

Regenerative Immunotherapy for Cancer: Transcription Factor Reprogramming of Tumor-Specific T Cells

{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. T Cell Differentiation and Loss of Stemness
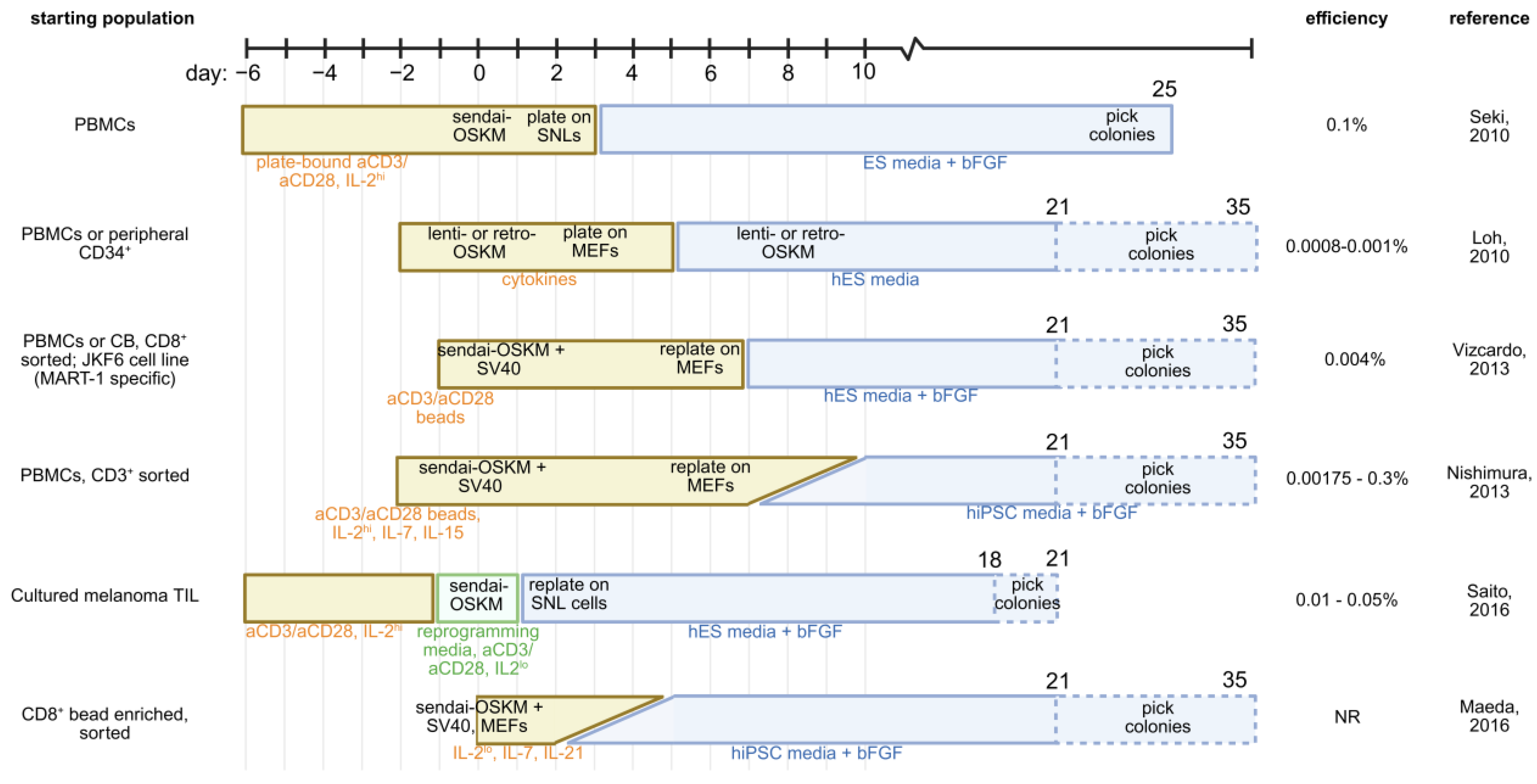
3. Early Reprogramming Strategies
4. Contemporary Techniques for T Cell Reprogramming
5. The Vision of T Cell to iPSC Reprogramming
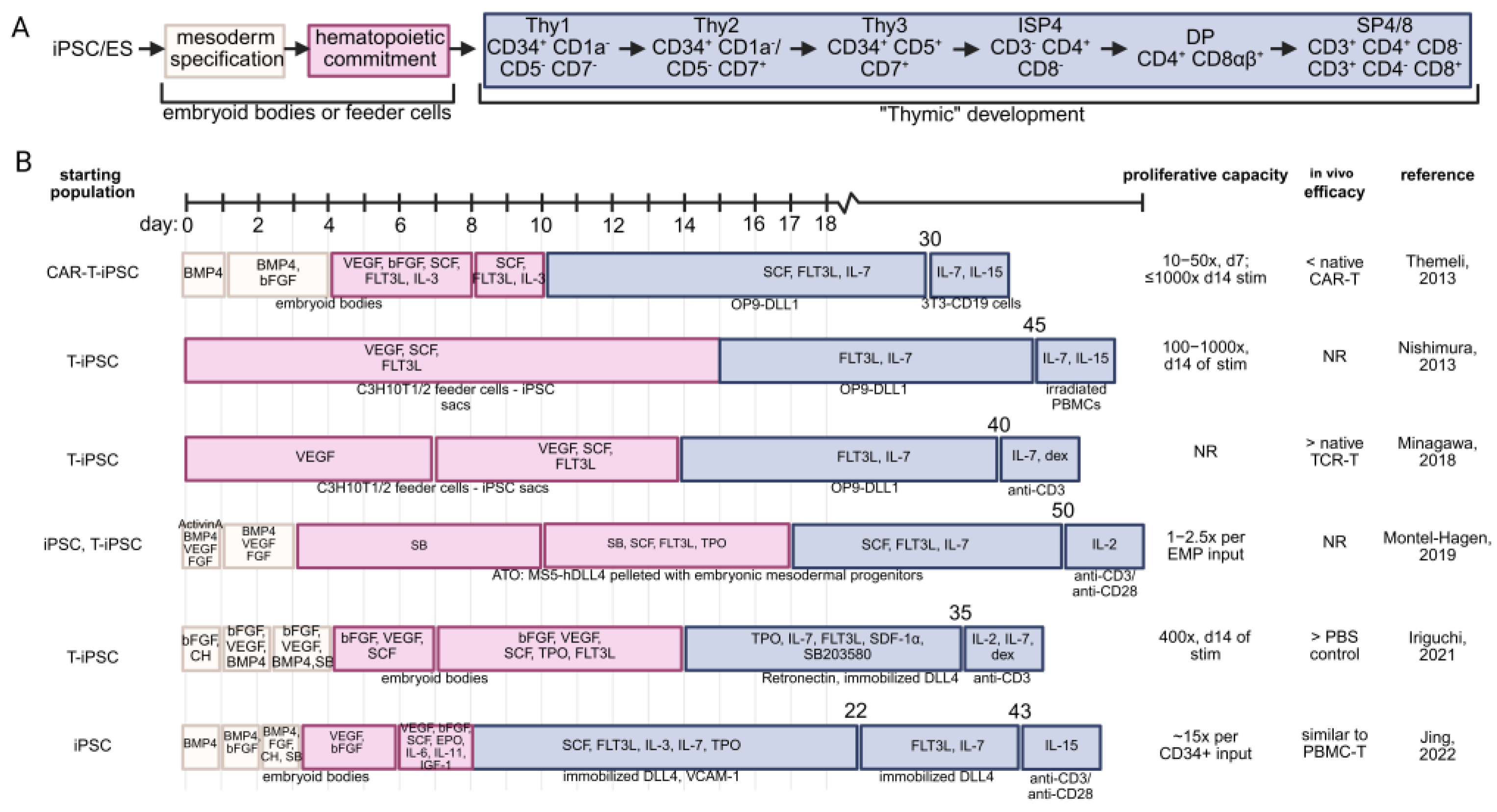
6. T Cell Reprogramming Necessitates Subsequent Differentiation and Maturation Techniques
7. Current Models and Applications for T Cell Reprogramming
7.1. Personalized Versus “Off the Shelf”
7.2. Use of Immune Lineages with Restricted Antigen Recognition
7.3. Directing Antigen Specificity Using TCR Transgenes
7.4. Directing Antigen Specificity Using Chimeric Antigen Receptors
8. Techniques for CD4 T Cell Reprogramming and Re-Maturation
9. Progress as a Clinical Therapy and Major Roadblocks to Translation
10. Future Directions
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Savy, T.; Flanders, L.; Karpanasamy, T.; Sun, M.; Gerlinger, M. Cancer evolution: From Darwin to the Extended Evolutionary Synthesis. Trends Cancer 2025, 11, 204–215. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef]
- Rückert, M.; Flohr, A.S.; Hecht, M.; Gaipl, U.S. Radiotherapy and the immune system: More than just immune suppression. Stem Cells 2021, 39, 1155–1165. [Google Scholar] [CrossRef]
- Charpentier, M.; Spada, S.; Van Nest, S.J.; Demaria, S. Radiation therapy-induced remodeling of the tumor immune microenvironment. Semin. Cancer Biol. 2022, 86, 737–747. [Google Scholar] [CrossRef]
- Gu, A.; Li, J.; Li, M.Y.; Liu, Y. Patient-derived xenograft model in cancer: Establishment and applications. MedComm (2020) 2025, 6, e70059. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Tsunoda, M.; Ogiwara, H.; Shimizu, H.; Abe, H.; Ogawa, T.; Abe, T.; Shichino, S.; Matsushima, K.; Ueha, S. Clonal Spreading of Tumor-Infiltrating T Cells Underlies the Robust Antitumor Immune Responses. Cancer Immunol. Res. 2023, 11, 847–862. [Google Scholar] [CrossRef]
- Mao, Y.; Xie, H.; Lv, M.; Yang, Q.; Shuang, Z.; Gao, F.; Li, S.; Zhu, L.; Wang, W. The landscape of objective response rate of anti-PD-1/L1 monotherapy across 31 types of cancer: A system review and novel biomarker investigating. Cancer Immunol. Immunother. 2023, 72, 2483–2498. [Google Scholar] [CrossRef]
