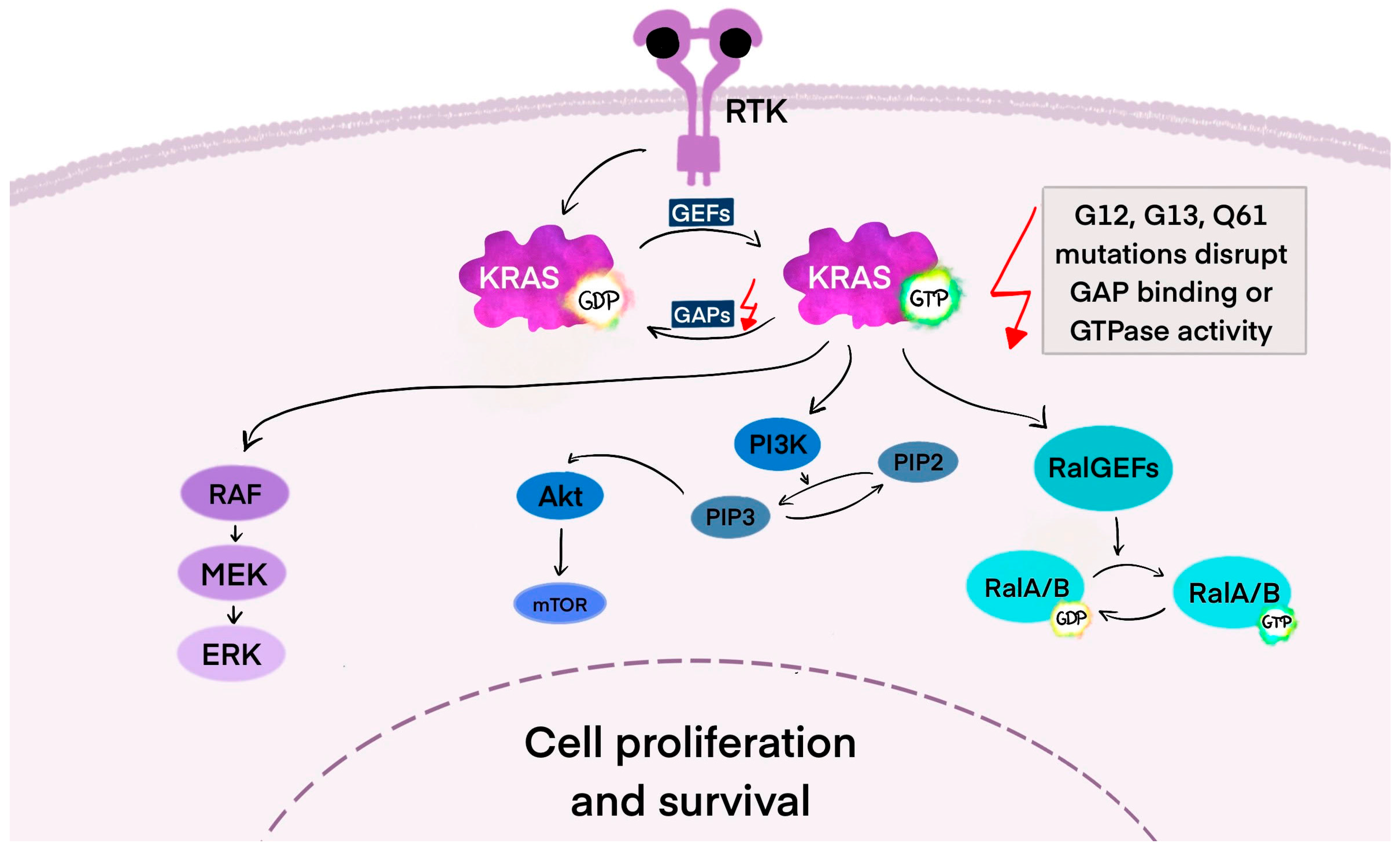
3. KRAS Mutation—Biomolecular Introduction
3.1. KRAS—The Mechanism of Action in the Cancer Cell
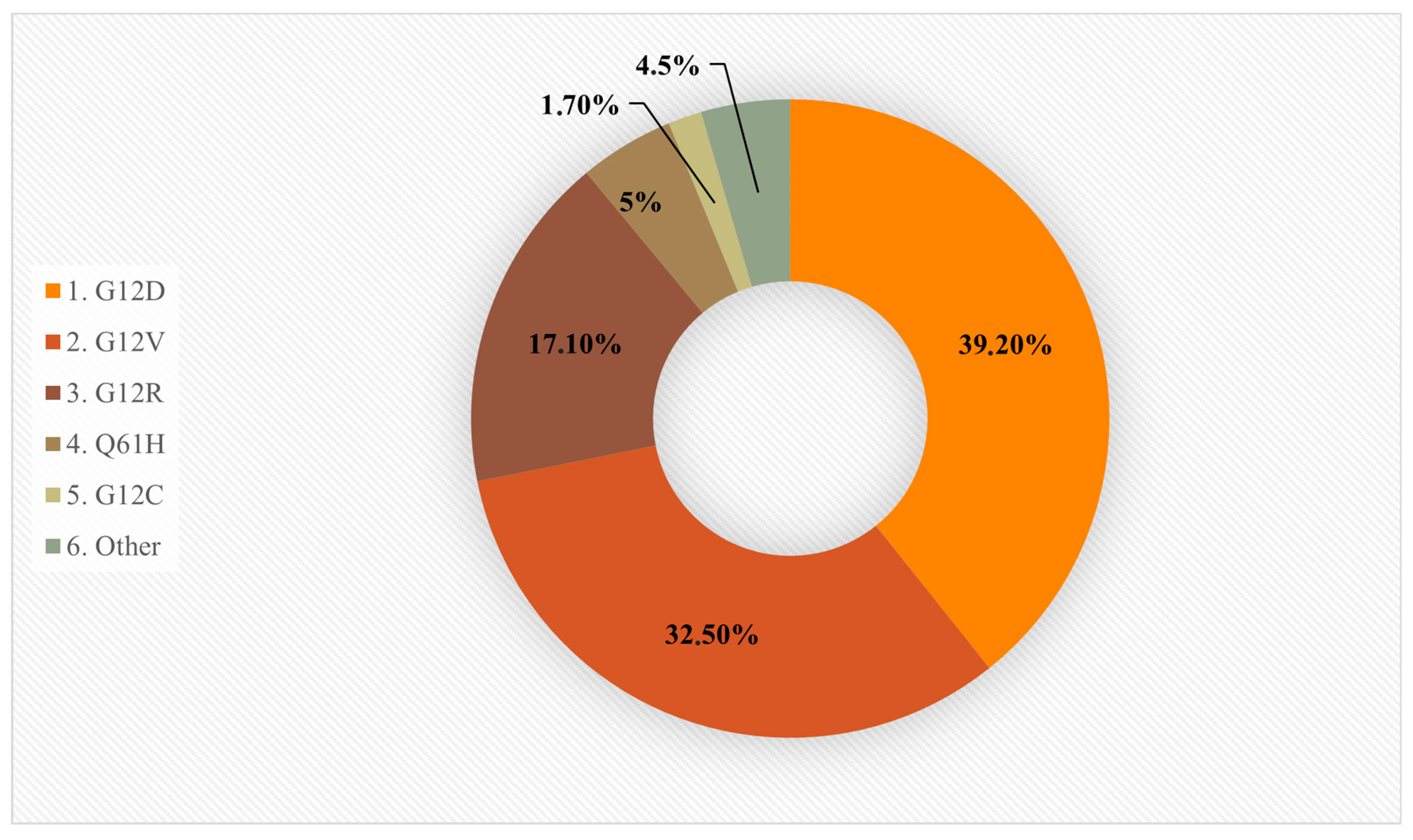
3.2. KRAS-Mutant Cancers and Specific Codon Mutations
3.3. Common Co-Mutations
3.4. KRAS Wild-Type
3.5. Amplification of KRAS
3.6. KRAS-Driven Initiation and Immune Modulation in PDAC
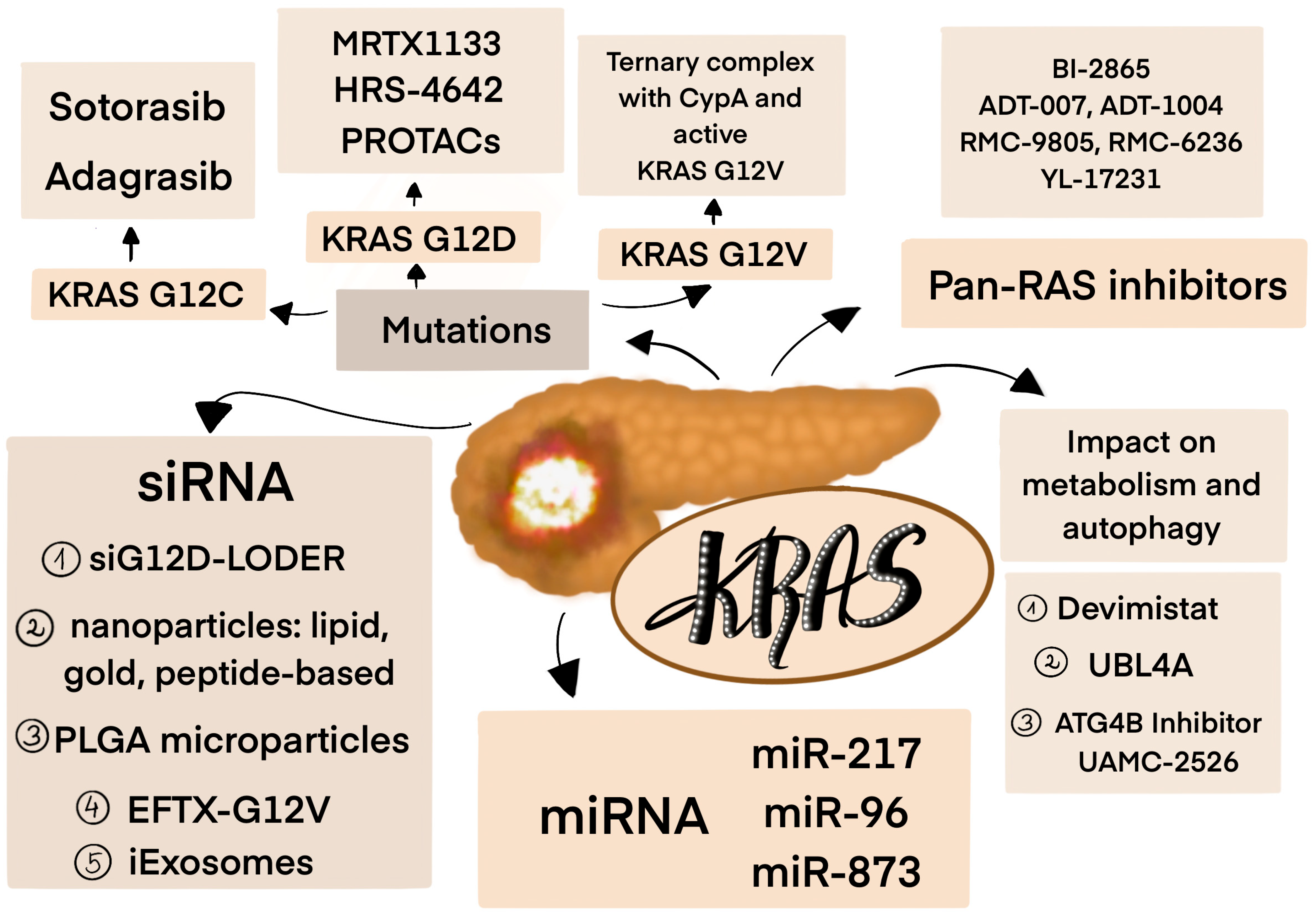
4. Targeting KRAS Mutations
4.1. KRAS Inhibitors
4.2. Pan-RAS Inhibitors
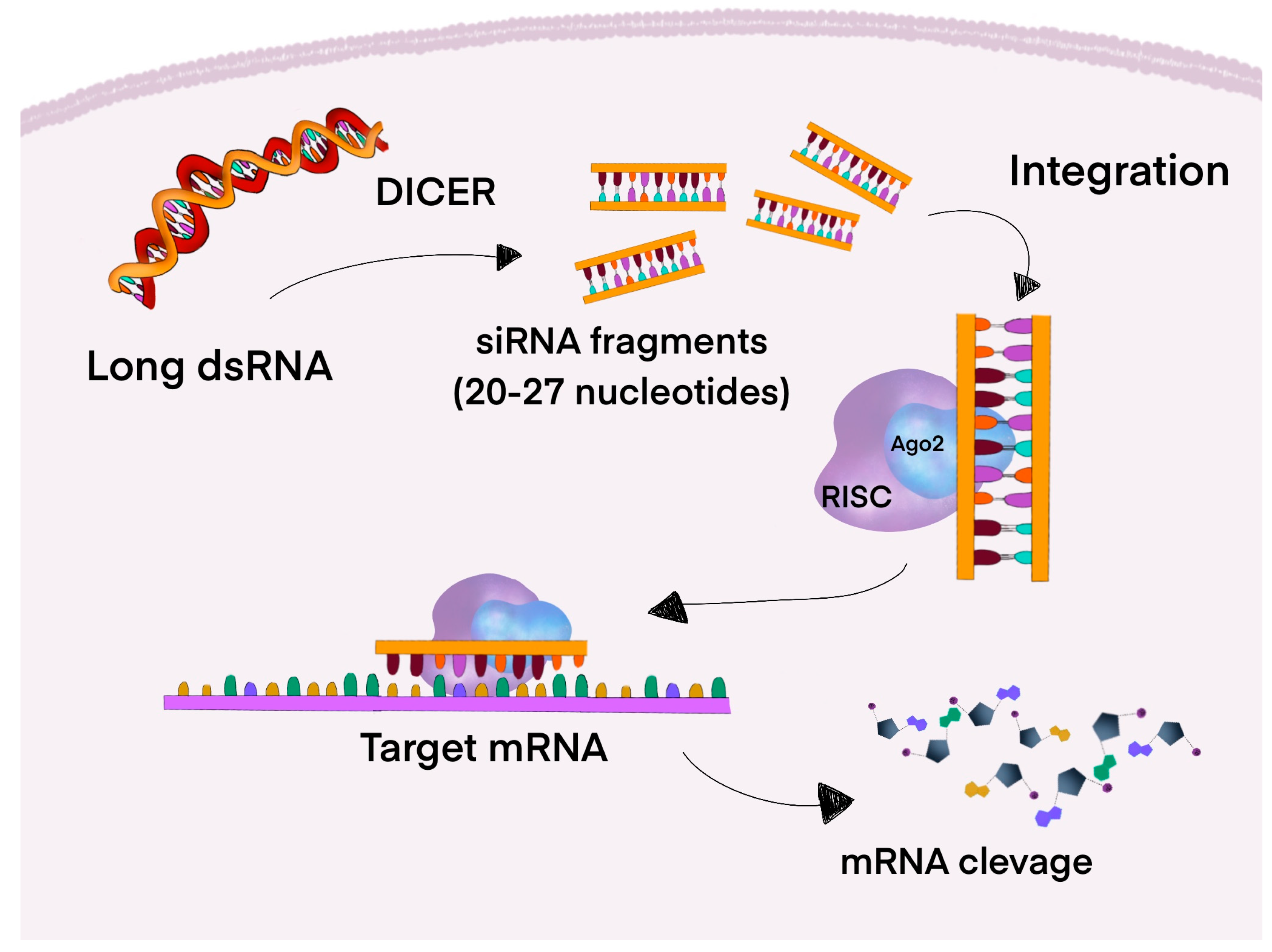
4.3. RNA Interference (RNAi)
4.3.1. Small Interfering RNA
4.3.2. MicroRNA
4.3.3. Challenges of RNAi-Based Therapies
5. Resistance and Targeting Ras Effector Pathways
6. Immunotherapy in Pancreatic Cancer
7. Cancer Cell Metabolism
8. Future Management of KRAS-Mutated PDAC
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADM | Acinar-to-ductal metaplasia |
| AEs | Adverse effects |
| AGO2 | Argonaute 2 |
| AJCC | The American Joint Committee on Cancer |
| ALK | Anaplastic lymphoma kinase |
| ALT | Alanine aminotransferase |
| ARID1A | AT-rich interaction domain 1A |
| AST | Aspartate aminotransferase |
| ATM | Ataxia telangiectasia mutated |
| AuNP | Gold nanoparticles |
| BCAAs | Branched-chain amino acids |
| Bcl-2 | B-cell leukemia/lymphoma 2 protein |
| BRCA2 | BReast CAncer gene |
| CCL4 | Carbon tetrachloride |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CFPAC-1 | Human pancreatic cell line harboring a KRAS G12V mutation |
| COX2 | Cyclooxygenase-2 |
| CRC | Colorectal cancer |
| ctDNA | Circulating Tumor DNA |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein 4 |
| CXCL8 | Chemokine (CXC motif) ligand 8 |
| CXCR2 | C-X-C motif chemokine receptor 2 |
| CypA | Cyclophilin A |
| DCR | Disease control rate |
| DNA | Deoxyribonucleic acid |
| dsRNA | Double-stranded RNA |
| EGF | Epidermal growth factor |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial-to-mesenchymal transition |
| ERBB2 | Erythroblastic leukemia viral oncogene homologue 2 |
| ERK | Extracellular signal-regulated kinase |
| ERK1/2 | Extracellular signal-regulated kinase 1 and 2 |
| FDA | Food and Drug Administration |
| FGF3 | Fibroblast Growth Factor 3 |
| FGFR3 | Fibroblast Growth Factor Receptor 3 |
| GAP | GTPase-activating protein |
| GATA6 | GATA-binding factor 6 |
| G-CSF | Granulocyte colony-stimulating factor |
| GDP/GTP | Guanosine Diphosphate/Guanosine Triphosphate |
| GEF | Guanine nucleotide exchange factor |
| GEFs | Guanine nucleotide exchange factors |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GRB2 | Growth factor receptor-bound protein 2 |
| GTPase | Guanosine triphosphatase |
| HCQ | Hydroxychloroquine |
| HER2 | human epidermal growth factor receptor 2 |
| HRAS | Harvey rat sarcoma viral oncogene |
| ICIs | Immune checkpoint inhibitors |
| IFN | Interferon |
| IL-10 | Interleukin-10 |
| IPMN | Intraductal papillary mucinous neoplasms |
| JNK | Jun N-terminal kinase |
| KDM6A | Lysine demethylase 6A |
| KRAS | Kirsten rat sarcoma viral oncogene |
| LNPs | Lipid nanoparticles |
| MAPK | Mitogen-activated protein kinase |
| MDSCs | Myeloid-derived suppressor cells |
| MHC-I | Major Histocompatibility Complex class I |
| miRNA | MicroRNA |
| MLL2 | Mixed Lineage Leukemia 2 |
| MMP9 | Matrix metalloproteinase-9 |
| mPFS | Median progression-free survival |
| mTOR | Mammalian target of rapamycin |
| ncRNA | Non-coding RNA |
| NET | Neuroendocrine tumor |
| NF1 | Neurofibrin-1 |
| NF-κB | Nuclear Factor kappa B |
| NR5A2 | Nuclear Receptor Subfamily 5 Group A Member 2 |
| NRAS | Neuroblastoma rat sarcoma viral oncogene |
| NRG1 | Neuregulin 1 |
| NSCLC | Non-small cell lung cancer |
| NTRK | Neurotrophic tyrosine receptor kinase |
| ORR | Objective response rate |
| OS | Overall survival |
| Pan-IN | Pancreatic intraepithelial neoplasia |
| PARP | Poly(ADP-ribose) polymerase |
| PC | Pancreatic cancer |
| PCOS | Polycystic ovarian syndrome |
| PD-1 | Programmed Death 1 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PD-L1 | Programmed Death-Ligand 1 |
| PEI | Pancreatic exocrine insufficiency |
| PI3K | Phosphoinositide 3-kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PK | Pharmacokinetics |
| PLGA | Poly9lactic-co-glycolic-acid |
| PPP | Pentose phosphate pathway |
| PROTACs | Proteolysis targeting chimeras |
| PROX1 | Prospero Homeobox 1 |
| RAF | Rapid fibrosarcoma |
| RASA1 | p120-RasGAP protein |
| RASGRF2 | RAS protein-specific guanine nucleotide-releasing factor 2 |
| RISC | RNA-induced silencing complex |
