

Advancing Antibody–Drug Conjugates: Precision Oncology Approaches for Breast and Pancreatic Cancers


Simple Summary
Abstract
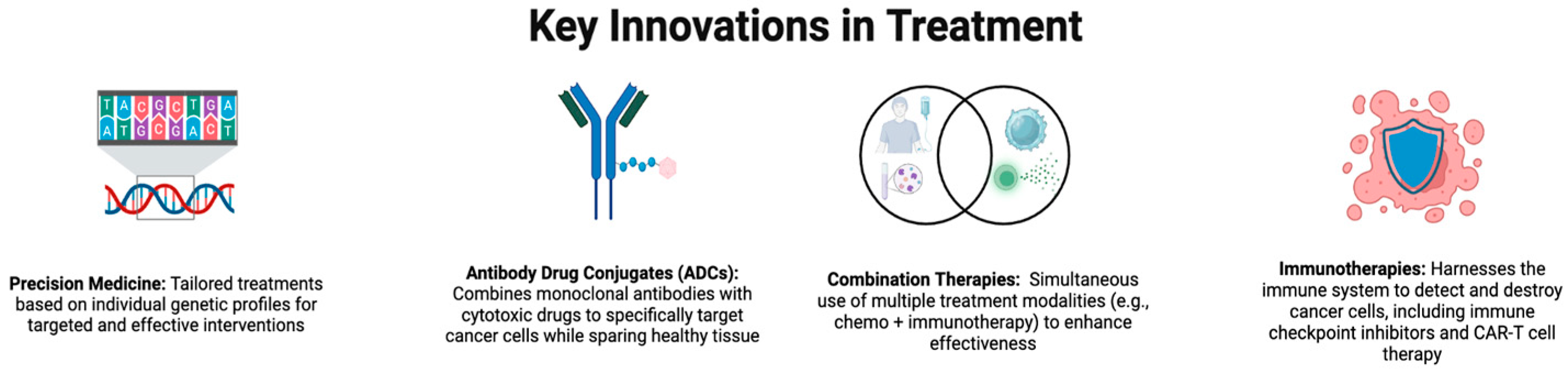
1. Introduction
1.1. Overview of Cancer
1.2. Pancreatic Cancer
1.3. Breast Cancer
1.4. Antibodies and Cytotoxic Drugs in Cancer Therapy
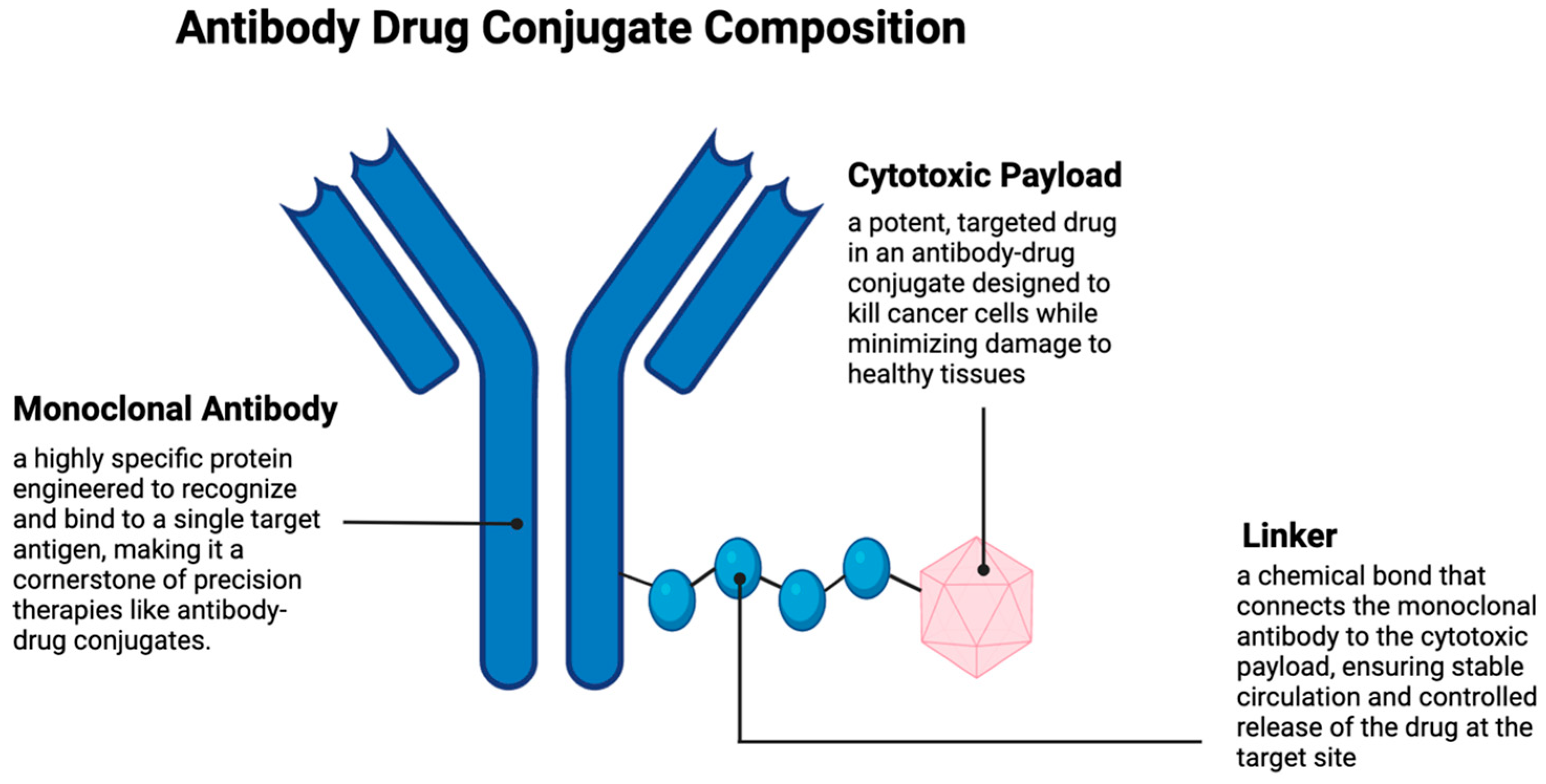
2. Antibody–Drug Conjugates
- Be highly expressed on cancer cells with minimal presence in normal tissue, such as membrane proteins like HER2 (for breast cancer) and CD30 (for certain lymphomas), or TROP-2 (for breast/pancreatic cancer), which have demonstrated clinical success;
- Be internalized efficiently after the ADC binds, ensuring the cytotoxic drug is delivered into the cancer cell [47].