- Osipov, A.; Lim, S.J.; Popovic, A.; Azad, N.S.; Laheru, D.A.; Zheng, L.; Jaffee, E.M.; Wang, H.; Yarchoan, M. Tumor Mutational Burden, Toxicity, and Response of Immune Checkpoint Inhibitors Targeting PD(L)1, CTLA-4, and Combination: A Meta-regression Analysis. Clin. Cancer Res. 2020, 26, 4842–4851. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell–like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Biasco, L.; Izotova, N.; Rivat, C.; Ghorashian, S.; Richardson, R.; Guvenel, A.; Hough, R.; Wynn, R.; Popova, B.; Lopes, A.; et al. Clonal expansion of T memory stem cells determines early anti-leukemic responses and long-term CAR T cell persistence in patients. Nat. Cancer 2021, 2, 629–642. [Google Scholar] [CrossRef]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef]
- Henning, A.N.; Roychoudhuri, R.; Restifo, N.P. Epigenetic control of CD8+ T cell differentiation. Nat. Rev. Immunol. 2018, 18, 340–356. [Google Scholar] [CrossRef]
- Byers, V.S.; Sercarz, E.E. The X-Y-Z scheme of immunocyte maturation. IV. The exhaustion of memory cells. J. Exp. Med. 1968, 127, 307–325. [Google Scholar] [CrossRef]
- Sercarz, E.; Coons, A.H. Specific Inhibition of Antibody Formation During Immunological Paralysis and Unresponsiveness. Nature 1959, 184, 1080–1082. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Huang, Y.J.; Ngiow, S.F.; Baxter, A.E.; Manne, S.; Park, S.L.; Wu, J.E.; Khan, O.; Giles, J.R.; Wherry, E.J. Continuous expression of TOX safeguards exhausted CD8 T cell epigenetic fate. Sci. Immunol. 2025, 10, eado3032. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef]
- Rudloff, M.W.; Zumbo, P.; Favret, N.R.; Roetman, J.J.; Detrés Román, C.R.; Erwin, M.M.; Murray, K.A.; Jonnakuti, S.T.; Dündar, F.; Betel, D.; et al. Hallmarks of CD8+ T cell dysfunction are established within hours of tumor antigen encounter before cell division. Nat. Immunol. 2023, 24, 1527–1539. [Google Scholar] [CrossRef]
- Abdel-Hakeem, M.S.; Manne, S.; Beltra, J.-C.; Stelekati, E.; Chen, Z.; Nzingha, K.; Ali, M.-A.; Johnson, J.L.; Giles, J.R.; Mathew, D.; et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat. Immunol. 2021, 22, 1008–1019. [Google Scholar] [CrossRef]
- Gurdon, J.B.; Laskey, R.A.; Reeves, O.R. The developmental capacity of nuclei transplanted from keratinized skin cells of adult frogs. J. Embryol. Exp. Morphol. 1975, 34, 93–112. [Google Scholar] [CrossRef]
- Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H. Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef]
- Wakayama, T.; Perry, A.C.; Zuccotti, M.; Johnson, K.R.; Yanagimachi, R. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 1998, 394, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.A.; Atienza, J.; Melton, D.A.; Eggan, K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science 2005, 309, 1369–1373. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; He, P.; Slukvin, I.I.; Thomson, J.A. Human embryonic stem cells reprogram myeloid precursors following cell-cell fusion. Stem Cells 2006, 24, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Hochedlinger, K.; Jaenisch, R. Monoclonal mice generated by nuclear transfer from mature B and T donor cells. Nature 2002, 415, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Wakao, H.; Ogonuki, N.; Miki, H.; Seino, K.; Nambu-Wakao, R.; Noda, S.; Miyoshi, H.; Koseki, H.; Taniguchi, M.; et al. Generation of cloned mice by direct nuclear transfer from natural killer T cells. Curr. Biol. 2005, 15, 1114–1118. [Google Scholar] [CrossRef]
- Hochedlinger, K.; Jaenisch, R. Nuclear reprogramming and pluripotency. Nature 2006, 441, 1061–1067. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Maherali, N.; Sridharan, R.; Xie, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachechko, R.; Tchieu, J.; Jaenisch, R.; et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef]
- Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007, 448, 318–324. [Google Scholar] [CrossRef]
- Loh, Y.H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef]
- Nishimura, K.; Sano, M.; Ohtaka, M.; Furuta, B.; Umemura, Y.; Nakajima, Y.; Ikehara, Y.; Kobayashi, T.; Segawa, H.; Takayasu, S.; et al. Development of defective and persistent Sendai virus vector: A unique gene delivery/expression system ideal for cell reprogramming. J. Biol. Chem. 2011, 286, 4760–4771. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Crompton, J.G.; Clever, D.; Vizcardo, R.; Rao, M.; Restifo, N.P. Reprogramming antitumor immunity. Trends Immunol. 2014, 35, 178–185. [Google Scholar] [CrossRef]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef]
- Saito, H.; Okita, K.; Fusaki, N.; Sabel, M.S.; Chang, A.E.; Ito, F. Reprogramming of Melanoma Tumor-Infiltrating Lymphocytes to Induced Pluripotent Stem Cells. Stem Cells Int. 2016, 2016, 8394960. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Islam, S.M.R.; Maeda, T.; Tamaoki, N.; Good, M.L.; Kishton, R.J.; Paria, B.C.; Yu, Z.; Bosch-Marce, M.; Bedanova, N.M.; Liu, C.; et al. Reprogramming of Tumor-reactive Tumor-infiltrating Lymphocytes to Human-induced Pluripotent Stem Cells. Cancer Res. Commun. 2023, 3, 917–932. [Google Scholar] [CrossRef] [PubMed]
- Eminli, S.; Foudi, A.; Stadtfeld, M.; Maherali, N.; Ahfeldt, T.; Mostoslavsky, G.; Hock, H.; Hochedlinger, K. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat. Genet. 2009, 41, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Churko, J.M.; Lee, J.; Ameen, M.; Gu, M.; Venkatasubramanian, M.; Diecke, S.; Sallam, K.; Im, H.; Wang, G.; Gold, J.D.; et al. Transcriptomic and epigenomic differences in human induced pluripotent stem cells generated from six reprogramming methods. Nat. Biomed. Eng. 2017, 1, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Bulliard, Y.; Andersson, B.S.; Baysal, M.A.; Damiano, J.; Tsimberidou, A.M. Reprogramming T cell differentiation and exhaustion in CAR-T cell therapy. J. Hematol. Oncol. 2023, 16, 108. [Google Scholar] [CrossRef]
