
Oncogenic Activity and Sorafenib Sensitivity of ARAF p.S214C Mutation in Lung Cancer

 , , , , ,
, , , , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Resources
2.2. Mutation Database Acquisition
2.3. The Cell Cultures and Drugs
2.4. Retroviral Vectors and Infection
2.5. Western Blotting
2.6. The MTT Assay
2.7. The Migration Assay
2.8. The Invasion Assay
2.9. The Colony Formation Assay
2.10. The Spheroid Formation Assay
2.11. RNA Sequencing
2.12. The Reverse-Phase Protein Array Analysis
2.13. The Animal Experiments
2.14. Immunohistochemistry and Hematoxylin and Eosin Staining
2.15. The Statistical Analysis
3. Results
3.1. ARAF p.S214C Is a Rare but Important Mutation in Lung Adenocarcinoma
3.2. ARAF p.S214C Activates the MEK-ERK Pathway and Enhances the Proliferation, Colony and Spheroid Formation, Migration, and Invasion of Cells In Vitro
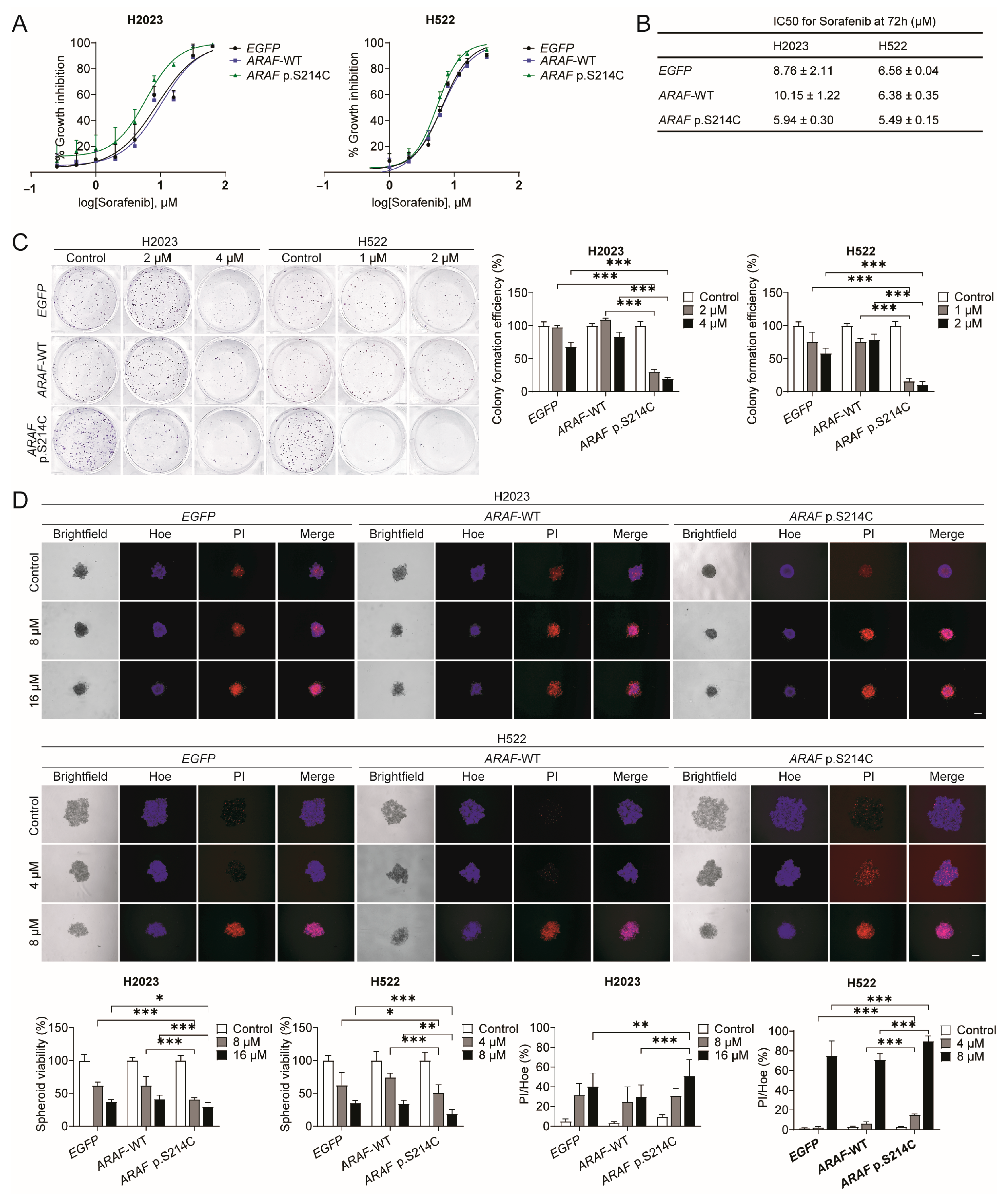
3.3. ARAF p.S214C Is Sensitive to Sorafenib In Vitro
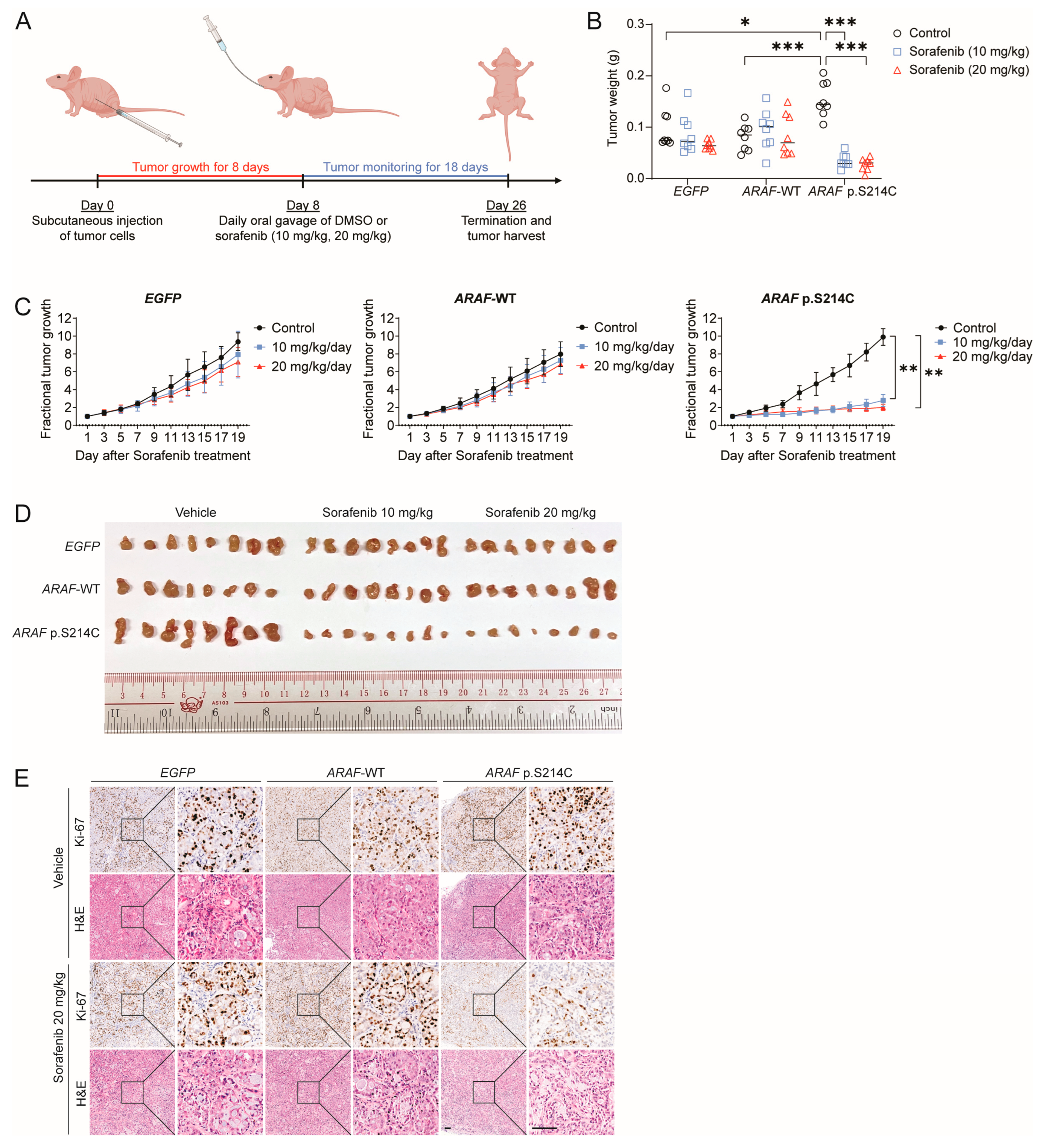
3.4. ARAF p.S214C Is Sensitive to Sorafenib In Vivo
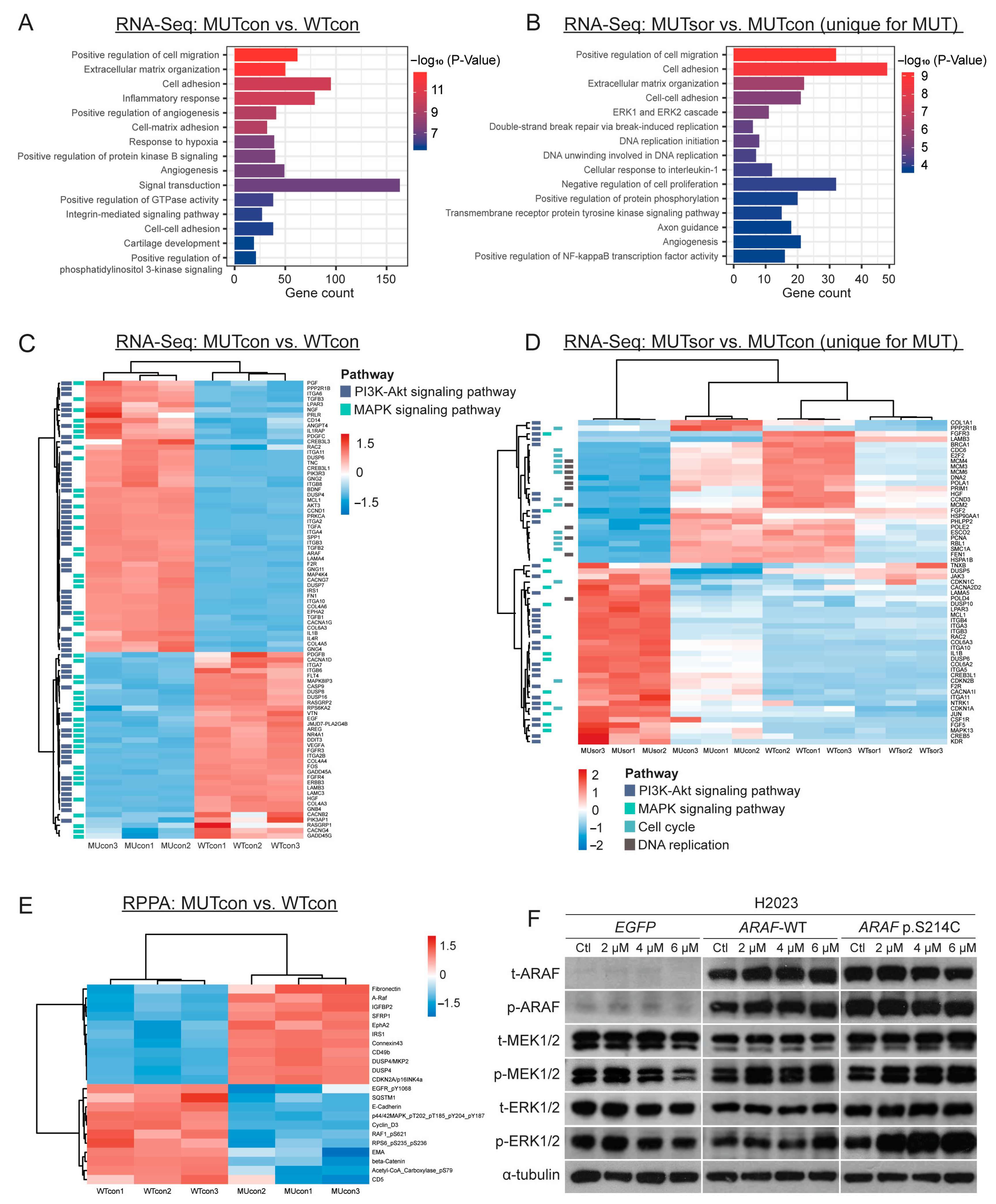
3.5. Transcriptomic and Proteomic Analyses of the ARAF-WT and ARAF p.S214C Cells in the Absence or Presence of Sorafenib Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALK | Anaplastic lymphoma kinase |
| ANOVA | Analysis of variance |
| CDK | Cyclin-dependent kinase |
| CKI | Cyclin-dependent kinase inhibitor |
| CMG | Cdc45-MCM-GINS |
| CR | Conserved region |
| DEG | Differentially expressed gene |
| ECL | Enhanced chemiluminescence |
| EGFR | Epidermal growth factor receptor |
| EMA | Epithelial membrane antigen |
| ER | Exceptional responder |
| ERK | Extracellular-signal-regulated kinase |
| FBS | Fetal bovine serum |
| H&E | Hematoxylin and eosin |
| IHC | Immunohistochemistry |
| KRAS | Kirsten rat sarcoma virus |
| MEK | Mitogen-activated protein kinase |
| MTT | 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide |
| NSCLC | Non-small-cell lung cancer |