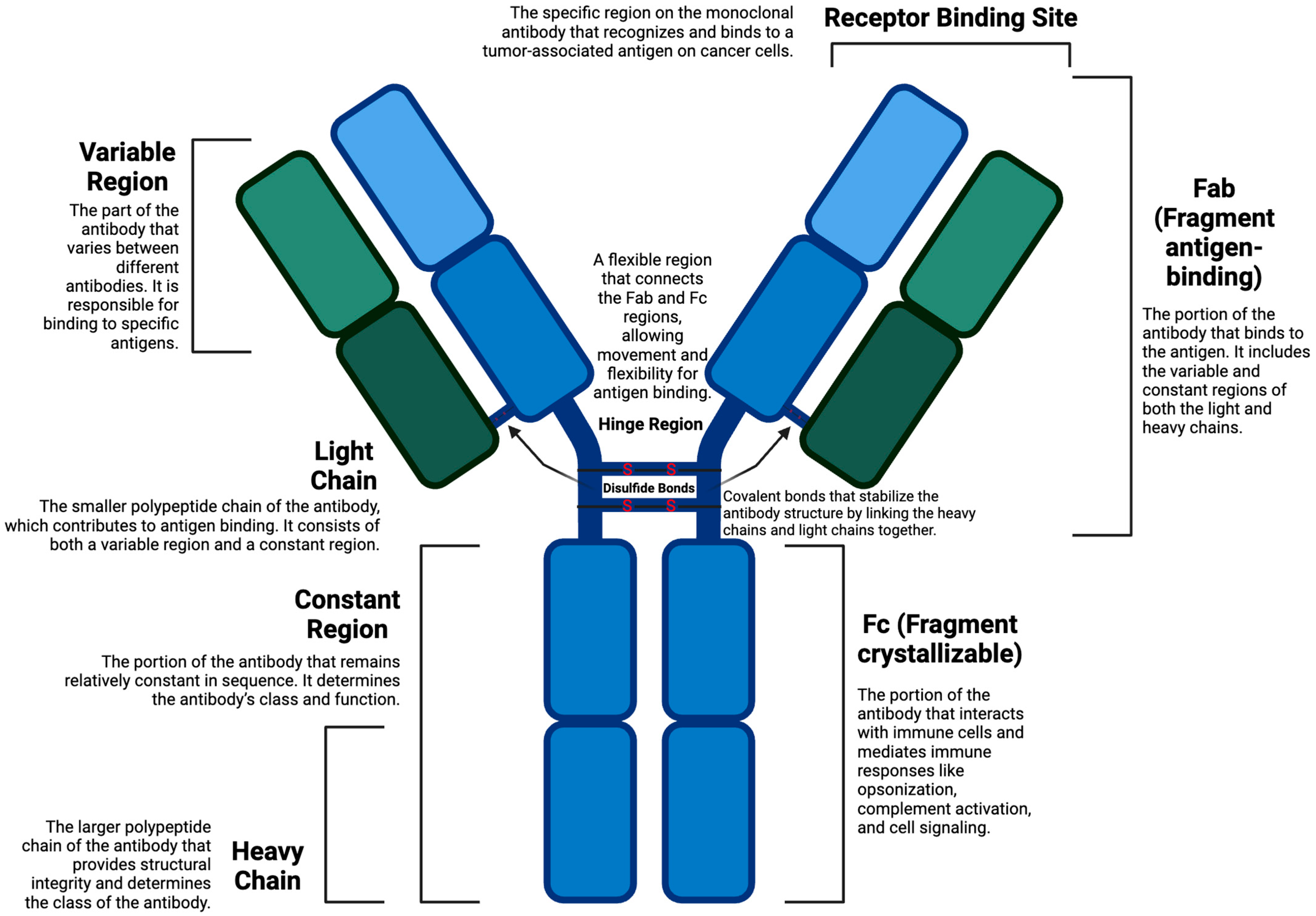
2.1. Antibodies
2.2. Cytotoxic Payload
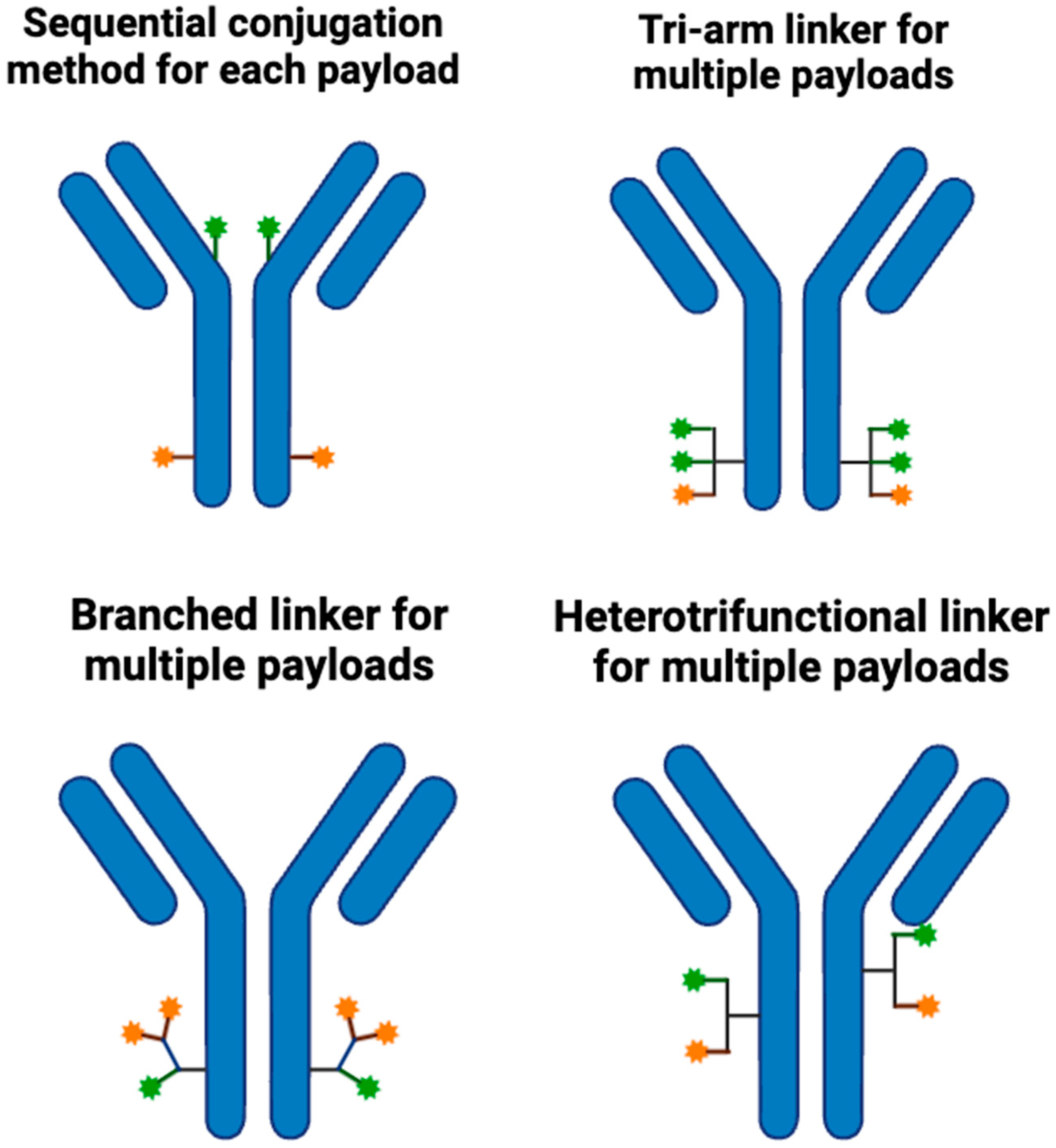
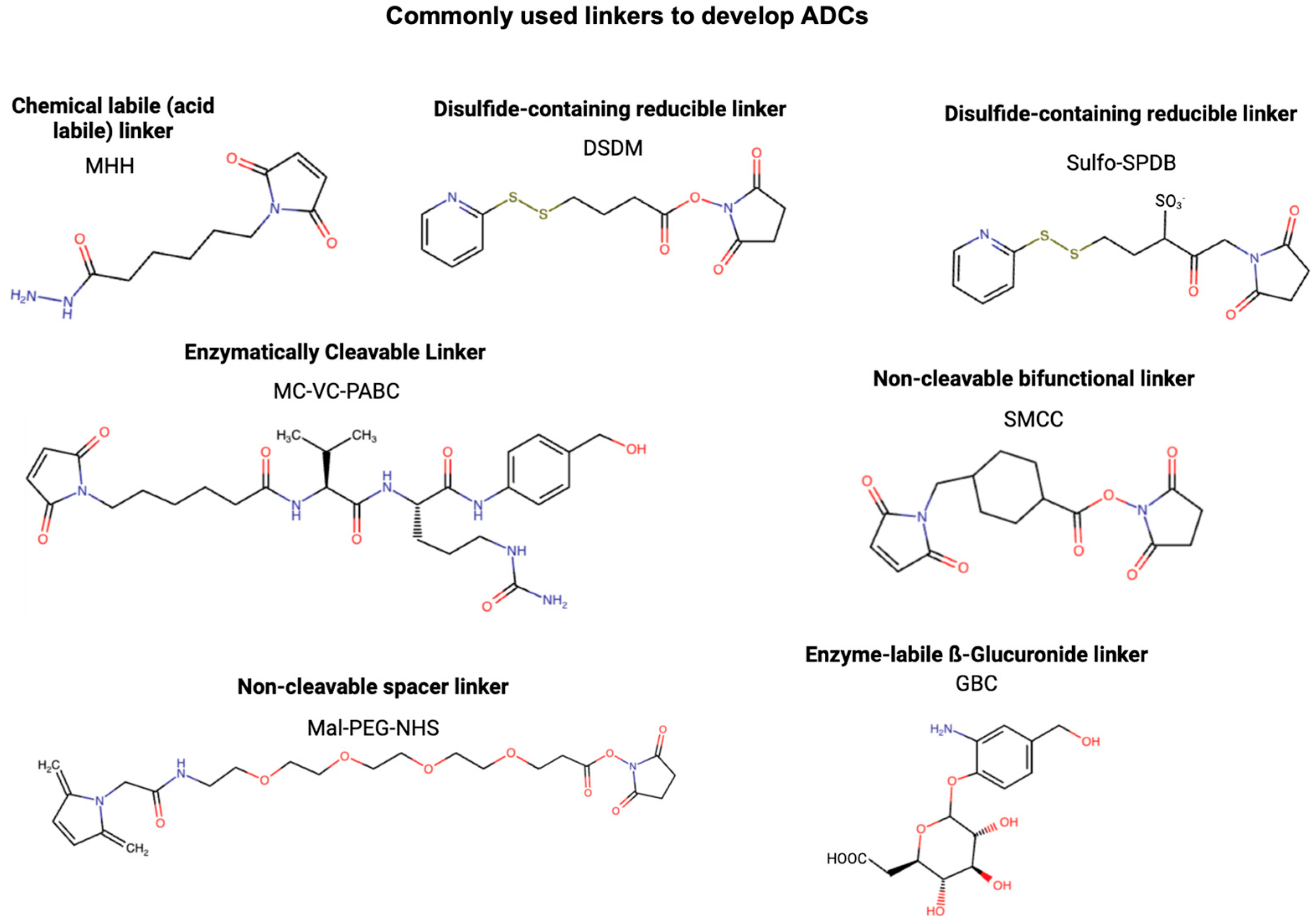
2.3. Linker
2.4. Internalization of Antibodies
3. Preclinical Studies and Lab Research
4. Overview of Selected ADCs
4.1. Comparison of Outcomes in Pancreatic vs. Breast Cancer
4.2. Sequencing Antibody–Drug Conjugates (ADCs) in Cancer Therapy
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5T4 | Oncofetal Antigen 5T4 |
| ADC | Antibody–Drug Conjugate |
| ADCC | Antibody-Dependent Cellular Cytotoxicity |
| ADAM9 | A Disintegrin and Metalloproteinase Domain 9 |
| ADEX | Aberrantly Differentiated Endocrine Exocrine |
| AML | Acute Myeloid Leukemia |
| B7-H4 | B7 Homolog 4 |
| CA9 | Carbonic Anhydrase IX |
| CanAg | Cancer Antigen |
| CEA | Carcinoembryonic Antigen |
| CD142 | Cluster of Differentiation 142 |
| CD276 (B7H3) | Cluster of Differentiation 276, B7 Homolog 3 |
| CD71 | Cluster of Differentiation 71 |
| CDC | Complement-Dependent Cytotoxicity |
| CFC1B | Cripto, CFC1B |
| CHO | Chinese Hamster Ovary |
| DAR | Drug-to-Antibody Ratio |
| ED-B | Extra-Domain B |
| EFNA4 | Ephrin A4 |
| EGFR | Epidermal Growth Factor Receptor |
| ER | Estrogen Receptor |
| EphA2 | Ephrin Type-A Receptor 2 |
| FDA | Food and Drug Administration |
| FRα | Folate Receptor Alpha |
| FXYD5 | FXYD Domain-Containing Ion Transport Regulator 5 |
| GCC | Guanylate Cyclase C |
| GPNMB | Glycoprotein Non-Metastatic Melanoma Protein B |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HER3 | Human Epidermal Growth Factor Receptor 3 |
| IL | Interleukin |
| ILD | Interstitial Lung Disease |
| LAMP1 | Lysosomal-Associated Membrane Protein 1 |
| Le(y) Antigen | Lewis Y Antigen |
| Ly75 | Lymphocyte Antigen 75 |
| mAb | Monoclonal Antibody |
| MUC1 | Mucin 1 |
| MSLN | Mesothelin |
| MT1-MMP; MMP14 | Membrane-Type 1 Matrix Metalloproteinase |
| Nectin 4 | Nectin Cell Adhesion Molecule 4 |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PRLR | Prolactin Recepto |
| PTK7; CCK4 | Protein Tyrosine Kinase 7, Cholecystokinin Tetrapeptide |
| ROR1 | Receptor Tyrosine Kinase-Like Orphan Receptor 1 |
| ROR2 | Receptor Tyrosine Kinase-Like Orphan Receptor 2 |
| SAIL | Secreted and Acidic Lysine-Rich Protein |
| SORT1 | Sortilin |
| TAG-72 | Tumor-Associated Glycoprotein 72 |
| TNBC | Triple-Negative Breast Cancer |
| Trop-2 | Trophoblast Antigen 2 |
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.B.; Anderson, R.N. The leading causes of death in the US for 2020. JAMA 2021, 325, 1829–1830. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Zavala, V.A.; Bracci, P.M.; Carethers, J.M.; Carvajal-Carmona, L.; Coggins, N.B.; Cruz-Correa, M.R.; Davis, M.; de Smith, A.J.; Dutil, J.; Figueiredo, J.C. Cancer health disparities in racial/ethnic minorities in the United States. Br. J. Cancer 2021, 124, 315–332. [Google Scholar] [CrossRef]
- Winn, R.; Winkfield, K.; Mitchell, E. Addressing disparities in cancer care and incorporating precision medicine for minority populations. J. Natl. Med. Assoc. 2023, 115, S2–S7. [Google Scholar] [CrossRef]