- Ji, S.; Xiong, M.; Chen, H.; Liu, Y.; Zhou, L.; Hong, Y.; Wang, M.; Wang, C.; Fu, X.; Sun, X. Cellular rejuvenation: Molecular mechanisms and potential therapeutic interventions for diseases. Signal Transduct. Target. Ther. 2023, 8, 116. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef]
- Maeda, T.; Nagano, S.; Ichise, H.; Kataoka, K.; Yamada, D.; Ogawa, S.; Koseki, H.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; et al. Regeneration of CD8αβ T Cells from T-cell-Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Res. 2016, 76, 6839–6850. [Google Scholar] [CrossRef]
- Galic, Z.; Kitchen, S.G.; Kacena, A.; Subramanian, A.; Burke, B.; Cortado, R.; Zack, J.A. T lineage differentiation from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11742–11747. [Google Scholar] [CrossRef]
- Deftos, M.L.; Bevan, M.J. Notch signaling in T cell development. Curr. Opin. Immunol. 2000, 12, 166–172. [Google Scholar] [CrossRef]
- Izon, D.J.; Punt, J.A.; Xu, L.; Karnell, F.G.; Allman, D.; Myung, P.S.; Boerth, N.J.; Pui, J.C.; Koretzky, G.A.; Pear, W.S. Notch1 regulates maturation of CD4+ and CD8+ thymocytes by modulating TCR signal strength. Immunity 2001, 14, 253–264. [Google Scholar] [CrossRef]
- Schmitt, T.M.; Zúñiga-Pflücker, J.C. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 2002, 17, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, F.; Velghe, I.; Vanwalleghem, L.; De Smedt, M.; Van Coppernolle, S.; Taghon, T.; Moore, H.D.; Leclercq, G.; Langerak, A.W.; Kerre, T.; et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. J. Immunol. 2009, 182, 6879–6888. [Google Scholar] [CrossRef]
- Chang, C.W.; Lai, Y.S.; Lamb, L.S., Jr.; Townes, T.M. Broad T-cell receptor repertoire in T-lymphocytes derived from human induced pluripotent stem cells. PLoS ONE 2014, 9, e97335. [Google Scholar] [CrossRef]
- Huijskens, M.J.; Walczak, M.; Koller, N.; Briedé, J.J.; Senden-Gijsbers, B.L.; Schnijderberg, M.C.; Bos, G.M.; Germeraad, W.T. Technical advance: Ascorbic acid induces development of double-positive T cells from human hematopoietic stem cells in the absence of stromal cells. J. Leukoc. Biol. 2014, 96, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Kawana-Tachikawa, A.; Kitayama, S.; Ueda, T.; Miki, S.; Watanabe, A.; Kaneko, S. Generation of highly proliferative, rejuvenated cytotoxic T cell clones through pluripotency reprogramming for adoptive immunotherapy. Mol. Ther. 2021, 29, 3027–3041. [Google Scholar] [CrossRef]