| PCNA | Proliferating cell nuclear antigen |
| PDGFR | Platelet-derived growth factor receptor |
| PI3K | Phosphatidylinositol 3-kinase |
| PLAT-A | Platinum-A |
| P/S | Penicillin/streptomycin |
| RBS | RAS-binding domain |
| RET | Rearranged during transfection |
| RNA-Seq | RNA sequencing |
| RPPA | Reverse-phase protein array |
| VEGFR | Vascular endothelial growth factor receptor |
| WT | Wild-type |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, M.; Borgeaud, M.; Addeo, A.; Friedlaender, A. Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. World J. Clin. Oncol. 2021, 12, 217–237. [Google Scholar] [CrossRef]
- Zhu, C.; Guan, X.; Zhang, X.; Luan, X.; Song, Z.; Cheng, X.; Zhang, W.; Qin, J.-J. Targeting KRAS mutant cancers: From druggable therapy to drug resistance. Mol. Cancer 2022, 21, 159. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Tkacik, E.; Eck, M.J. Signaling from RAS to RAF: The molecules and their mechanisms. Ann. Rev. Biochem. 2024, 93, 289–316. [Google Scholar] [CrossRef]
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. Raf-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 2022, 85, 123–154. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Facchinetti, F.; Rossi, G.; Minari, R.; Conti, A.; Friboulet, L.; Tiseo, M.; Planchard, D. BRAF in non-small cell lung cancer (NSCLC): Pickaxing another brick in the wall. Cancer Treat. Rev. 2018, 66, 82–94. [Google Scholar] [CrossRef]
- Harada, G.; Yang, S.-R.; Cocco, E.; Drilon, A. Rare molecular subtypes of lung cancer. Nat. Rev. Clin. Oncol. 2023, 20, 229–249. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both RAF and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef]
- Lang, L. FDA approves sorafenib for patients with inoperable liver cancer. Gastroenterology 2008, 134, 379. [Google Scholar] [CrossRef]