| RNAi | RNA interference |
| RNF43 | Ring Finger Protein 43 |
| SHP2 | Src homology 2 domain-containing protein tyrosine phosphatase 2 |
| siRNA | Small interfering RNA |
| SMAD4 | Mothers against decapentaplegic homolog 4 |
| SOC | Standard of care |
| SOS1/SOS2 | Son of sevenless 1 and 2 |
| STING | Stimulators of interferon genes |
| TAMs | Tumor-associated macrophages |
| TGF-β1 | Transforming Growth Factor beta 1 |
| TIBC | Triple-negative breast cancer |
| TIGIT | T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain |
| TME | Tumor microenvironment |
| TP53 | Tumor protein p53 |
| Tregs | T regulatory cells |
| TTM | Time to metastasis |
| UBL4A | Ubiquitin-like protein 4A |
| VEGF | Vascular endothelial growth factor |
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Qian, L.C.; Cao, Y.Y.; Daniels, M.J.; Song, L.N.; Tian, Y.; Wang, Z.Q. Computed tomography-based radiomics diagnostic approach for differential diagnosis between early- and late-stage pancreatic ductal adenocarcinoma. World J. Gastrointest. Oncol. 2024, 16, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Tumas, J.; Tumiene, B.; Jurkeviciene, J.; Jasiunas, E.; Sileikis, A. Nutritional and immune impairments and their effects on outcomes in early pancreatic cancer patients undergoing pancreatoduodenectomy. Clin. Nutr. 2020, 39, 3385–3394. [Google Scholar] [CrossRef] [PubMed]
- Kamarajah, S.K.; Bundred, J. Comments on: Sarcopenia and sarcopenic obesity are significantly associated with poorer overall survival in patients with pancreatic cancer: Systematic review and meta-analysis. Int. J. Surg. 2019, 66, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Loubnane, G.; Sleiman, C.; Rizk, S. Tocotrienol isoforms: The molecular mechanisms underlying their effects in cancer therapy and their implementation in clinical trials. J. Integr. Med. 2024, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lok, V.; Ngai, C.H.; Zhang, L.; Yuan, J.; Lao, X.Q.; Ng, K.; Chong, C.; Zheng, Z.J.; Wong, M.C.S. Worldwide Burden of, Risk Factors for, and Trends in Pancreatic Cancer. Gastroenterology 2021, 160, 744–754. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Mękal, D.; Sobocki, J.; Badowska-Kozakiewicz, A.; Sygit, K.; Cipora, E.; Bandurska, E.; Czerw, A.; Deptała, A. Evaluation of Nutritional Status and the Impact of Nutritional Treatment in Patients with Pancreatic Cancer. Cancers 2023, 15, 3816. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, Y.; Wang, Y.; Yang, Z.; Yan, X.; Li, S.; Jiang, Y.; Li, Y.; Zhao, S.; Zhao, H.; et al. No Association of Polycystic Ovary Syndrome with Pancreatic Cancer: A Mendelian Randomization Study. Phenomics 2024, 4, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, T.M.; Villafane-Ferriol, N.; Shah, K.P.; Shah, R.M.; Tran Cao, H.S.; Massarweh, N.N.; Silberfein, E.J.; Choi, E.A.; Hsu, C.; McElhany, A.L.; et al. Nutritional and Metabolic Derangements in Pancreatic Cancer and Pancreatic Resection. Nutrients 2017, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Santos, I.; Mendes, L.; Mansinho, H.; Santos, C.A. Nutritional status and functional status of the pancreatic cancer patients and the impact of adjacent symptoms. Clin. Nutr. 2021, 40, 5486–5493. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Conroy, T.; et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v56–v68. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.J.; Kuk, D.; Castillo, C.F.; Basturk, O.; Wolfgang, C.L.; Cameron, J.L.; Lillemoe, K.D.; Ferrone, C.R.; Morales-Oyarvide, V.; He, J.; et al. Multi-institutional Validation Study of the American Joint Commission on Cancer (8th Edition) Changes for T and N Staging in Patients With Pancreatic Adenocarcinoma. Ann. Surg. 2017, 265, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Isaji, S.; Mizuno, S.; Windsor, J.A.; Bassi, C.; Fernández-del Castillo, C.; Hackert, T.; Hayasaki, A.; Katz, M.H.G.; Kim, S.-W.; Kishiwada, M.; et al. International consensus on definition and criteria of borderline resectable pancreatic ductal adenocarcinoma 2017. Pancreatology 2018, 18, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.B. What Makes a Pancreatic Cancer Resectable? Am. Soc. Clin. Oncol. Educ. Book. 2018, 38, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, C.L.; Herman, J.M.; Laheru, D.A.; Klein, A.P.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent progress in pancreatic cancer. CA Cancer J. Clin. 2013, 63, 318–348. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Kwaśniewska, D.; Fudalej, M.; Nurzyński, P.; Badowska-Kozakiewicz, A.; Czerw, A.; Cipora, E.; Sygit, K.; Bandurska, E.; Deptała, A. How A Patient with Resectable or Borderline Resectable Pancreatic Cancer should Be Treated-A Comprehensive Review. Cancers 2023, 15, 4275. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.; Shinde, S.; Saxena, S.; Thakur, S.; Walia, T.; Dixit, V.; Tiwari, A.K.; Vishvakarma, N.K.; Dwivedi, M.; Shukla, D. A Comprehensive Review of Diagnostic and Therapeutic Strategies for the Management of Pancreatic Cancer. Crit. Rev. Oncog. 2020, 25, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Fudalej, M.; Kwaśniewska, D.; Nurzyński, P.; Badowska-Kozakiewicz, A.; Mękal, D.; Czerw, A.; Sygit, K.; Deptała, A. New Treatment Options in Metastatic Pancreatic Cancer. Cancers 2023, 15, 2327. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sohal, D.P.S. Pancreatic Adenocarcinoma Management. JCO Oncol. Pract. 2023, 19, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Schorn, S.; Demir, I.E.; Samm, N.; Scheufele, F.; Calavrezos, L.; Sargut, M.; Schirren, R.M.; Friess, H.; Ceyhan, G.O. Meta-analysis of the impact of neoadjuvant therapy on patterns of recurrence in pancreatic ductal adenocarcinoma. BJS Open 2018, 2, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Wang, J.; Wang, J.; Zhang, Q.; Liang, T. Cell of Origin of Pancreatic cancer: Novel Findings and Current Understanding. Pancreas 2024, 53, e288–e297. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Juiz, N.; Elkaoutari, A.; Bigonnet, M.; Gayet, O.; Roques, J.; Nicolle, R.; Iovanna, J.; Dusetti, N. Basal-like and classical cells coexist in pancreatic cancer revealed by single-cell analysis on biopsy-derived pancreatic cancer organoids from the classical subtype. FASEB J. 2020, 34, 12214–12228. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Dance, M.; Montagner, A.; Salles, J.P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal 2008, 20, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Trahey, M.; McCormick, F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 1987, 238, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Terrell, E.M.; Morrison, D.K. Ras-Mediated Activation of the Raf Family Kinases. Cold Spring Harb. Perspect. Med. 2019, 9, a033746. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Hofer, F.; Fields, S.; Schneider, C.; Martin, G.S. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc. Natl. Acad. Sci. USA 1994, 91, 11089–11093. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M.; Kendall, K.R.; Wang, Y.; Cheung, A.; Haigis, M.C.; Glickman, J.N.; Niwa-Kawakita, M.; Sweet-Cordero, A.; Sebolt-Leopold, J.; Shannon, K.M.; et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 2008, 40, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Poulin, E.J.; Bera, A.K.; Lu, J.; Lin, Y.J.; Strasser, S.D.; Paulo, J.A.; Huang, T.Q.; Morales, C.; Yan, W.; Cook, J.; et al. Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov. 2019, 9, 738–755. [Google Scholar] [CrossRef] [PubMed]
- Stickler, S.; Rath, B.; Hamilton, G. Targeting KRAS in pancreatic cancer. Oncol. Res. 2024, 32, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Sivakumar, S.; Schrock, A.B.; Madison, R.; Fabrizio, D.; Gjoerup, O.; Ross, J.S.; Frampton, G.M.; Napalkov, P.; Montesion, M.; et al. Comprehensive pan-cancer genomic landscape of KRAS altered cancers and real-world outcomes in solid tumors. NPJ Precis. Oncol. 2022, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes With Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018, 4, e173420. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.M.; Ying, H.; Juan, J.; Jenkins, N.A.; Copeland, N.G. KRAS-related proteins in pancreatic cancer. Pharmacol. Ther. 2016, 168, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Chen, X.; Xu, X.; Zheng, C.; Huang, W.; Zhou, Y.; Akuetteh, P.D.P.; Yang, H.; Shi, K.; Chen, B.; et al. STRAP as a New Therapeutic Target for Poor Prognosis of Pancreatic Ductal Adenocarcinoma Patients Mainly Caused by TP53 Mutation. Front. Oncol. 2020, 10, 594224. [Google Scholar] [CrossRef] [PubMed]