- Mohtashami, M.; Brauer, P.M.; Zúñiga-Pflücker, J.C. Induction of Human T Cell Development In Vitro with OP9-DL4-7FS Cells Expressing Human Cytokines. Methods Mol. Biol. 2023, 2580, 249–260. [Google Scholar] [CrossRef]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat. Commun. 2021, 12, 430. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, A.; Yoshikawa, T.; Yasukawa, M.; Hotta, A.; Kunitomo, M.; Iriguchi, S.; Takiguchi, M.; Kassai, Y.; Imai, E.; Yasui, Y.; et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell 2018, 23, 850–858.e854. [Google Scholar] [CrossRef]
- van Oers, N.S.; Teh, S.J.; Garvin, A.M.; Forbush, K.A.; Perlmutter, R.M.; Teh, H.S. CD8 inhibits signal transduction through the T cell receptor in CD4-CD8- thymocytes from T cell receptor transgenic mice reconstituted with a transgenic CD8 alpha molecule. J. Immunol. 1993, 151, 777–790. [Google Scholar] [CrossRef]
- Nakayama, K.; Nakayama, K.; Negishi, I.; Kuida, K.; Louie, M.C.; Kanagawa, O.; Nakauchi, H.; Loh, D.Y. Requirement for CD8 beta chain in positive selection of CD8-lineage T cells. Science 1994, 263, 1131–1133. [Google Scholar] [CrossRef]
- Vizcardo, R.; Klemen, N.D.; Islam, S.M.R.; Gurusamy, D.; Tamaoki, N.; Yamada, D.; Koseki, H.; Kidder, B.L.; Yu, Z.; Jia, L.; et al. Generation of Tumor Antigen-Specific iPSC-Derived Thymic Emigrants Using a 3D Thymic Culture System. Cell Rep. 2018, 22, 3175–3190. [Google Scholar] [CrossRef] [PubMed]
- Seet, C.S.; He, C.; Bethune, M.T.; Li, S.; Chick, B.; Gschweng, E.H.; Zhu, Y.; Kim, K.; Kohn, D.B.; Baltimore, D.; et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 2017, 14, 521–530. [Google Scholar] [CrossRef]
- Montel-Hagen, A.; Seet, C.S.; Li, S.; Chick, B.; Zhu, Y.; Chang, P.; Tsai, S.; Sun, V.; Lopez, S.; Chen, H.C.; et al. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell 2019, 24, 376–389.e378. [Google Scholar] [CrossRef] [PubMed]
- Montel-Hagen, A.; Sun, V.; Casero, D.; Tsai, S.; Zampieri, A.; Jackson, N.; Li, S.; Lopez, S.; Zhu, Y.; Chick, B.; et al. In Vitro Recapitulation of Murine Thymopoiesis from Single Hematopoietic Stem Cells. Cell Rep. 2020, 33, 108320. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Scarfo, I.; Najia, M.A.; Lummertz da Rocha, E.; Han, A.; Sanborn, M.; Bingham, T.; Kubaczka, C.; Jha, D.K.; Falchetti, M.; et al. EZH1 repression generates mature iPSC-derived CAR T cells with enhanced antitumor activity. Cell Stem Cell 2022, 29, 1181–1196.e1186. [Google Scholar] [CrossRef]