- Eissa, M.A.; Lerner, L.; Abdelfatah, E.; Shankar, N.; Canner, J.K.; Hasan, N.M.; Yaghoobi, V.; Huang, B.; Kerner, Z.; Takaesu, F. Promoter methylation of ADAMTS1 and BNC1 as potential biomarkers for early detection of pancreatic cancer in blood. Clin. Epigenet. 2019, 11, 59. [Google Scholar] [CrossRef]
- Cai, J.; Chen, H.; Lu, M.; Zhang, Y.; Lu, B.; You, L.; Zhang, T.; Dai, M.; Zhao, Y. Advances in the epidemiology of pancreatic cancer: Trends, risk factors, screening, and prognosis. Cancer Lett. 2021, 520, 1–11. [Google Scholar] [CrossRef]
- Gheorghe, G.; Bungau, S.; Ilie, M.; Behl, T.; Vesa, C.M.; Brisc, C.; Bacalbasa, N.; Turi, V.; Costache, R.S.; Diaconu, C.C. Early diagnosis of pancreatic cancer: The key for survival. Diagnostics 2020, 10, 869. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Khan, A.A.; Liu, X.; Yan, X.; Tahir, M.; Ali, S.; Huang, H. An overview of genetic mutations and epigenetic signatures in the course of pancreatic cancer progression. Cancer Metastasis Rev. 2021, 40, 245–272. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Gordon, E.M.; Anderson, W.F.; Parekh, D. Gene therapy for primary and metastatic pancreatic cancer with intraperitoneal retroviral vector bearing the wild-type p53 gene. Surgery 1998, 124, 143–151. [Google Scholar] [CrossRef]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Eshleman, J.R. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lim, D.H.; Jang, K.-T.; Lim, T.; Lee, J.; Choi, Y.-L.; Jang, H.-L.; Yi, J.H.; Baek, K.K.; Park, S.H. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol. Cancer Ther. 2011, 10, 1993–1999. [Google Scholar] [CrossRef]
- Tirosh, A.; Kebebew, E. Genetic and epigenetic alterations in pancreatic neuroendocrine tumors. J. Gastrointest. Oncol. 2020, 11, 567. [Google Scholar] [CrossRef] [PubMed]
- Gujarathi, R.; Abou Azar, S.; Tobias, J.; Polite, B.N.; Setia, N.; Feinberg, N.; Appelbaum, D.E.; Keutgen, X.M.; Liao, C.-Y. MEN1/DAXX/ATRX mutations enhance progression-free survival in gastroenteropancreatic neuroendocrine tumors treated with peptide receptor radionuclide therapy. Endocr.-Relat. Cancer 2024, 31, e240065. [Google Scholar] [CrossRef]
- Testini, M.; Gurrado, A.; Lissidini, G.; Venezia, P.; Greco, L.; Piccinni, G. Management of mucinous cystic neoplasms of the pancreas. World J. Gastroenterol. 2010, 16, 5682. [Google Scholar] [CrossRef]
- Thompson, E.D.; Wood, L.D. Pancreatic neoplasms with acinar differentiation: A review of pathologic and molecular features. Arch. Pathol. Lab. Med. 2020, 144, 808–815. [Google Scholar] [CrossRef]
- Das, K.K.; Early, D. Pancreatic cancer screening. Curr. Treat. Options Gastroenterol. 2017, 15, 562–575. [Google Scholar] [CrossRef]
- Gutiérrez, M.L.; Muñoz-Bellvís, L.; Orfao, A. Genomic heterogeneity of pancreatic ductal adenocarcinoma and its clinical impact. Cancers 2021, 13, 4451. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.A.; Gallinger, S. Pancreatic cancer evolution and heterogeneity: Integrating omics and clinical data. Nat. Rev. Cancer 2022, 22, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mavros, M.N.; Moris, D.; Karanicolas, P.J.; Katz, M.H.; O’Reilly, E.M.; Pawlik, T.M. Clinical trials of systemic chemotherapy for resectable pancreatic cancer: A review. JAMA Surg. 2021, 156, 663–672. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef]