- Kane, R.C.; Farrell, A.T.; Saber, H.; Tang, S.; Williams, G.; Jee, J.M.; Liang, C.; Booth, B.; Chidambaram, N.; Morse, D.; et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 2006, 12, 7271–7278. [Google Scholar] [CrossRef] [PubMed]
- Pitoia, F.; Jerkovich, F. Selective use of sorafenib in the treatment of thyroid cancer. Drug Des. Devel. Ther. 2016, 10, 1119–1131. [Google Scholar] [CrossRef]
- Hendrixson, M.; Gladkiy, Y.; Thyagarajan, A.; Sahu, R.P. Efficacy of sorafenib-based therapies for non-small cell lung cancer. Med. Sci. 2024, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Greulich, H.; Kaplan, B.; Araujo, L.; Amann, J.; Horn, L.; Schiller, J.; Villalona-Calero, M.A.; Meyerson, M.; Carbone, D.P. Oncogenic and sorafenib-sensitive ARAF mutations in lung adenocarcinoma. J. Clin. Investig. 2014, 124, 1582–1586. [Google Scholar] [CrossRef]
- Herrera-Juárez, M.; Serrano-Gómez, C.; Bote-de-Cabo, H.; Paz-Ares, L. Targeted therapy for lung cancer: Beyond EGFR and ALK. Cancer 2023, 129, 1803–1820. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.C.; Wang, W.; Zhu, B.; Wang, X. Lung cancer heterogeneity and new strategies for drug therapy. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 531–546. [Google Scholar] [CrossRef]
- Wu, F.; Fan, J.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; Zhag, J.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Odogwu, L.; Mathieu, L.; Blumenthal, G.; Larkins, E.; Goldberg, K.B.; Griffin, N.; Bijwaard, K.; Lee, E.Y.; Philip, R.; Jiang, X.; et al. FDA approval summary: Dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring BRAF V600E mutations. Oncologist 2018, 23, 740–745. [Google Scholar] [CrossRef]
- Riely, G.J.; Ahn, M.-J.; Clarke, J.; Dagogo-Jack, I.; Felip, E.; Gelsomino, F.; Goldman, J.W.; Hussein, M.; Johnson, M.L.; Morgensztern, D.; et al. LBA56 updated efficacy and safety from the phase II pharos study of encorafenib plus binimetinib in patients with BRAF V600E-mutant metastatic NSCLC (mNSCLC). Ann. Oncol. 2024, 35, S1246–S1247. [Google Scholar] [CrossRef]