- Jing, R.; Falchetti, M.; Han, T.; Najia, M.; Hensch, L.T.; Meader, E.; Lummertz da Rocha, E.; Kononov, M.; Wang, S.; Bingham, T.; et al. Maturation and persistence of CAR T cells derived from human pluripotent stem cells via chemical inhibition of G9a/GLP. Cell Stem Cell 2025, 32, 71–85.e75. [Google Scholar] [CrossRef]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef]
- Leidner, R.; Sanjuan Silva, N.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Peri, A.; Salomon, N.; Wolf, Y.; Kreiter, S.; Diken, M.; Samuels, Y. The landscape of T cell antigens for cancer immunotherapy. Nat. Cancer 2023, 4, 937–954. [Google Scholar] [CrossRef] [PubMed]
- de Rham, C.; Villard, J. Potential and limitation of HLA-based banking of human pluripotent stem cells for cell therapy. J. Immunol. Res. 2014, 2014, 518135. [Google Scholar] [CrossRef]
- Kitayama, S.; Zhang, R.; Liu, T.Y.; Ueda, N.; Iriguchi, S.; Yasui, Y.; Kawai, Y.; Tatsumi, M.; Hirai, N.; Mizoro, Y.; et al. Cellular Adjuvant Properties, Direct Cytotoxicity of Re-differentiated Vα24 Invariant NKT-like Cells from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 213–227. [Google Scholar] [CrossRef]
- Zeng, J.; Tang, S.Y.; Wang, S. Derivation of mimetic γδ T cells endowed with cancer recognition receptors from reprogrammed γδ T cell. PLoS ONE 2019, 14, e0216815. [Google Scholar] [CrossRef]
- Maeda, T.; Nagano, S.; Kashima, S.; Terada, K.; Agata, Y.; Ichise, H.; Ohtaka, M.; Nakanishi, M.; Fujiki, F.; Sugiyama, H.; et al. Regeneration of Tumor-Antigen-Specific Cytotoxic T Lymphocytes from iPSCs Transduced with Exogenous TCR Genes. Mol. Ther. Methods Clin. Dev. 2020, 19, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Nagano, S.; Maeda, T.; Ichise, H.; Kashima, S.; Ohtaka, M.; Nakanishi, M.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; Masuda, K.; et al. High Frequency Production of T Cell-Derived iPSC Clones Capable of Generating Potent Cytotoxic T Cells. Mol. Ther. Methods Clin. Dev. 2020, 16, 126–135. [Google Scholar] [CrossRef]
- Niizuma, K.; Nishimura, T.; Villanueva, J.; Amaya, L.; Fowler, J.L.; Isobe, T.; Nakauchi, Y.; Saavedra, B.; Xu, H.; Nakanishi, M.; et al. Development of iPSC-Derived T Cells Targeting EGFR Neoantigens in Non-Small Cell Lung Cancer. Mol. Ther. Methods Clin. Dev. 2025, 101517. [Google Scholar] [CrossRef]
- Kashima, S.; Maeda, T.; Masuda, K.; Nagano, S.; Inoue, T.; Takeda, M.; Kono, Y.; Kobayashi, T.; Saito, S.; Higuchi, T.; et al. Cytotoxic T Lymphocytes Regenerated from iPS Cells Have Therapeutic Efficacy in a Patient-Derived Xenograft Solid Tumor Model. iScience 2020, 23, 100998. [Google Scholar] [CrossRef]