- Pittella-Silva, F.; Kimura, Y.; Low, S.K.; Nakamura, Y.; Motoya, M. Amplification of mutant KRAS(G12D) in a patient with advanced metastatic pancreatic adenocarcinoma detected by liquid biopsy: A case report. Mol. Clin. Oncol. 2021, 15, 172. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shao, C.; Liu, X.; Lu, X.; Jia, X.; Zheng, X.; Wang, S.; Zhu, L.; Li, K.; Pang, Y.; et al. Oncogenic ERBB2 aberrations and KRAS mutations cooperate to promote pancreatic ductal adenocarcinoma progression. Carcinogenesis 2020, 41, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Barzi, A.; Weipert, C.M.; Espenschied, C.R.; Raymond, V.M.; Wang-Gillam, A.; Nezami, M.A.; Gordon, E.J.; Mahadevan, D.; Mody, K. ERBB2 (HER2) amplifications and co-occurring KRAS alterations in the circulating cell-free DNA of pancreatic ductal adenocarcinoma patients and response to HER2 inhibition. Front. Oncol. 2024, 14, 1339302. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Azar, I.; Xiu, J.; Hall, M.J.; Hendifar, A.E.; Lou, E.; Hwang, J.J.; Gong, J.; Feldman, R.; Ellis, M.; et al. Molecular Characterization of KRAS Wild-type Tumors in Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. 2022, 28, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Topham, J.T.; Tsang, E.S.; Karasinska, J.M.; Metcalfe, A.; Ali, H.; Kalloger, S.E.; Csizmok, V.; Williamson, L.M.; Titmuss, E.; Nielsen, K.; et al. Integrative analysis of KRAS wildtype metastatic pancreatic ductal adenocarcinoma reveals mutation and expression-based similarities to cholangiocarcinoma. Nat. Commun. 2022, 13, 5941. [Google Scholar] [CrossRef] [PubMed]
- Linehan, A.; O’Reilly, M.; McDermott, R.; O’Kane, G.M. Targeting KRAS mutations in pancreatic cancer: Opportunities for future strategies. Front Med. 2024, 11, 1369136. [Google Scholar] [CrossRef] [PubMed]
- Dilly, J.; Hoffman, M.T.; Abbassi, L.; Li, Z.; Paradiso, F.; Parent, B.D.; Hennessey, C.J.; Jordan, A.C.; Morgado, M.; Dasgupta, S.; et al. Mechanisms of Resistance to Oncogenic KRAS Inhibition in Pancreatic Cancer. Cancer Discov. 2024, 14, 2135–2161. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Beatty, G.L.; Vonderheide, R.H. Immunosurveillance of pancreatic adenocarcinoma: Insights from genetically engineered mouse models of cancer. Cancer Lett. 2009, 279, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA Grants Accelerated Approval to Sotorasib for KRAS G12C-Mutated NSCLC. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-sotorasib-kras-g12c-mutated-nsclc (accessed on 1 April 2025).
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Satake, H.; George, T.J.; Yaeger, R.; Hollebecque, A.; Garrido-Laguna, I.; Schuler, M.; Burns, T.F.; Coveler, A.L.; Falchook, G.S.; et al. Sotorasib in KRAS p.G12C–Mutated Advanced Pancreatic Cancer. N. Engl. J. Med. 2023, 388, 33–43. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA Grants Accelerated Approval to Adagrasib for KRAS G12C-Mutated NSCLC. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-adagrasib-kras-g12c-mutated-nsclc (accessed on 1 April 2025).
- U.S. Food and Drug Administration. FDA Grants Accelerated Approval to Adagrasib with Cetuximab for KRAS G12C-Mutated Colorectal Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-adagrasib-cetuximab-kras-g12c-mutated-colorectal-cancer (accessed on 1 April 2025).
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRAS(G12C) Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.S.; Spira, A.I.; Yaeger, R.; Buchschacher, G.L.; McRee, A.J.; Sabari, J.K.; Johnson, M.L.; Barve, M.A.; Hafez, N.; Velastegui, K.; et al. KRYSTAL-1: Updated activity and safety of adagrasib (MRTX849) in patients (Pts) with unresectable or metastatic pancreatic cancer (PDAC) and other gastrointestinal (GI) tumors harboring a KRASG12C mutation. J. Clin. Oncol. 2022, 40, 519. [Google Scholar] [CrossRef]
- Pant, S.; Yaeger, R.; Spira, A.I.; Pelster, M.; Sabari, J.K.; Hafez, N.; Barve, M.A.; Velastegui, K.; Yan, X.; Der-Torossian, H.; et al. KRYSTAL-1: Activity and safety of adagrasib (MRTX849) in patients with advanced solid tumors harboring a KRASG12C mutation. J. Clin. Oncol. 2023, 41, 425082. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Lawler, W.E.; Shum, M.K.; Dakhil, S.R.; Spira, A.I.; Barlesi, F.; Reck, M.; Garassino, M.C.; Spigel, D.R.; Alvarez, D.; et al. KRYSTAL-12: A randomized phase 3 study of adagrasib (MRTX849) versus docetaxel in patients (pts) with previously treated non-small-cell lung cancer (NSCLC) with KRASG12C mutation. J. Clin. Oncol. 2021, 39, TPS9129. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Wang, L.; Zuo, X.; Maitra, A.; Bresalier, R.S. A Small Molecule with Big Impact: MRTX1133 Targets the KRASG12D Mutation in Pancreatic Cancer. Clin. Cancer Res. 2024, 30, 655–662. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Study of MRTX1133 in Patients With Advanced Solid Tumors Harboring a KRAS G12D Mutation. Available online: https://clinicaltrials.gov/study/NCT05737706 (accessed on 1 April 2025).