- Bonacquisti, E.E.; Nguyen, J. Connexin 43 (CX43) in cancer: Implications for therapeutic approaches via gap junctions. Cancer Lett. 2019, 442, 439–444. [Google Scholar] [CrossRef]
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EPHA2 in cancer. J. Hematol. Oncol. 2020, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-C.; Yang, C.-H.; Cheng, L.-H.; Chang, W.-T.; Lin, Y.-R.; Cheng, H.-C. Fibronectin in cancer: Friend or foe. Cells 2019, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Schröder, D.C.; Nenkov, M.; Rizwan, M.N.; Abubrig, M.; Sonnemann, J.; Murrieta-Coxca, J.M.; Morales-Prieto, D.M.; Westermann, M.; Gaßler, N.; et al. Epithelial membrane protein 2 suppresses non-small cell lung cancer cell growth by inhibition of MAPK pathway. Int. J. Mol. Sci. 2021, 22, 2944. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Zhang, Z.; Westover, D.; Tang, Z.; Liu, Y.; Sun, J.; Sun, Y.; Zhang, R.; Wang, Z.; Zhou, S.; Hesilaiti, N.; et al. Wnt/β-catenin signaling in the development and therapeutic resistance of non-small cell lung cancer. J. Transl. Med. 2024, 22, 565. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. P21(WAF1) mediates cell-cycle inhibition, relevant to cancer suppression and therapy. Cancer Res. 2016, 76, 5189–5191. [Google Scholar] [CrossRef]
- Xiang, S.; Reed, D.R.; Alexandrow, M.G. The CMG helicase and cancer: A tumor “engine” and weakness with missing mutations. Oncogene 2023, 42, 473–490. [Google Scholar] [CrossRef]
- Xiang, S.; Craig, K.C.; Luo, X.; Welch, D.L.; Ferreira, R.B.; Lawrence, H.R.; Lawrence, N.J.; Reed, D.R.; Alexandrow, M.G. Identification of ATP-competitive human CMG helicase inhibitors for cancer intervention that disrupt CMG-replisome function. Mol. Cancer Ther. 2024, 23, 1568–1585. [Google Scholar] [CrossRef]