- Chang, P.C.; Yuan, X.; Zampieri, A.; Towns, C.; Yoo, S.P.; Engstrom, C.; Tsai, S.; Robles, C.R.; Zhu, Y.; Lopez, S.; et al. Generation of antigen-specific mature T cells from RAG1−/−RAG2−/−B2M−/− stem cells by engineering their microenvironment. Nat. Biomed. Eng. 2024, 8, 461–478. [Google Scholar] [CrossRef]
- Minguet, S.; Maus, M.V.; Schamel, W.W. From TCR fundamental research to innovative chimeric antigen receptor design. Nat. Rev. Immunol. 2025, 25, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Ohoka, Y.; Kuwata, T.; Tozawa, Y.; Zhao, Y.; Mukai, M.; Motegi, Y.; Suzuki, R.; Yokoyama, M.; Iwata, M. In vitro differentiation and commitment of CD4+ CD8+ thymocytes to the CD4 lineage, without TCR engagement. Int. Immunol. 1996, 8, 297–306. [Google Scholar] [CrossRef]
- Fong, H.; Mendel, M.; Jascur, J.; Najmi, L.; Kim, K.; Lew, G.; Garimalla, S.; Schock, S.; Hu, J.; Villegas, A.G.; et al. A Serum- and Feeder-Free System to Generate CD4 and Regulatory T Cells from Human iPSCs. Stem Cells 2025, 43, sxaf001. [Google Scholar] [CrossRef]
- Wildt, K.F.; Sun, G.; Grueter, B.; Fischer, M.; Zamisch, M.; Ehlers, M.; Bosselut, R. The transcription factor Zbtb7b promotes CD4 expression by antagonizing Runx-mediated activation of the CD4 silencer. J. Immunol. 2007, 179, 4405–4414. [Google Scholar] [CrossRef]
- Ishiguro, Y.; Iriguchi, S.; Asano, S.; Shinohara, T.; Shiina, S.; Arima, S.; Kassai, Y.; Sakai, Y.; Obama, K.; Kaneko, S. Lineage tracing of T cell differentiation from T-iPSC by 2D feeder-free culture and 3D organoid culture. Front. Immunol. 2023, 14, 1303713. [Google Scholar] [CrossRef]
- Hoang, D.M.; Pham, P.T.; Bach, T.Q.; Ngo, A.T.L.; Nguyen, Q.T.; Phan, T.T.K.; Nguyen, G.H.; Le, P.T.T.; Hoang, V.T.; Forsyth, N.R.; et al. Stem cell-based therapy for human diseases. Signal Transduct. Target. Ther. 2022, 7, 272. [Google Scholar] [CrossRef]
- Hosking, M.P.; Shirinbak, S.; Omilusik, K.; Chandra, S.; Kaneko, M.K.; Gentile, A.; Yamamoto, S.; Shrestha, B.; Grant, J.; Boyett, M.; et al. Preferential tumor targeting of HER2 by iPSC-derived CAR T cells engineered to overcome multiple barriers to solid tumor efficacy. Cell Stem Cell 2025. online ahead of print. [Google Scholar] [CrossRef]
- Baulu, E.; Gardet, C.; Chuvin, N.; Depil, S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci. Adv. 2023, 9, eadf3700. [Google Scholar] [CrossRef]
- Kirkeby, A.; Main, H.; Carpenter, M. Pluripotent stem-cell-derived therapies in clinical trial: A 2025 update. Cell Stem Cell 2025, 32, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Moy, A.B.; Kamath, A.; Ternes, S.; Kamath, J. The Challenges to Advancing Induced Pluripotent Stem Cell-Dependent Cell Replacement Therapy. Med. Res. Arch. 2023, 11, 4784. [Google Scholar] [CrossRef] [PubMed]