- Zhou, C.; Li, W.; Song, Z.; Zhang, Y.; Zhang, Y.; Huang, D.; Yang, Z.; Zhou, M.; Mao, R.; Huang, C.; et al. LBA33 A first-in-human phase I study of a novel KRAS G12D inhibitor HRS-4642 in patients with advanced solid tumors harboring KRAS G12D mutation. Ann. Oncol. 2023, 34, S1273. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Inamura, K.; Nishizono, Y.; Suzuki, A.; Tanaka, H.; Yoshinari, T.; Yamanaka, Y. ASP3082, a First-in-class novel KRAS G12D degrader, exhibits remarkable anti-tumor activity in KRAS G12D mutated cancer models. Eur. J. Cancer 2022, 174, S30. [Google Scholar] [CrossRef]
- Park, W.; Kasi, A.; Spira, A.I.; Berlin, J.D.; Wang, J.S.; Herzberg, B.; Kuboki, Y.; Kitano, S.; Pelster, M.; Goldman, J.W.; et al. Preliminary safety and clinical activity of ASP3082, a first-in-class, KRAS G12D selective protein degrader in adults with advanced pancreatic (PC), colorectal (CRC), and non-small cell lung cancer (NSCLC). Ann. Oncol. 2024, 35, S482–S535. [Google Scholar] [CrossRef]
- Ji, X.; Li, H.; Wu, G.; Zhang, Q.; He, X.; Wu, Y.; Zong, B.; Xu, X.; Liang, C.; Wang, B.; et al. Discovery and Characterization of RP03707: A Highly Potent and Selective KRASG12D PROTAC. J. Med. Chem. 2025, 68, 10238–10254. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Jo, Y.S.; Nale, S.D.; Kim, C.J.; Park, T.H.; Bok, J.H.; Kim, D.M.; Kim, H.M.; Park, J.M.; Dong, J.J.; et al. Abstract 7010: HDB-82: A promising PROTAC degrader for KRAS G12D mutant cancers. Cancer Res. 2025, 85, 7010. [Google Scholar] [CrossRef]
- Guedeney, N.; Cornu, M.; Schwalen, F.; Kieffer, C.; Voisin-Chiret, A.S. PROTAC technology: A new drug design for chemical biology with many challenges in drug discovery. Drug Discov. Today 2023, 28, 103395. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto-Sato, E.; Imanishi, S.; Huang, L.; Itakura, S.; Iwasaki, Y.; Ishizaka, M. A First-Class Degrader Candidate Targeting Both KRAS G12D and G12V Mediated by CANDDY Technology Independent of Ubiquitination. Molecules 2023, 28, 5600. [Google Scholar] [CrossRef] [PubMed]
- Koltun, E.; Cregg, J.; Rice, M.A.; Whalen, D.M.; Freilich, R.; Jiang, J.; Hansen, R.; Bermingham, A.; Knox, J.E.; Dinglasan, J.; et al. Abstract 1260: First-in-class, orally bioavailable KRASG12V(ON) tri-complex inhibitors, as single agents and in combinations, drive profound anti-tumor activity in preclinical models of KRASG12V mutant cancers. Cancer Res. 2021, 81, 1260. [Google Scholar] [CrossRef]
- Li, D.; Geng, K.; Hao, Y.; Gu, J.; Kumar, S.; Olson, A.T.; Kuismi, C.C.; Kim, H.M.; Pan, Y.; Sherman, F.; et al. Targeted degradation of oncogenic KRASG12V triggers antitumor immunity in lung cancer models. J. Clin. Investig. 2024, 135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Morstein, J.; Ecker, A.K.; Guiley, K.Z.; Shokat, K.M. Chemoselective Covalent Modification of K-Ras(G12R) with a Small Molecule Electrophile. J. Am. Chem. Soc. 2022, 144, 15916–15921. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Coley, A.B.; Ward, A.; Keeton, A.B.; Chen, X.; Maxuitenko, Y.; Prakash, A.; Li, F.; Foote, J.B.; Buchsbaum, D.J.; Piazza, G.A. Chapter Five—Pan-RAS inhibitors: Hitting multiple RAS isozymes with one stone. In Advances in Cancer Research; O’Bryan, J.P., Piazza, G.A., Eds.; Academic Press: Cambridge, MA, USA, 2022; Volume 153, pp. 131–168. [Google Scholar]
- Kim, D.; Herdeis, L.; Rudolph, D.; Zhao, Y.; Böttcher, J.; Vides, A.; Ayala-Santos, C.I.; Pourfarjam, Y.; Cuevas-Navarro, A.; Xue, J.Y.; et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature 2023, 619, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Foote, J.B.; Mattox, T.E.; Keeton, A.B.; Chen, X.; Smith, F.T.; Berry, K.; Holmes, T.W.; Wang, J.; Huang, C.-h.; Ward, A.; et al. A Pan-RAS Inhibitor with a Unique Mechanism of Action Blocks Tumor Growth and Induces Antitumor Immunity in Gastrointestinal Cancer. Cancer Res. 2025, 85, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Bandi, D.S.R.; Nagaraju, G.P.; Sarvesh, S.; Carstens, J.L.; Foote, J.B.; Graff, E.C.; Fang, Y.D.; Keeton, A.B.; Chen, X.; Valiyaveettil, J.; et al. ADT-1004: A first-in-class, oral pan-RAS inhibitor with robust antitumor activity in preclinical models of pancreatic ductal adenocarcinoma. Mol. Cancer 2025, 24, 76. [Google Scholar] [CrossRef] [PubMed]
- Cregg, J.; Edwards, A.V.; Chang, S.; Lee, B.J.; Knox, J.E.; Tomlinson, A.C.A.; Marquez, A.; Liu, Y.; Freilich, R.; Aay, N.; et al. Discovery of Daraxonrasib (RMC-6236), a Potent and Orally Bioavailable RAS(ON) Multi-selective, Noncovalent Tri-complex Inhibitor for the Treatment of Patients with Multiple RAS-Addicted Cancers. J. Med. Chem. 2025, 68, 6064–6083. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Menard, M.; Weller, C.; Wang, Z.; Burnett, L.; Aronchik, I.; Steele, S.; Flagella, M.; Zhao, R.; Evans, J.W.W.; et al. Abstract 526: RMC-9805, a first-in-class, mutant-selective, covalent and oral KRASG12D(ON) inhibitor that induces apoptosis and drives tumor regression in preclinical models of KRASG12D cancers. Cancer Res. 2023, 83, 526. [Google Scholar] [CrossRef]
- Jiang, J.; Jiang, L.; Maldonato, B.J.; Wang, Y.; Holderfield, M.; Aronchik, I.; Winters, I.P.; Salman, Z.; Blaj, C.; Menard, M.; et al. Translational and Therapeutic Evaluation of RAS-GTP Inhibition by RMC-6236 in RAS-Driven Cancers. Cancer Discov. 2024, 14, 994–1017. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Laguna, I.; Wolpin, B.M.; Park, W.; Azad, N.S.; Spira, A.I.; Starodub, A.; Sommerhalder, D.; Punekar, S.R.; Herzberg, B.; Barve, M.A.; et al. Safety, efficacy, and on-treatment circulating tumor DNA (ctDNA) changes from a phase 1 study of RMC-6236, a RAS(ON) multi-selective, tri-complex inhibitor, in patients with RAS mutant pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2025, 43, 722. [Google Scholar] [CrossRef]
- Revolution Medicines, Inc. Study of RMC-6236 in Patients with Advanced Solid Tumors Harboring Specific Mutations in RAS. Available online: https://clinicaltrials.gov/study/NCT05379985 (accessed on 1 April 2025).
- Revolution Medicines, Inc. Study of RMC-6291 in Combination with RMC-6236 in Participants with Advanced KRAS G12C Mutant Solid Tumors. Available online: https://clinicaltrials.gov/study/NCT06128551 (accessed on 1 April 2025).