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk factors and preventions of breast cancer. Int. J. Biol. Sci. 2017, 13, 1387. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.-C.; Yu, B.-L.; Horng, C.-F.; Tsai, S.Y.; Chen, C.-M.; Chu, N.-M.; Tsou, M.-H.; Lin, C.K.; Shih, L.-S.; Liu, M.-C. Long-term survival and stage I breast cancer subtypes. J. Cancer Res. Pract. 2016, 3, 1–8. [Google Scholar] [CrossRef]
- Rej, R.K.; Roy, J.; Allu, S.R. Therapies for the treatment of advanced/metastatic estrogen receptor-positive breast cancer: Current situation and future directions. Cancers 2024, 16, 552. [Google Scholar] [CrossRef]
- Iacopetta, D.; Ceramella, J.; Baldino, N.; Sinicropi, M.S.; Catalano, A. Targeting breast cancer: An overlook on current strategies. Int. J. Mol. Sci. 2023, 24, 3643. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef]
- Farah, R.A.; Clinchy, B.; Herrera, L.; Vitetta, E.S. The development of monoclonal antibodies for the therapy of cancer. Crit. Rev. ™ Eukaryot. Gene Expr. 1998, 8, 321–356. [Google Scholar] [CrossRef] [PubMed]
- Augustyniak, D.; Majkowska-Skrobek, G.; Roszkowiak, J.; Dorotkiewicz-Jach, A. Defensive and offensive cross-reactive antibodies elicited by pathogens: The good, the bad and the ugly. Curr. Med. Chem. 2017, 24, 4002–4037. [Google Scholar] [CrossRef]
- Cassinelli, G.; Zuco, V.; Gatti, L.; Lanzi, C.; Zaffaroni, N.; Colombo, D.; Perego, P. Targeting the Akt kinase to modulate survival, invasiveness and drug resistance of cancer cells. Curr. Med. Chem. 2013, 20, 1923–1945. [Google Scholar] [CrossRef] [PubMed]
- Raghani, N.R.; Chorawala, M.R.; Mahadik, M.; Patel, R.B.; Prajapati, B.G.; Parekh, P.S. Revolutionizing cancer treatment: Comprehensive insights into immunotherapeutic strategies. Med. Oncol. 2024, 41, 51. [Google Scholar] [CrossRef]
- Nurgalieva, Z.; Liu, C.-C.; Du, X.L. Chemotherapy use and risk of bone marrow suppression in a large population-based cohort of older women with breast and ovarian cancer. Med. Oncol. 2011, 28, 716–725. [Google Scholar] [CrossRef]
- Akbarali, H.I.; Muchhala, K.H.; Jessup, D.K.; Cheatham, S. Chemotherapy induced gastrointestinal toxicities. Adv. Cancer Res. 2022, 155, 131–166. [Google Scholar]
- Windebank, A.J.; Grisold, W. Chemotherapy-induced neuropathy. J. Peripher. Nerv. Syst. 2008, 13, 27–46. [Google Scholar] [CrossRef]
- Xue, X.; Liang, X.-J. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin. J. Cancer 2012, 31, 100. [Google Scholar] [CrossRef]
- Salehan, M.; Morse, H. DNA damage repair and tolerance: A role in chemotherapeutic drug resistance. Br. J. Biomed. Sci. 2013, 70, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Izzo, D.; Ascione, L.; Guidi, L.; Marsicano, R.M.; Koukoutzeli, C.; Trapani, D.; Curigliano, G. Innovative payloads for ADCs in cancer treatment: Moving beyond the selective delivery of chemotherapy. Ther. Adv. Med. Oncol. 2025, 17, 17588359241309461. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC toxicity and strategies to increase ADC tolerability. Cancers 2023, 15, 713. [Google Scholar] [CrossRef] [PubMed]
- Pysz, I.; Jackson, P.J.; Thurston, D.E. Introduction to antibody–drug conjugates (ADCs). 2019. Available online: https://aacrjournals.org/mct/article/3/6/661/234887/Inactivation-of-the-mitotic-checkpoint-as-a (accessed on 16 April 2025).