- Sugiura, R.; Satoh, R.; Takasaki, T. ERK: A double-edged sword in cancer. ERK-dependent apoptosis as a potential therapeutic strategy for cancer. Cells 2021, 10, 2509. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.-C. ERK and cell death: Mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Timofeev, O.; Giron, P.; Lawo, S.; Pichler, M.; Noeparast, M. ERK pathway agonism for cancer therapy: Evidence, insights, and a target discovery framework. NPJ Precis. Oncol. 2024, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR project genie biopharma collaborative in cbioportal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Berger, A.H.; Brooks, A.N.; Wu, X.; Shrestha, Y.; Chouinard, C.; Piccioni, F.; Bagul, M.; Kamburov, A.; Imielinski, M.; Hogstrom, L.; et al. High-throughput phenotyping of lung cancer somatic mutations. Cancer Cell 2016, 30, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA drug approval summary: Gefitinib (ZD1839) (Iressa®) tablets. Oncologist 2003, 8, 303–306. [Google Scholar] [CrossRef]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.-Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA approval summary: Crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014, 19, e5–e11. [Google Scholar] [CrossRef]
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 2020, 12, 939–947. [Google Scholar] [CrossRef]
- Takebe, N.; McShane, L.; Conley, B. Exceptional responders—Discovering predictive biomarkers. Nat. Rev. Clin. Oncol. 2015, 12, 132–134. [Google Scholar] [CrossRef]
- Nishikawa, G.; Luo, J.; Prasad, V. A comprehensive review of exceptional responders to anticancer drugs in the biomedical literature. Eur. J. Cancer 2018, 101, 143–151. [Google Scholar] [CrossRef]
- Lim, S.M.; Cho, B.C.; Kim, S.-W.; Kang, S.Y.; Heo, D.S.; Kim, H.T.; Lee, D.H.; Kim, D.-W.; Jung, M.; Choi, J.-H.; et al. A multicenter phase II study of sorafenib in combination with erlotinib in patients with advanced non-small cell lung cancer (KCSG-0806). Lung Cancer 2016, 93, 1–8. [Google Scholar] [CrossRef]