- Revolution Medicines, Inc. Study of RMC-9805 in Participants with KRAS G12D-Mutant Solid Tumors. Available online: https://clinicaltrials.gov/study/NCT06040541 (accessed on 1 April 2025).
- Revolution Medicines, Inc. Study of RAS(ON) Inhibitor Combinations in Patients with Advanced RAS-mutated NSCLC. Available online: https://clinicaltrials.gov/study/NCT06162221 (accessed on 1 April 2025).
- Revolution Medicines, Inc. Study of RAS(ON) Inhibitors in Patients with Gastrointestinal Solid Tumors. Available online: https://clinicaltrials.gov/study/NCT06445062 (accessed on 1 April 2025).
- Wolpin, B.M.; Wainberg, Z.A.; Garrido-Laguna, I.; Manji, G.A.; Spira, A.I.; Azad, N.S.; Pant, S.; Zhang, Y.; Trang, J.; Salman, Z.; et al. Trial in progress: RASolute 302—A phase 3, multicenter, global, open-label, randomized study of daraxonrasib (RMC-6236), a RAS(ON) multi-selective inhibitor, versus standard of care chemotherapy in patients with previously treated metastatic pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2025, 43, TPS4230. [Google Scholar] [CrossRef]
- Spira, A.I.; Papadopoulos, K.P.; Kim, D.W.; Parikh, A.R.; Barve, M.A.; Powderly, J.D.; Starodub, A.; Strickler, J.H.; Li, B.T.; Oberstein, P.E.; et al. Preliminary safety, antitumor activity, and circulating tumor DNA (ctDNA) changes with RMC-9805, an oral, RAS(ON) G12D-selective tri-complex inhibitor in patients with KRAS G12D pancreatic ductal adenocarcinoma (PDAC) from a phase 1 study in advanced solid tumors. J. Clin. Oncol. 2025, 43, 724. [Google Scholar] [CrossRef]
- A Study of YL-17231 in Patients with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/study/NCT06078800 (accessed on 21 July 2025).
- Xu, Z.; Weaver, D.; Drakas, R.; Lou, Y. AACR Abstract 2627: The small molecule KRAS inhibitor, TEB-17231, blocks tumor Progression and Overcomes KRAS G12C Inhibitor Mediated Resistance. Cancer Res. 2023, 83, 2627. [Google Scholar] [CrossRef]
- Kim, M.J.; Chang, H.; Nam, G.; Ko, Y.; Kim, S.H.; Roberts, T.M.; Ryu, J.H. RNAi-Based Approaches for Pancreatic Cancer Therapy. Pharmaceutics 2021, 13, 1638. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Khvorova, A. RNAi-based drug design: Considerations and future directions. Nat. Rev. Drug Discov. 2024, 23, 341–364. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Liang, G.; Cui, K.; Liang, Y.; Wang, Q.; Lv, S.; Cheng, X.; Zhang, L. Insight Into the Prospects for RNAi Therapy of Cancer. Front. Pharmacol. 2021, 12, 644718. [Google Scholar] [CrossRef] [PubMed]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Kara, G.; Calin, G.A.; Ozpolat, B. RNAi-based therapeutics and tumor targeted delivery in cancer. Adv. Drug Deliv. Rev. 2022, 182, 114113. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-C.; Di Trani, N.; Conte, M.; Nguyen, D.C.; Jokonya, S.; Wu, A.; Vander Pol, R.; Joubert, A.L.; Facchi, I.; Wood, A.M.; et al. Nanofluidic delivery implant sustains localization and maximizes efficacy of intratumoral immunotherapy. Nano Today 2024, 56, 102258. [Google Scholar] [CrossRef]
- Surana, R.; Lebleu, V.; Lee, J.; Smaglo, B.; Zhao, D.; Lee, M.; Wolff, R.; Overman, M.; Mendt, M.; McAndrews, K.; et al. Phase I study of mesenchymal stem cell (MSC)-derived exosomes with KRAS G12D siRNA in patients with metastatic pancreatic cancer harboring a KRAS G12D mutation. J. Clin. Oncol. 2022, 40, TPS633. [Google Scholar] [CrossRef]
- Anthiya, S.; Öztürk, S.C.; Yanik, H.; Tavukcuoglu, E.; Şahin, A.; Datta, D.; Charisse, K.; Álvarez, D.M.; Loza, M.I.; Calvo, A.; et al. Targeted siRNA lipid nanoparticles for the treatment of KRAS-mutant tumors. J. Control Release 2023, 357, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Küçükekmekci, B.; Budak Yıldıran, F.A. Investigation of the efficacy of siRNA-mediated KRAS gene silencing in pancreatic cancer therapy. PeerJ 2024, 12, e18214. [Google Scholar] [CrossRef] [PubMed]
- Khlebtsov, N.; Dykman, L. Biodistribution and toxicity of engineered gold nanoparticles: A review of in vitro and in vivo studies. Chem. Soc. Rev. 2011, 40, 1647–1671. [Google Scholar] [CrossRef] [PubMed]
- Strand, M.S.; Krasnick, B.A.; Pan, H.; Zhang, X.; Bi, Y.; Brooks, C.; Wetzel, C.; Sankpal, N.; Fleming, T.; Goedegebuure, S.P.; et al. Precision delivery of RAS-inhibiting siRNA to KRAS driven cancer via peptide-based nanoparticles. Oncotarget 2019, 10, 4761–4775. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Shirvan, M.; M, R.; Yaron, M.; Zorde, E.; Shragai, O.; Aloni, R.M.; Bernstein, A.; Shinar, D.; Nyska, A.; Hadar, I.; et al. SIL-204 siRNA encapsulated in extended release microparticles for the treatment of localized cancer that harbors a KRAS G12x or G13D mutation. J. Clin. Oncol. 2025, 43, 745. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Stanland, L.; Huggins, H.; Porrello, A.; Chareddy, Y.; Azam, S.; Perry, J.; Pallan, P.; Whately, K.; Edatt, L.; Fleming, M.; et al. Abstract PR012: A first-in-class EGFR-directed KRAS G12V selective inhibitor. Mol. Cancer Ther. 2024, 23, PR012. [Google Scholar] [CrossRef]
- Robbins, M.; Judge, A.; MacLachlan, I. siRNA and innate immunity. Oligonucleotides 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Yonemori, K.; Kurahara, H.; Maemura, K.; Natsugoe, S. MicroRNA in pancreatic cancer. J. Hum. Genet. 2017, 62, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Wei, D.; Liu, X.; Long, D.; Tian, X.; Yang, Y. MicroRNAs as potential therapeutic targets for pancreatic cancer. Chin. Med. J. 2022, 135, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Daoud, A.Z.; Mulholland, E.J.; Cole, G.; McCarthy, H.O. MicroRNAs in Pancreatic Cancer: Biomarkers, prognostic, and therapeutic modulators. BMC Cancer 2019, 19, 1130. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.G.; Yu, S.N.; Lu, Z.H.; Ma, Y.H.; Gu, Y.M.; Chen, J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis 2010, 31, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.U.; Prieto-Vila, M.; Hironaka, A.; Ochiya, T. The role of extracellular vesicle microRNAs in cancer biology. Clin. Chem. Lab. Med. 2017, 55, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Lu, Z.; Liu, C.; Meng, Y.; Ma, Y.; Zhao, W.; Liu, J.; Yu, J.; Chen, J. miRNA-96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res. 2010, 70, 6015–6025. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Trepotec, Z.; Lichtenegger, E.; Plank, C.; Aneja, M.K.; Rudolph, C. Delivery of mRNA Therapeutics for the Treatment of Hepatic Diseases. Mol. Ther. 2019, 27, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Mokhlis, H.A.; Bayraktar, R.; Kabil, N.N.; Caner, A.; Kahraman, N.; Rodriguez-Aguayo, C.; Zambalde, E.P.; Sheng, J.; Karagoz, K.; Kanlikilicer, P.; et al. The Modulatory Role of MicroRNA-873 in the Progression of KRAS-Driven Cancers. Mol. Ther. Nucleic Acids 2019, 14, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2021, 28, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Garibaldi-Ríos, A.F.; Figuera, L.E.; Zúñiga-González, G.M.; Gómez-Meda, B.C.; García-Verdín, P.M.; Carrillo-Dávila, I.A.; Gutiérrez-Hurtado, I.A.; Torres-Mendoza, B.M.; Gallegos-Arreola, M.P. In Silico Identification of Dysregulated miRNAs Targeting KRAS Gene in Pancreatic Cancer. Diseases 2024, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Brown, D.; Winkler, M. The promise of microRNA replacement therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef] [PubMed]