- Giugliano, F.; Corti, C.; Tarantino, P.; Michelini, F.; Curigliano, G. Bystander effect of antibody–drug conjugates: Fact or fiction? Curr. Oncol. Rep. 2022, 24, 809–817. [Google Scholar] [CrossRef]
- Tang, H.; Liu, Y.; Yu, Z.; Sun, M.; Lin, L.; Liu, W.; Han, Q.; Wei, M.; Jin, Y. The analysis of key factors related to ADCs structural design. Front. Pharmacol. 2019, 10, 373. [Google Scholar] [CrossRef]
- Abdollahpour-Alitappeh, M.; Lotfinia, M.; Gharibi, T.; Mardaneh, J.; Farhadihosseinabadi, B.; Larki, P.; Faghfourian, B.; Sepehr, K.S.; Abbaszadeh-Goudarzi, K.; Abbaszadeh-Goudarzi, G. Antibody–drug conjugates (ADCs) for cancer therapy: Strategies, challenges, and successes. J. Cell. Physiol. 2019, 234, 5628–5642. [Google Scholar] [CrossRef]
- Adams, G.P.; Weiner, L.M. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005, 23, 1147–1157. [Google Scholar] [CrossRef]
- Parit, S.; Manchare, A.; Gholap, A.D.; Mundhe, P.; Hatvate, N.; Rojekar, S.; Patravale, V. Antibody-drug conjugates: A promising breakthrough in cancer therapy. Int. J. Pharm. 2024, 659, 124211. [Google Scholar] [CrossRef]
- Tashima, T. Delivery of Drugs into Cancer Cells Using Antibody–Drug Conjugates Based on Receptor-Mediated Endocytosis and the Enhanced Permeability and Retention Effect. Antibodies 2022, 11, 78. [Google Scholar] [CrossRef]
- Ponziani, S.; Di Vittorio, G.; Pitari, G.; Cimini, A.M.; Ardini, M.; Gentile, R.; Iacobelli, S.; Sala, G.; Capone, E.; Flavell, D.J. Antibody-drug conjugates: The new frontier of chemotherapy. Int. J. Mol. Sci. 2020, 21, 5510. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Román, V.R.G.; Murray, J.C.; Weiner, L.M. Antibody-dependent cellular cytotoxicity (ADCC). In Antibody Fc; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1–27. [Google Scholar]
- Gancz, D.; Fishelson, Z. Cancer resistance to complement-dependent cytotoxicity (CDC): Problem-oriented research and development. Mol. Immunol. 2009, 46, 2794–2800. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, M.C.; Minute, L.; Rodriguez, I.; Garasa, S.; Perez-Ruiz, E.; Inogés, S.; Melero, I.; Berraondo, P. Antibody-dependent cell cytotoxicity: Immunotherapy strategies enhancing effector NK cells. Immunol. Cell Biol. 2017, 95, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Lowman, H.B. Antibody humanization and affinity maturation using phage display. In Phage Display in Biotechnology and Drug Discovery; CRC Press: Boca Raton, FL, USA, 2005; pp. 513–548. [Google Scholar]
- Kamakura, D.; Asano, R.; Yasunaga, M. T cell bispecific antibodies: An antibody-based delivery system for inducing antitumor immunity. Pharmaceuticals 2021, 14, 1172. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, J. Progresses of T-cell-engaging bispecific antibodies in treatment of solid tumors. Int. Immunopharmacol. 2024, 138, 112609. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, Y.; Wang, F. Trispecific antibodies for cancer immunotherapy. Immunology 2023, 169, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Wu, D. Advances in therapeutic Fc engineering–modulation of IgG-associated effector functions and serum half-life. Front. Immunol. 2016, 7, 580. [Google Scholar] [CrossRef]
- Segués, A.; Huang, S.; Sijts, A.; Berraondo, P.; Zaiss, D.M. Opportunities and challenges of bi-specific antibodies. Int. Rev. Cell Mol. Biol. 2022, 369, 45–70. [Google Scholar]
- Ye, X.; Yu, Y.; Zheng, X.; Ma, H. Clinical immunotherapy in pancreatic cancer. Cancer Immunol. Immunother. 2024, 73, 64. [Google Scholar] [CrossRef]
- Kiem, D.; Ocker, M.; Greil, R.; Neureiter, D.; Melchardt, T. Enhancing anti-CD274 (PD-L1) targeting through combinatorial immunotherapy with bispecific antibodies and fusion proteins: From preclinical to phase II clinical trials. Expert Opin. Investig. Drugs 2024, 33, 229–242. [Google Scholar] [CrossRef]
- Lum, L.G.; Thakur, A.; Choi, M.; Deol, A.; Kondadasula, V.; Schalk, D.; Fields, K.; Dufrense, M.; Philip, P.; Dyson, G. Clinical and immune responses to anti-CD3 x anti-EGFR bispecific antibody armed activated T cells (EGFR BATs) in pancreatic cancer patients. Oncoimmunology 2020, 9, 1773201. [Google Scholar] [CrossRef]
- Vahidi, S.; Touchaei, A.Z.; Samadani, A.A. IL-15 as a key regulator in NK cell-mediated immunotherapy for cancer: From bench to bedside. Int. Immunopharmacol. 2024, 133, 112156. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wang, Z.; Wang, Y. Bispecific antibody drug conjugates: Making 1+ 1> 2. Acta Pharm. Sin. B 2024, 14, 1965–1986. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, D.; Cai, L.; Yao, H.; Yan, M.; Wang, X.; Shen, W.; Du, Y.; Pang, H.; Lai, X. First-in-human HER2-targeted bispecific antibody KN026 for the treatment of patients with HER2-positive metastatic breast cancer: Results from a phase I study. Clin. Cancer Res. 2022, 28, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Beishenaliev, A.; Loke, Y.L.; Goh, S.J.; Geo, H.N.; Mugila, M.; Misran, M.; Chung, L.Y.; Kiew, L.V.; Roffler, S.; Teo, Y.Y. Bispecific antibodies for targeted delivery of anti-cancer therapeutic agents: A review. J. Control. Release 2023, 359, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, Y.; Peer, C.J.; Landsman, R.; Skorupan, N.; Cao, L.; Alewine, C. Low serum mesothelin in pancreatic cancer patients results from retention of shed mesothelin in the tumor microenvironment. Transl. Oncol. 2022, 21, 101440. [Google Scholar] [CrossRef]
- Faust, J.R.; Hamill, D.; Kolb, E.A.; Gopalakrishnapillai, A.; Barwe, S.P. Mesothelin: An immunotherapeutic target beyond solid tumors. Cancers 2022, 14, 1550. [Google Scholar] [CrossRef]
- Duivelshof, B.L.; Jiskoot, W.; Beck, A.; Veuthey, J.-L.; Guillarme, D.; D’Atri, V. Glycosylation of biosimilars: Recent advances in analytical characterization and clinical implications. Anal. Chim. Acta 2019, 1089, 1–18. [Google Scholar] [CrossRef]
- Reusch, D.; Tejada, M.L. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology 2015, 25, 1325–1334. [Google Scholar] [CrossRef]
- Hayes, J.M.; Cosgrave, E.F.; Struwe, W.B.; Wormald, M.; Davey, G.P.; Jefferis, R.; Rudd, P.M. Glycosylation and Fc receptors. In Fc Receptors; Springer: Berlin/Heidelberg, Germany, 2014; pp. 165–199. [Google Scholar]
- Wang, L.-X.; Tong, X.; Li, C.; Giddens, J.P.; Li, T. Glycoengineering of antibodies for modulating functions. Annu. Rev. Biochem. 2019, 88, 433–459. [Google Scholar] [CrossRef]