- Manegold, C.; Dingemans, A.M.C.; Gray, J.E.; Nakagawa, K.; Nicolson, M.; Peters, S.; Reck, M.; Wu, Y.L.; Brustugun, O.T.; Crinò, L.; et al. The potential of combined immunotherapy and antiangiogenesis for the synergistic treatment of advanced NSCLC. J. Thorac. Oncol. 2017, 12, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Qiang, H.; Chang, Q.; Xu, J.; Qian, J.; Zhang, Y.; Lei, Y.; Han, B.; Chu, T. New advances in antiangiogenic combination therapeutic strategies for advanced non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; March, M.E.; Gutierrez-Uzquiza, A.; Kao, C.; Seiler, C.; Pinto, E.; Matsuoka, L.S.; Battig, M.R.; Bhoj, E.J.; Wenger, T.L.; et al. ARAF recurrent mutation causes central conducting lymphatic anomaly treatable with a MEK inhibitor. Nat. Med. 2019, 25, 1116–1122. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.; Mu, W.; Chen, X.J.; Chan, M.S.M.; Chen, Z.; Yeung, S.F.; Chan, H.H.Y.; Chow, S.T.; Ko, B.C.B.; Chan, D.W.; et al. Oncogenic Activity and Sorafenib Sensitivity of ARAF p.S214C Mutation in Lung Cancer. Cancers 2025, 17, 2246. https://doi.org/10.3390/cancers17132246
Lee C, Mu W, Chen XJ, Chan MSM, Chen Z, Yeung SF, Chan HHY, Chow ST, Ko BCB, Chan DW, et al. Oncogenic Activity and Sorafenib Sensitivity of ARAF p.S214C Mutation in Lung Cancer. Cancers. 2025; 17(13):2246. https://doi.org/10.3390/cancers17132246
Chicago/Turabian StyleLee, Carol, Weixue Mu, Xi July Chen, Mandy Sze Man Chan, Zhishan Chen, Sai Fung Yeung, Helen Hoi Yin Chan, Sin Ting Chow, Ben Chi Bun Ko, David Wai Chan, and et al. 2025. "Oncogenic Activity and Sorafenib Sensitivity of ARAF p.S214C Mutation in Lung Cancer" Cancers 17, no. 13: 2246. https://doi.org/10.3390/cancers17132246
APA StyleLee, C., Mu, W., Chen, X. J., Chan, M. S. M., Chen, Z., Yeung, S. F., Chan, H. H. Y., Chow, S. T., Ko, B. C. B., Chan, D. W., Cho, W. C., Lui, V. W. Y., & Tsui, S. K. W. (2025). Oncogenic Activity and Sorafenib Sensitivity of ARAF p.S214C Mutation in Lung Cancer. Cancers, 17(13), 2246. https://doi.org/10.3390/cancers17132246