- Mainini, F.; Eccles, M.R. Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy. Molecules 2020, 25, 2692. [Google Scholar] [CrossRef] [PubMed]
- Gurreri, E.; Genovese, G.; Perelli, L.; Agostini, A.; Piro, G.; Carbone, C.; Tortora, G. KRAS-Dependency in Pancreatic Ductal Adenocarcinoma: Mechanisms of Escaping in Resistance to KRAS Inhibitors and Perspectives of Therapy. Int. J. Mol. Sci. 2023, 24, 9313. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Cahill, M.A.; Neubauer, H. PGRMC Proteins Are Coming of Age: A Special Issue on the Role of PGRMC1 and PGRMC2 in Metabolism and Cancer Biology. Cancers 2021, 13, 512. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Nucleic Acid-Based Approaches to Tackle KRAS Mutant Cancers. Int. J. Mol. Sci. 2023, 24, 16933. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.-P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.-Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Bodoky, G.; Timcheva, C.; Spigel, D.R.; La Stella, P.J.; Ciuleanu, T.E.; Pover, G.; Tebbutt, N.C. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig. New Drugs 2012, 30, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Hidalgo, M.; Canon, J.L.; Macarulla, T.; Bazin, I.; Poddubskaya, E.; Manojlovic, N.; Radenkovic, D.; Verslype, C.; Raymond, E.; et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int. J. Cancer 2018, 143, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.K.; Park, H.; Tolcher, A.W.; Ou, S.-H.I.; Garon, E.B.; George, B.; Janne, P.A.; Moody, S.E.; Tan, E.Y.; Sen, S.K.; et al. KRYSTAL-2: A phase I/II trial of adagrasib (MRTX849) in combination with TNO155 in patients with advanced solid tumors with KRAS G12C mutation. J. Clin. Oncol. 2021, 39, TPS146. [Google Scholar] [CrossRef]
- Brana, I.; Shapiro, G.; Johnson, M.L.; Yu, H.A.; Robbrecht, D.; Tan, D.S.-W.; Siu, L.L.; Minami, H.; Steeghs, N.; Hengelage, T.; et al. Initial results from a dose finding study of TNO155, a SHP2 inhibitor, in adults with advanced solid tumors. J. Clin. Oncol. 2021, 39, 3005. [Google Scholar] [CrossRef]
- LaMarche, M.J.; Acker, M.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.H.-T.; Chen, Y.-N.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 13578–13594. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.A.; Mainardi, S.; Dias, M.H.; Bosma, A.; van Dijk, E.; Selig, R.; Albrecht, W.; Laufer, S.A.; Zender, L.; Bernards, R. Small-molecule inhibition of MAP2K4 is synergistic with RAS inhibitors in KRAS-mutant cancers. Proc. Natl. Acad. Sci. USA 2024, 121, e2319492121. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Hobor, S.; Bertotti, A.; Zecchin, D.; Huang, S.; Galimi, F.; Cottino, F.; Prahallad, A.; Grernrum, W.; Tzani, A.; et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014, 7, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.S.; McDonald, P.C.; Nemirovsky, O.; Awrey, S.; Chafe, S.C.; Schaeffer, D.F.; Li, J.; Renouf, D.J.; Stanger, B.Z.; Dedhar, S. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100131. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, N.; Yan, L.; Qiryaqos, F.; Engstrom, L.D.; Laguer, J.; Calinisan, A.; Hebbert, A.; Waters, L.; Moya, K.; Bowcut, V.; et al. The SOS1 Inhibitor MRTX0902 Blocks KRAS Activation and Demonstrates Antitumor Activity in Cancers Dependent on KRAS Nucleotide Loading. Mol. Cancer Ther. 2024, 23, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J.; et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Gort, E.; Johnson, M.L.; Hwang, J.J.; Pant, S.; Dünzinger, U.; Riemann, K.; Kitzing, T.; Janne, P.A. A phase I, open-label, dose-escalation trial of BI 1701963 as monotherapy and in combination with trametinib in patients with KRAS mutated advanced or metastatic solid tumors. J. Clin. Oncol. 2020, 38, TPS3651. [Google Scholar] [CrossRef]
- Johnson, M.L.; Gort, E.; Pant, S.; Lolkema, M.P.; Sebastian, M.; Scheffler, M.; Hwang, J.; Dünzinger, U.; Riemann, K.; Kitzing, T.; et al. 524P A phase I, open-label, dose-escalation trial of BI 1701963 in patients (pts) with KRAS mutated solid tumours: A snapshot analysis. Ann. Oncol. 2021, 32, S591–S592. [Google Scholar] [CrossRef]
- Norgard, R.J.; Budhani, P.; O’Brien, S.A.; Xia, Y.; Egan, J.N.; Flynn, B.; Tagore, J.R.; Seco, J.; Peet, G.W.; Mikucka, A.; et al. Reshaping the Tumor Microenvironment of KRASG12D Pancreatic Ductal Adenocarcinoma with Combined SOS1 and MEK Inhibition for Improved Immunotherapy Response. Cancer Res. Commun. 2024, 4, 1548–1560. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Xu, D.; Liao, M.-m.; Sun, Y.; Bao, W.-d.; Yao, F.; Ma, L. Barriers and opportunities in pancreatic cancer immunotherapy. npj Precis. Oncol. 2024, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hwang, R.F.; Logsdon, C.D.; Ullrich, S.E. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 2013, 73, 3927–3937. [Google Scholar] [CrossRef] [PubMed]
- Renouf, D.J.; Loree, J.M.; Knox, J.J.; Topham, J.T.; Kavan, P.; Jonker, D.; Welch, S.; Couture, F.; Lemay, F.; Tehfe, M.; et al. The CCTG PA.7 phase II trial of gemcitabine and nab-paclitaxel with or without durvalumab and tremelimumab as initial therapy in metastatic pancreatic ductal adenocarcinoma. Nat. Commun. 2022, 13, 5020. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, K.K.; McAndrews, K.M.; LeBleu, V.S.; Yang, S.; Lyu, H.; Li, B.; Sockwell, A.M.; Kirtley, M.L.; Morse, S.J.; Moreno Diaz, B.A.; et al. KRASG12D inhibition reprograms the microenvironment of early and advanced pancreatic cancer to promote FAS-mediated killing by CD8+ T cells. Cancer Cell 2023, 41, 1606–1620.e1608. [Google Scholar] [CrossRef] [PubMed]
- Ghukasyan, R.; Liang, K.; Chau, K.; Li, L.; Chan, C.; Abt, E.R.; Le, T.; Park, J.Y.; Wu, N.; Premji, A.; et al. MEK Inhibition Sensitizes Pancreatic Cancer to STING Agonism by Tumor Cell-intrinsic Amplification of Type I IFN Signaling. Clin. Cancer Res. 2023, 29, 3130–3141. [Google Scholar] [CrossRef] [PubMed]
- Chamma, H.; Vila, I.K.; Taffoni, C.; Turtoi, A.; Laguette, N. Activation of STING in the pancreatic tumor microenvironment: A novel therapeutic opportunity. Cancer Lett. 2022, 538, 215694. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; McAllister, D.; Vonderhaar, E.P.; Palen, K.; Riese, M.J.; Gershan, J.; Johnson, B.D.; Dwinell, M.B. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J. Immunother. Cancer 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Courau, T.; Borison, J.; Ritchie, A.J.; Mayer, A.T.; Krummel, M.F.; Collisson, E.A. Activating Immune Recognition in Pancreatic Ductal Adenocarcinoma via Autophagy Inhibition, MEK Blockade, and CD40 Agonism. Gastroenterology 2022, 162, 590–603.e514. [Google Scholar] [CrossRef] [PubMed]