- Ivanova, A.; Falcioni, F. Challenges and opportunities for the large-scale chemoenzymatic glycoengineering of therapeutic N-glycosylated monoclonal antibodies. Front. Catal. 2022, 1, 810779. [Google Scholar] [CrossRef]
- Koehn, F.E. Natural product cytotoxins as payloads for antibody drug conjugates. In Natural Products and Cancer Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2012; pp. 97–119. [Google Scholar]
- Lin, K.; Tibbitts, J. Pharmacokinetic considerations for antibody drug conjugates. Pharm. Res. 2012, 29, 2354–2366. [Google Scholar] [CrossRef]
- Kostova, V.; Désos, P.; Starck, J.-B.; Kotschy, A. The chemistry behind ADCs. Pharmaceuticals 2021, 14, 442. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sineh Sepehr, K.; Khatami, M.; Bagheri, N. Potential drugs used in the antibody–drug conjugate (ADC) architecture for cancer therapy. J. Cell. Physiol. 2020, 235, 31–64. [Google Scholar] [CrossRef]
- Weber, G.F.; Weber, G.F. DNA damaging drugs. Mol. Ther. Cancer 2015, 9–112. [Google Scholar] [CrossRef]
- Wang, X.; Gigant, B.; Zheng, X.; Chen, Q. Microtubule-targeting agents for cancer treatment: Seven binding sites and three strategies. MedComm–Oncol. 2023, 2, e46. [Google Scholar] [CrossRef]
- Fu, Y.; Ho, M. DNA damaging agent-based antibody-drug conjugates for cancer therapy. Antib. Ther. 2018, 1, 43–53. [Google Scholar] [CrossRef]
- Gandullo-Sánchez, L.; Ocaña, A.; Pandiella, A. Generation of antibody-drug conjugate resistant models. Cancers 2021, 13, 4631. [Google Scholar] [CrossRef]
- Yu, S.-F.; Zheng, B.; Go, M.; Lau, J.; Spencer, S.; Raab, H.; Soriano, R.; Jhunjhunwala, S.; Cohen, R.; Caruso, M. A novel anti-CD22 anthracycline-based antibody–drug conjugate (ADC) that overcomes resistance to auristatin-based ADCs. Clin. Cancer Res. 2015, 21, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Bahot, A.; Sekar, G.; Bansode, M.; Khunteta, K.; Sonar, P.V.; Hebale, A.; Salokhe, V.; Sinha, B.K. Understanding cancer’s defense against topoisomerase-active drugs: A comprehensive review. Cancers 2024, 16, 680. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, H.; Gou, L.; Li, W.; Wang, Y. Antibody–drug conjugates: Recent advances in payloads. Acta Pharm. Sin. B 2023, 13, 4025–4059. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Lin, X.; Gao, X.; Khan, R.U.; Liao, J.-Y.; Du, S.; Ge, J.; Zeng, S.; Yao, S.Q. The dawn of a new era: Targeting the “undruggables” with antibody-based therapeutics. Chem. Rev. 2023, 123, 7782–7853. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Bai, H.; Peng, B.; Fang, B.; Baell, J.; Li, L.; Huang, W.; Voelcker, N.H. Stimulus-cleavable chemistry in the field of controlled drug delivery. Chem. Soc. Rev. 2021, 50, 4872–4931. [Google Scholar] [CrossRef] [PubMed]
- Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An assurance for controlled delivery of antibody-drug conjugate. Pharmaceutics 2022, 14, 396. [Google Scholar] [CrossRef]
- Leyton, J.V. The endosomal-lysosomal system in ADC design and cancer therapy. Expert Opin. Biol. Ther. 2023, 23, 1067–1076. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Bargh, J. The Development of Sulfatase-Cleavable Linkers for Antibody-Drug Conjugates. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2021. [Google Scholar]
- Ashman, N.; Bargh, J.D.; Spring, D.R. Non-internalising antibody–drug conjugates. Chem. Soc. Rev. 2022, 51, 9182–9202. [Google Scholar] [CrossRef]
- Slastnikova, T.A.; Ulasov, A.; Rosenkranz, A.; Sobolev, A. Targeted intracellular delivery of antibodies: The state of the art. Front. Pharmacol. 2018, 9, 1208. [Google Scholar] [CrossRef]
- Chalouni, C.; Doll, S. Fate of antibody-drug conjugates in cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 20. [Google Scholar] [CrossRef]
- Yang, C.; He, B.; Zhang, H.; Wang, X.; Zhang, Q.; Dai, W. IgG Fc affinity ligands and their applications in antibody-involved drug delivery: A brief review. Pharmaceutics 2023, 15, 187. [Google Scholar] [CrossRef]
- Brandsma, A.M.; Jacobino, S.R.; Meyer, S.; ten Broeke, T.; Leusen, J.H. Fc receptor inside-out signaling and possible impact on antibody therapy. Immunol. Rev. 2015, 268, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Koenderman, L. Inside-out control of Fc-receptors. Front. Immunol. 2019, 10, 544. [Google Scholar] [CrossRef]
- Presta, L.G. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv. Drug Deliv. Rev. 2006, 58, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J. Treatment of HER2-overexpressing breast cancer. Ann. Oncol. 2010, 21, vii36–vii40. [Google Scholar] [CrossRef]
- Damelin, M.; Zhong, W.; Myers, J.; Sapra, P. Evolving strategies for target selection for antibody-drug conjugates. Pharm. Res. 2015, 32, 3494–3507. [Google Scholar] [CrossRef]
- Sassoon, I.; Blanc, V. Antibody–drug conjugate (ADC) clinical pipeline: A review. In Antibody-Drug Conjugates; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–27. [Google Scholar]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Hegg, R.; Chung, W.-P.; Im, S.-A.; Jacot, W.; Ganju, V.; Chiu, J.W.Y.; Xu, B.; Hamilton, E.; Madhusudan, S. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: Updated results from DESTINY-Breast03, a randomised, open-label, phase 3 trial. Lancet 2023, 401, 105–117. [Google Scholar] [CrossRef]
- Liu, F.; Li, Y.; Yang, D.; Tang, L.; Yang, Q.; Jiang, M.; Tian, L.; An, J. Meta-analysis of the clinical efficacy and safety of T-DM1 in the treatment of HER2-positive breast cancer. Indian J. Cancer 2024, 61, 146–155. [Google Scholar] [CrossRef]
- Chiu, J.W.Y.; Lee, S.C.; Ho, J.C.-m.; Park, Y.H.; Chao, T.-C.; Kim, S.-B.; Lim, E.; Lin, C.-H.; Loi, S.; Low, S.Y. Clinical Guidance on the monitoring and management of Trastuzumab Deruxtecan (T-DXd)-Related adverse events: Insights from an Asia-Pacific Multidisciplinary Panel. Drug Saf. 2023, 46, 927–949. [Google Scholar] [CrossRef]
- Sasso, J.M.; Tenchov, R.; Bird, R.; Iyer, K.A.; Ralhan, K.; Rodriguez, Y.; Zhou, Q.A. The evolving landscape of antibody–drug conjugates: In depth analysis of recent research progress. Bioconjug. Chem. 2023, 34, 1951–2000. [Google Scholar] [CrossRef]