- Llorente, I.L.; Hatanaka, E.A.; Meadow, M.E.; Xie, Y.; Lowry, W.E.; Carmichael, S.T. Reliable generation of glial enriched progenitors from human fibroblast-derived iPSCs. Stem Cell Res. 2021, 55, 102458. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef]
- Rubio-Casillas, A.; Cowley, D.; Raszek, M.; Uversky, V.N.; Redwan, E.M. Review: N1-methyl-pseudouridine (m1Ψ): Friend or foe of cancer? Int. J. Biol. Macromol. 2024, 267, 131427. [Google Scholar] [CrossRef]
- Mulroney, T.E.; Pöyry, T.; Yam-Puc, J.C.; Rust, M.; Harvey, R.F.; Kalmar, L.; Horner, E.; Booth, L.; Ferreira, A.P.; Stoneley, M.; et al. N1-methylpseudouridylation of mRNA causes +1 ribosomal frameshifting. Nature 2024, 625, 189–194. [Google Scholar] [CrossRef]
- Morais, P.; Adachi, H.; Yu, Y.T. The Critical Contribution of Pseudouridine to mRNA COVID-19 Vaccines. Front. Cell Dev. Biol. 2021, 9, 789427. [Google Scholar] [CrossRef]
- Pepini, T.; Pulichino, A.M.; Carsillo, T.; Carlson, A.L.; Sari-Sarraf, F.; Ramsauer, K.; Debasitis, J.C.; Maruggi, G.; Otten, G.R.; Geall, A.J.; et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J. Immunol. 2017, 198, 4012–4024. [Google Scholar] [CrossRef]
- Wang, J.; Sun, S.; Deng, H. Chemical reprogramming for cell fate manipulation: Methods, applications, and perspectives. Cell Stem Cell 2023, 30, 1130–1147. [Google Scholar] [CrossRef]
- Ando, M.; Nishimura, T.; Yamazaki, S.; Yamaguchi, T.; Kawana-Tachikawa, A.; Hayama, T.; Nakauchi, Y.; Ando, J.; Ota, Y.; Takahashi, S.; et al. A Safeguard System for Induced Pluripotent Stem Cell-Derived Rejuvenated T Cell Therapy. Stem Cell Rep. 2015, 5, 597–608. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCaw, T.R.; Restifo, N.P.; Plath, K.; Crompton, J.G. Regenerative Immunotherapy for Cancer: Transcription Factor Reprogramming of Tumor-Specific T Cells. Cancers 2025, 17, 2225. https://doi.org/10.3390/cancers17132225
McCaw TR, Restifo NP, Plath K, Crompton JG. Regenerative Immunotherapy for Cancer: Transcription Factor Reprogramming of Tumor-Specific T Cells. Cancers. 2025; 17(13):2225. https://doi.org/10.3390/cancers17132225
Chicago/Turabian StyleMcCaw, Tyler R., Nicholas P. Restifo, Kathrin Plath, and Joseph G. Crompton. 2025. "Regenerative Immunotherapy for Cancer: Transcription Factor Reprogramming of Tumor-Specific T Cells" Cancers 17, no. 13: 2225. https://doi.org/10.3390/cancers17132225
APA StyleMcCaw, T. R., Restifo, N. P., Plath, K., & Crompton, J. G. (2025). Regenerative Immunotherapy for Cancer: Transcription Factor Reprogramming of Tumor-Specific T Cells. Cancers, 17(13), 2225. https://doi.org/10.3390/cancers17132225