- Donahue, K.L.; Watkoske, H.R.; Kadiyala, P.; Du, W.; Brown, K.; Scales, M.K.; Elhossiny, A.M.; Espinoza, C.E.; Lasse Opsahl, E.L.; Griffith, B.D.; et al. Oncogenic KRAS-Dependent Stromal Interleukin-33 Directs the Pancreatic Microenvironment to Promote Tumor Growth. Cancer Discov. 2024, 14, 1964–1989. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Neuzillet, C.; Tijeras-Raballand, A.; Faivre, S.; de Gramont, A.; Raymond, E. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 2015, 6, 16832–16847. [Google Scholar] [CrossRef] [PubMed]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, X.; Kang, R.; Zeh, H.; Klionsky, D.J.; Tang, D. Regulation and function of autophagy in pancreatic cancer. Autophagy 2021, 17, 3275–3296. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Wang, J.; Chang, S.; Liu, M.; Pang, X. The greedy nature of mutant RAS: A boon for drug discovery targeting cancer metabolism? Acta Biochim. Biophys. Sin. 2016, 48, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Takhsha, F.S.; Vangestel, C.; Tanc, M.; De Bruycker, S.; Berg, M.; Pintelon, I.; Stroobants, S.; De Meyer, G.R.Y.; Van Der Veken, P.; Martinet, W. ATG4B Inhibitor UAMC-2526 Potentiates the Chemotherapeutic Effect of Gemcitabine in a Panc02 Mouse Model of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 750259. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Iwadate, D.; Kato, H.; Nakai, Y.; Tateishi, K.; Fujishiro, M. Targeting the Metabolic Rewiring in Pancreatic Cancer and Its Tumor Microenvironment. Cancers 2022, 14, 4351. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517. [Google Scholar] [CrossRef] [PubMed]
- White, E. Blockade of RAF and autophagy is the one-two punch to take out Ras. Proc. Natl. Acad. Sci. USA 2019, 116, 3965–3967. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Baker, N.M.; Miermont, A.M.; Thurman, R.D.; Pierobon, M.; Tran, T.H.; Anderson, A.O.; Waters, A.M.; Diehl, J.N.; Papke, B.; et al. Atypical KRAS(G12R) Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020, 10, 104–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, L.; Hu, J.; Zhao, Z.; Ji, L.; Cheng, C.; Zhang, G.; Zhang, T.; Li, Y.; Chen, H.; et al. UBL4A inhibits autophagy-mediated proliferation and metastasis of pancreatic ductal adenocarcinoma via targeting LAMP1. J. Exp. Clin. Cancer Res. 2019, 38, 297. [Google Scholar] [CrossRef] [PubMed]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Selwan, E.M.; Edinger, A.L. Branched chain amino acid metabolism and cancer: The importance of keeping things in context. Transl. Cancer Res. 2017, 6, S578–S584. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Buyse, M.E.; Alistar, A.T.; Rocha Lima, C.M.; Luther, S.; Pardee, T.S.; Van Cutsem, E. A Phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Future Oncol. 2019, 15, 3189–3196. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Sahai, V.; Bahary, N.; Mahipal, A.; Kasi, A.; Rocha Lima, C.M.S.; Alistar, A.T.; Oberstein, P.E.; Golan, T.; Metges, J.P.; et al. Devimistat (CPI-613) With Modified Fluorouarcil, Oxaliplatin, Irinotecan, and Leucovorin (FFX) Versus FFX for Patients With Metastatic Adenocarcinoma of the Pancreas: The Phase III AVENGER 500 Study. J. Clin. Oncol. 2024, 42, 3692–3701. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial | Phase | Enrollment | Patients | Summary |
|---|---|---|---|---|
| NCT05379985 | I | 614 | Patients with advanced solid tumors (NSCLC, CRC, PDAC) harboring specific RAS mutations | To evaluate safety, tolerability, PK, and clinical activity of escalating doses of RMC-6236. To determine the MTD and/or recommended phase 2 dose. |
| NCT06040541 | I | 444 | Patients with KRAS G12D-mutant solid tumors | To evaluate the safety, tolerability, PK, and preliminary clinical activity of RMC-9805. Two arms: RMC-9805 monotherapy arm and RMC-9805 plus RMC-6236 combination arm. |
| NCT06162221 | I II | 484 | Patients with RAS-mutated solid tumors with a focus on NSCLC | To evaluate the safety, tolerability, PK, and preliminary antitumor activity of novel RAS(ON) inhibitors combined with SOC or with each other: RMC-6291 +/− RMC-6236 + SOC RMC-6236 + SOC RMC-9805 +/− RMC-6236 + SOC. |
| NCT06445062 | I II | 1130 | Patients with RAS-mutated solid tumors with a focus on GI cancers (PDAC, CRC) | To evaluate the safety, tolerability, PK, and preliminary antitumor activity of novel RAS(ON) inhibitors combined with SOC or with each other: RMC-6236 + 5-fluorouracil-based regimen RMC-6236 + cetuximab with or without mFOLFOX6 RMC-6236 + gemcitabine + nab-paclitaxel RMC-9805 with or without RMC-6236 + 5-fluorouracil-based regimens RMC-9805 with or without RMC-6236 + cetuximab with or without mFOLFOX6 RMC-9805 with or without RMC-6236 + gemcitabine + nab-paclitaxel. |
| NCT06128551 | I | 210 | Patients with KRAS G12C-mutated solid tumors (CRC, PDAC, NSCLC) | To evaluate safety, tolerability, and PK profiles of RMC-6291 and RMC-6236. |
| NCT06625320 RASolute 302 | III | 460 | Patients with metastatic PDAC who were previously treated with one prior line of therapy with a 5-FU-based or gemcitabine-based regimen | To evaluate the safety and efficacy of an RMC-6236 inhibitor compared to SOC treatment. |
| Inhibitor | Target Mutation | Phase | NCT Number(s) | Efficacy (ORR/mPFS/OS) | Pros | Cons |
|---|---|---|---|---|---|---|
| Sotorasib | KRAS G12C | II | NCT03600883 | 20%/4.0 months/6.9 months | FDA-approved for NSCLC, promising in PDAC | Limited to G12C (~1–3% in PDAC) |
| Adagrasib | KRAS G12C | II | 1. NCT03785249, 2. NCT04685135 | 1. 33%/5.4 months/8.0 months 2. - | Promising second-line treatment; in combination with cetuximab | Limited to G12C (~1–3% in PDAC) Lower OS and mPFS results in PDAC group |
| MRTX1133 | KRAS G12D | I | NCT05737706 | - | Strong preclinical efficacy High selectivity; | Resistance and low oral bioavailability |
| ASP3082 | KRAS G12D | I | NCT05382559 | 33.3% at 300 mg (early phase) | first-in-class PROTAC; safe profile | High MW limits cell penetration |
| RMC-9805 | KRAS G12D | I | NCT06040541 | Preliminary ctDNA reduction | Safety, with no grade 4 and 5 TRAEs | Early data only, need for further investigation |
| RMC-6236 | Pan-RAS | I I, II I, II I III | NCT05379985, NCT06162221, NCT06445062, NCT06128551, RASolute302 (NCT05379985) | NCT05379985 mPFS 8.1 months for KRAS G12X mutation 7.6 months for broadly RAS-mutant tumors | Pan-RAS(ON) inhibitor; promising efficacy and safety profile | Still in trials, tolerability profile evolving |
| ADT-007 | Pan-RAS | Preclinical | — | Better than sotorasib, adagrasib, MRTX1133 (preclinical) | Targets nucleotide-free RAS; preclinical efficacy superior to other agents | No data from clinical studies Does not impact the growth of wild-type RAS PDAC |
| BI-2865 | Pan-KRAS | Preclinical | — | Tumor growth inhibition (preclinical) | Blocks multiple mutants; spares HRAS/NRAS | Compensatory RAS isoform activation which limits effectiveness |
| YL-17231 | Pan-RAS | I | NCT06078800 | - | Effective in resistant lines; good PK profile | Early phase, need for further investigation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krupa, K.; Fudalej, M.; Włoszek, E.; Miski, H.; Badowska-Kozakiewicz, A.M.; Mękal, D.; Budzik, M.P.; Czerw, A.; Deptała, A. Treatment of KRAS-Mutated Pancreatic Cancer: New Hope for the Patients? Cancers 2025, 17, 2453. https://doi.org/10.3390/cancers17152453
Krupa K, Fudalej M, Włoszek E, Miski H, Badowska-Kozakiewicz AM, Mękal D, Budzik MP, Czerw A, Deptała A. Treatment of KRAS-Mutated Pancreatic Cancer: New Hope for the Patients? Cancers. 2025; 17(15):2453. https://doi.org/10.3390/cancers17152453
Chicago/Turabian StyleKrupa, Kamila, Marta Fudalej, Emilia Włoszek, Hanna Miski, Anna M. Badowska-Kozakiewicz, Dominika Mękal, Michał P. Budzik, Aleksandra Czerw, and Andrzej Deptała. 2025. "Treatment of KRAS-Mutated Pancreatic Cancer: New Hope for the Patients?" Cancers 17, no. 15: 2453. https://doi.org/10.3390/cancers17152453
APA StyleKrupa, K., Fudalej, M., Włoszek, E., Miski, H., Badowska-Kozakiewicz, A. M., Mękal, D., Budzik, M. P., Czerw, A., & Deptała, A. (2025). Treatment of KRAS-Mutated Pancreatic Cancer: New Hope for the Patients? Cancers, 17(15), 2453. https://doi.org/10.3390/cancers17152453