- Breier, A.; Barancík, M.; Sulová, Z.; Uhrík, B. P-glycoprotein-implications of metabolism of neoplastic cells and cancer therapy. Curr. Cancer Drug Targets 2005, 5, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Pasini, L.; Ulivi, P. Liquid biopsy for the detection of resistance mechanisms in NSCLC: Comparison of different blood biomarkers. J. Clin. Med. 2019, 8, 998. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutational Percentage | Location |
|---|---|---|
| SMAD4 | 19–50% | Chromosome #18 p21 |
| CDKN2A | 49–98% | Chromosome #9 p21 |
| TP53 | 20–75% | Chromosome #17 p13.1 |
| KRAS | 70–95% | Chromosome #12 p12.1 |
| Histological Subtype | Incidence (%) | Genetic Mutations |
|---|---|---|
| Pancreatic Ductal Adenocarcinoma | ~80% | KRAS, TP53, CDKN2A, SMAD4 |
| Pancreatic Neuroendocrine Tumors | ~5% | MEN1, ATRX, DAXX |
| Acinar Cell Carcinoma | Rare | Unknown |
| Cystic Pancreatic Tumors | Rare | Pre-malignant markers |
| Tumor Stage | PDAC % | PNET % |
|---|---|---|
| Stage IA | 14% | 61% |
| Stage IB | 12% | 61% |
| Stage II | 7% | 52% |
| Stage III | 3% | 41% |
| ER | PR | HER2 | Subtype | Incidence | Targeted Treatment | 5, 10-Year Relapse-Free Survival |
|---|---|---|---|---|---|---|
| + | + | − | ER + luminal A | 50–60% | Endocrine | 95.6%, 89.5% |
| + | + | +/− | ER + luminal B | 10–15% | Chemotherapy Endocrine + Anti-HER2 | 95.6%, 89.5% |
| − | − | + | HER2-Enriched | 15–20% | Chemotherapy + Anti-HER2 | 92.9%, 92.9% |
| − | − | − | Basal-like/TNBC | 15–20% | Chemotherapy | 93.0%, 91.1% |
| Category | Mechanism | Examples |
|---|---|---|
| Microtubule Inhibitors | These agents disrupt microtubule polymerization, essential structures for cell division. | Examples include MMAE and DM1, commonly used in ADCs like brentuximab vedotin and trastuzumab emtansine. |
| DNA Damaging Agents | These agents work by damaging the cancer cell DNA, preventing it from replicating. | Common examples include PBDs and calicheamicins, which cause DNA strand breaks leading to apoptosis. |
| ADC | Linker | Payload |
|---|---|---|
| AbGn-110 | Proprietary linker | Biobetter cytotoxic payload |
| ADCT-502 | A cathepsin B-cleavable valine–alanine linker | Tesirine, a clinically validated, potent pyrrolobenzodiazepine (PBD-based) dimer toxin (SG3249). |
| ARX788 HER2 ADC; ARX788 | A non-natural amino acid linker para-acetyl-phenylalanine (pAcF) | Monomethyl Auristatin F (MMAF) |
| BAT8001 | A novel uncleavable linker | A maytansine derivative |
| BB-1701 | REsidue-SPEcific Conjugation | Eribulin |
| DHES0815A; RG6148 | Disulfide linker | DNA minor groove crosslinking agent pyrrolo[2,1-c][1,4]benzodiazepine monoamide (PBD-MA) |
| Disitamab Vedotin; RC-48 | A cleavable mc-val-cit-PABC-type linker. | Monomethyl Auristatin E (MMAE) |
| LCB14-0110; Herceptin-LC-LBG-MMAF | Proprietary linker | Monomethyl auristatin F (MMAF) |
| MEDI4276 | Site-specific conjugation on mc-Lys-MMETA to 2 engineered cysteine residues on the heavy chain via a maleimidocaproyl linker. | MMETA, a Tubulysin Payload, also known as AZ13599185. |
| MI130004 | Linker containing a maleimide group to enable conjugation to Cys residues. | PM050489 |
| MM-302 | PEG–DSPE | Liposomal doxorubicin |
| PF-06804103; Anti-NG-HER2 ADC | Cleavable valine–citrulline linker | Anti-Trop2 Aur0101 |
| TAA013 | Lysine–SMCC | DM1 (Maytansine) |
| Trastuzumab deruxtecan; DS-8201; DS-8201a; ENHERTU | A tetrapeptide linker, Gly–Phe–Leu–Gly (GFLG) | DXd |
| Trastuzumab duocarmazine; SYD985; Trastuzumab vc-seco-DUBA | A cleavable linker N-[2-(2 maleimidoethoxy)ethoxycarbonyl]-L-valyl-L-citrullinyl-p-aminobenzyloxycarbonyl-N-[2-(2-hydroxyethoxy)ethyl]-N-[2-(methylamino)ethyl]carbamoyl | Duocarmycin/Seco-DUBA |
| Trastuzumab Emtansine; T-DM1; Kadcyla | Noncleavable succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) linker | Maytansine DM1, a microtubule inhibitor. |
| Trastuzumab Imbotolimod; BDC-1001 | A non-cleavable linker | A proprietary Toll-like receptor (TLR) 7/8 dual agonist |
| Trastuzumab rezetecan; SHR-A1811 | A stable and cleavable linker. | SHR9265 |
| Zanidatamab Zovodotin; ZW49 | A proprietary cleavable, 1-maleimido-3,6,9-trioxadodecan-12-oyl-valyl-citrullyl linker | A novel, proprietary, N-acyl sulfonamide auristatin cytotoxin designed to take advantage of the enhanced antibody-HER2 internalization of ZW25. |
| ADC Target | ADC | Linker | Payload |
|---|---|---|---|
| FRα | BAT8006 | Proprietary linker | A small molecule topoisomerase I inhibitor |
| Farletuzumab Ecteribulin; MORAb-202 | Cathepsin-cleavable linker | The microtubule-targeting agent (MTA), eribulin, a derivative of the macrocyclic polyether natural product halichondrin B. | |
| IMGN151 | Stable, cleavable peptide linker | Maytansinoid derivative DM21 | |
| Rinatabart Sesutecan; PRO1184; Rina-S | Proprietary linker | Exatecan payload | |
| HER3 | AMT-562 | A via valine–alanine cleavable linker and a modified self-immolative PABC spacer (T800) | Site specifically conjugated to exatecan |
| Patritumab Deruxtecan; U3-1402; HER3 ADC | Peptide cleavable linker (a tetrapeptide-based cleavable linker) | Deruxtecan, DX-8951 derivative (DXd, topoisomerase I inhibitor), a camptothecin derivative | |
| Trop-2 | BHV-1510 | Proprietary linker | Proprietary potential best-in-class Topolx, site-specifically conjugated via Enzymatic (non-cysteine) |
| RN927C | AcLys–VC–PABC; site-specific transglutaminase-mediated conjugation | A proprietary microtubule inhibitor (MTI) linker-payload, PF-06380101 | |
| Sacituzumab Tirumotecan; SKB264; MK-2870 | via site-specific conjugation and highly stable linker | A proprietary cytotoxic, belotecan-derived topoisomerase I inhibitor. |
| ADC Target | ADC | Linker |
|---|---|---|
| 5T4 | ASN-004 | Single-chain homo-dimer antibody, Fleximer linker technology; 3 Fleximer chains per antibody. |
| PF 06263507; A1-mcMMAF; Anti-5T4 monoclonal antibody | A non-cleavable maleimidocaproyl linker | |
| ADAM9 | IMGC936 | A stable tripeptide linker |
| AXL | Mecbotamab Vedotin; BA3011; CAB-Axl-ADC | A cleavable mc–val–cit–PABC-type linker on an average of 4 cysteinyl |
| Mipasetamab Uzoptirine; ADCT-601 | A cleavable (valine–alanine dipeptide as cathepsine B cleavage site) maleimide type linker | |
| B7-H4 | SGN-B7H4V | Protease-cleavable peptide linker, valine –citrulline |
| CA9 | BAY79-4620; 3ee9 | Valine–citrulline (vc) |
| CD142 | Tisotumab Vedotin; Tivdak; HuMax-TF-ADC | A cleavable mc–val–cit–PABC type linker on an average of 3–4 cysteinyl |
| XB002 ICON-2 Tissue Factor ADC | Protease cleavable valine–citrulline (vc) linker | |
| CD276 (B7H3) | Mirzotamab Clezutoclax; ABBV-155; Mirzo-C | A cleavable dipeptide (valine–alanine) solubilizing linker. |
| CFC1B (Cripto) | BIIB-015 | MCC |
| EphA2 | MEDI-547 | Stable linker maleimidocaproyl (mc) |
| ED-B | PYX-201 | A cathepsin B-cleavable linker. |
| EFNA4 | PF-06647263; anti-EFNA4-ADC | Hydrazone cleavable linker |
| GPNMB | Glembatumumab vedotin; CDX-011 | A cleavable mc–val–cit–PABC-type linker |
| LAMP1 | SAR428926 | A disulfide-containing cleavable linker N-succinimidyl-4-(2-pyridyldithio)butyrate (SPDB) |
| Le(y) antigen | SGN-15; BMS-182248; BR96-DOX | A hydrazone linker |
| Ly75/CD205 | MEN1309; OBT076 | Cleavable linker |
| Mucin 1 | SAR 566658 | SPDB |
| MT1-MMP; MMP14 | BT1718 | A hindered disulfide linker |
| Nectin 4 | Enfortumab Vedotin; Padcev; ASG-22ME; ASG-22MSE | A cleavable mc–val–cit–PABC-type linker |
| Zelenectide Pevedotin (BT8009) | A valine–citrulline cleavable linker | |
| PRLR | REGN2878-DM1; Anti-PRLR-ADC | Non-cleavable SMCC linker |
| PTK7; CCK4 | Cofetuzumab Pelidotin; PF-06647020; ABBV-647 | Cleavable valine–citrulline linker, a cleavable mc–val–cit–PABC-type linker |
| ROR1 | Cirmtuzumab Vedotin; UC-961ADC3 | Lysine-linker |
| STRO-003 | β-Glucuronidase-cleavable linkers | |
| ROR2 | Ozuriftamab Vedotin; BA3021; Anti-ROR2 ADC; CAB-ROR2-ADC | A cleavable mc–val–cit–PABC-type linker |
| SORT1 | Sudocetaxel Zendusortide; TH1902 | Cleavable linker |
| Zinc transporter LIV-1 | Ladiratuzumab vedotin; SGN-LIV1A; Anti-LIV-1 ADC | A cleavable, mc–val–cit–PABC-type linker |
| ADC Target | ADC | Linker | Payload |
|---|---|---|---|
| 5T4 | ZV05-ADC; 5T4-MMAF ADC; ZV05-mcMMAF; ZV0501 | Proprietary linker | Monomethyl Auristatin F (MMAF) |
| ADAM9 | Anti-ADAM9 ADC | Lysine-linked via a cleavable sulfo-SPDB linker | Maytansine-derived microtubule disruptor DM4 |
| Anti-ADAM9 ADC | Conjugated to engineered cysteine residues via a cleavable peptide linker | Indolinobenzodiazepine DNA-alkylating monoimine (DGN549) | |
| MGC028 | bicyclononyne carbamoyl sulfamide Val–Ala–PABC | Exatecan, a topoisomerase I inhibitor payload | |
| IMGC936 | A stable tripeptide linker. | DM21C | |
| AXL | Mecbotamab Vedotin; BA3011; CAB-Axl-ADC | A cleavable mc–val–cit–PABC-type linker on an average of 4 cysteinyl | Monomethyl auristatin E (MMAE) (Vedotin) |
| CA9 | BAY79-4620 | Valine–citrulline (vc) | Monomethyl Auristatin E (MMAE) |
| CanAg | Cantuzumab mertansine; huC242-DM1; SB-408075 | A stable thiopentanoate linker (or reducible SPP (N-succinimidyl 4-(2-pyridyldithio)) linker) | Maytansinoid antimicrotubule agent DM1|N(sup 2′)-deacetyl-N(sup 2′)-(3-sulfanylpropanoyl)maytansine |
| CD71 | AbGn-107; Ab1-18Hr1 | Cleavable linker | Tubulin inhibitor DM4 |
| CD142 | Tisotumab Vedotin; Tivdak; HuMax-TF-ADC | A cleavable mc–val–cit–PABC-type linker on an average of 3–4 cysteinyl | Microtubule disrupting agent monomethyl auristatin E (MMAE) |
| CFC1B | BIIB015 | MCC | Maytansinoid derivative, DM4 |
| Claudin 18.2 | ATG-022 | mc-vc-PABC-MMAE | Monomethyl auristatin E (MMAE) |
| CMG901 | A cleavable linker | ||
| EO-3021; SYSA1801 | A cleavable linker | ||
| IMAB362-vcMMAE | Valine–citrulline linker | ||
| SOT102; SO-N102 | Site Specific; non-cleavable Amide/Peptide Linker | PNU-159682, an anthracycline derivative. | |
| TQB2103 | An enzymatically cleavable linker | A small-molecule toxin | |
| EGFR and HER3 | Izalontamab Brengitecan; BL-B01D1 | A cathepsin B cleavable linker | A topoisomerase I inhibitor agent (Ed-04) |
| EphA2 | MEDI-547 | Stable linker maleimidocaproyl (mc) | Auristatin MMAF |
| FXYD5 | EDC1; DYS-ADC | Proprietary linker | CEN-106 |
| GCC | Indusatumab Vedotin; MLN-0264; TAK-264 | A cleavable mc–val–cit–PABC-type linker. | Monomethyl auristatin E (MMAE) |
| HER2 | ARX788 HER2 ADC | A non-natural amino acid linker para-acetyl-phenylalanine (pAcF) | Monomethyl Auristatin F (MMAF) |
| HER3 | HER3-ADC | A cleavable valine–citrulline linker | Monomethyl auristatin E (MMAE) |
| Mucin 16 | Sofituzumab vedotin; Anti-MUC16 ADC; RG7458; DMUC5754A | A cleavable mc–val–cit–PABC-type linker | Monomethyl auristatin E (MMAE) |
| MUC1 | Clivatuzumab tetraxetan; hPAM4; hPAM4 IgG-DOTA | Conjugated, on an average of 4 to 7 lysyl, linked to the chelator by their N6. | Yttrium-90-labeled (90Y); Chelator tetraxetan (DOTA) |
| MSLN | DMOT4039A; RG7600 | Protease-cleavable peptide linker. | Monomethyl auristatin E (MMAE) |
| Nectin 4 | Enfortumab Vedotin; Padcev; ASG-22ME; ASG-22MSE | A cleavable mc–val–cit–PABC-type linker | Monomethyl Auristatin E (MMAE) |
| ROR1 | Cirmtuzumab Vedotin; UC-961ADC3 | Lysine linker | Monomethyl Auristatin E (MMAE) |
| SAIL c15orf54 | IGN786 | A maleimidocaproyl (mc) linker | Monomethyl auristatin F (MMAF) |
| TAG-72 | Satumomab Penditide; OncoScint CR/OV | DTPA as a linker for the added In-111 | Indium-111 |
| Trop-2 | RN927C | AcLys-VC-PABC | PF-06380101 (a Dolastatin 10 analogue) |
| Sacituzumab Tirumotecan; SKB264; MK-2870 | via site-specific conjugation and highly stable linker | A proprietary cytotoxic, belotecan-derived topoisomerase I inhibitor. | |
| Sacituzumab Govitecan | CL2A (pH-sensitive linker) | SN-38 (Topoisomerase I inhibitor, active metabolite of irinotecan) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yajaman, D.R.; Oh, Y.; Trevino, J.G.; Harrell, J.C. Advancing Antibody–Drug Conjugates: Precision Oncology Approaches for Breast and Pancreatic Cancers. Cancers 2025, 17, 1792. https://doi.org/10.3390/cancers17111792
Yajaman DR, Oh Y, Trevino JG, Harrell JC. Advancing Antibody–Drug Conjugates: Precision Oncology Approaches for Breast and Pancreatic Cancers. Cancers. 2025; 17(11):1792. https://doi.org/10.3390/cancers17111792
Chicago/Turabian StyleYajaman, Dhanvin R., Youngman Oh, Jose G. Trevino, and J. Chuck Harrell. 2025. "Advancing Antibody–Drug Conjugates: Precision Oncology Approaches for Breast and Pancreatic Cancers" Cancers 17, no. 11: 1792. https://doi.org/10.3390/cancers17111792
APA StyleYajaman, D. R., Oh, Y., Trevino, J. G., & Harrell, J. C. (2025). Advancing Antibody–Drug Conjugates: Precision Oncology Approaches for Breast and Pancreatic Cancers. Cancers, 17(11), 1792. https://doi.org/10.3390/cancers17111792