
Integrative Molecular and Immune Profiling in Advanced Unresectable Melanoma: Tumor Microenvironment and Peripheral PD-1+ CD4+ Effector Memory T-Cells as Potential Markers of Response to Immune Checkpoint Inhibitor Therapy

 ,
,  , , , , , ,
, , , , , ,  , , and
, , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design, Patients, and Inclusion Criteria
2.2. Tumor Sample Preparation, Library Construction, Sequencing, and Data Processing
2.3. Survival Analysis
2.4. Hierarchical Clustering Analysis (HCA)
2.5. Differential Expression Analysis (DEA)
2.6. Gene Set Enrichment Analysis (GSEA)
2.7. Immune, Stroma, and Tumor Microenvironment Signatures
2.8. Blood Sample Preparation, PBMCs Mass Cytometry Staining and Analysis
2.9. Statistical Analysis
3. Results
3.1. Patient Demographics and Clinical Characteristics
3.2. Histopathological and Tumoral Molecular Findings
3.3. Log-Rank Analysis Identifies Candidate Genes Associated with PFS
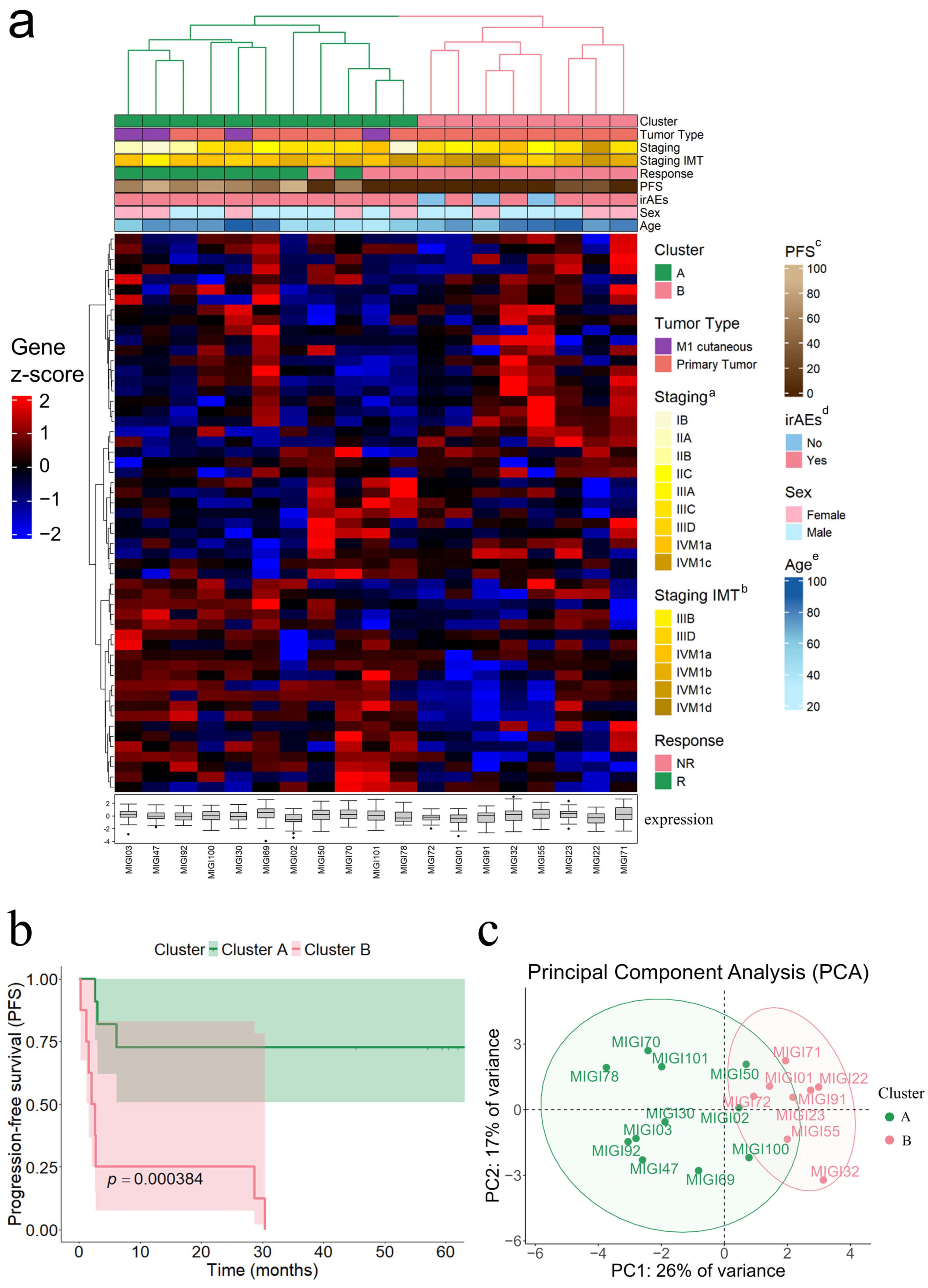
3.4. Hierarchical Clustering Analysis Identifies Molecular Subgroups Associated with PFS
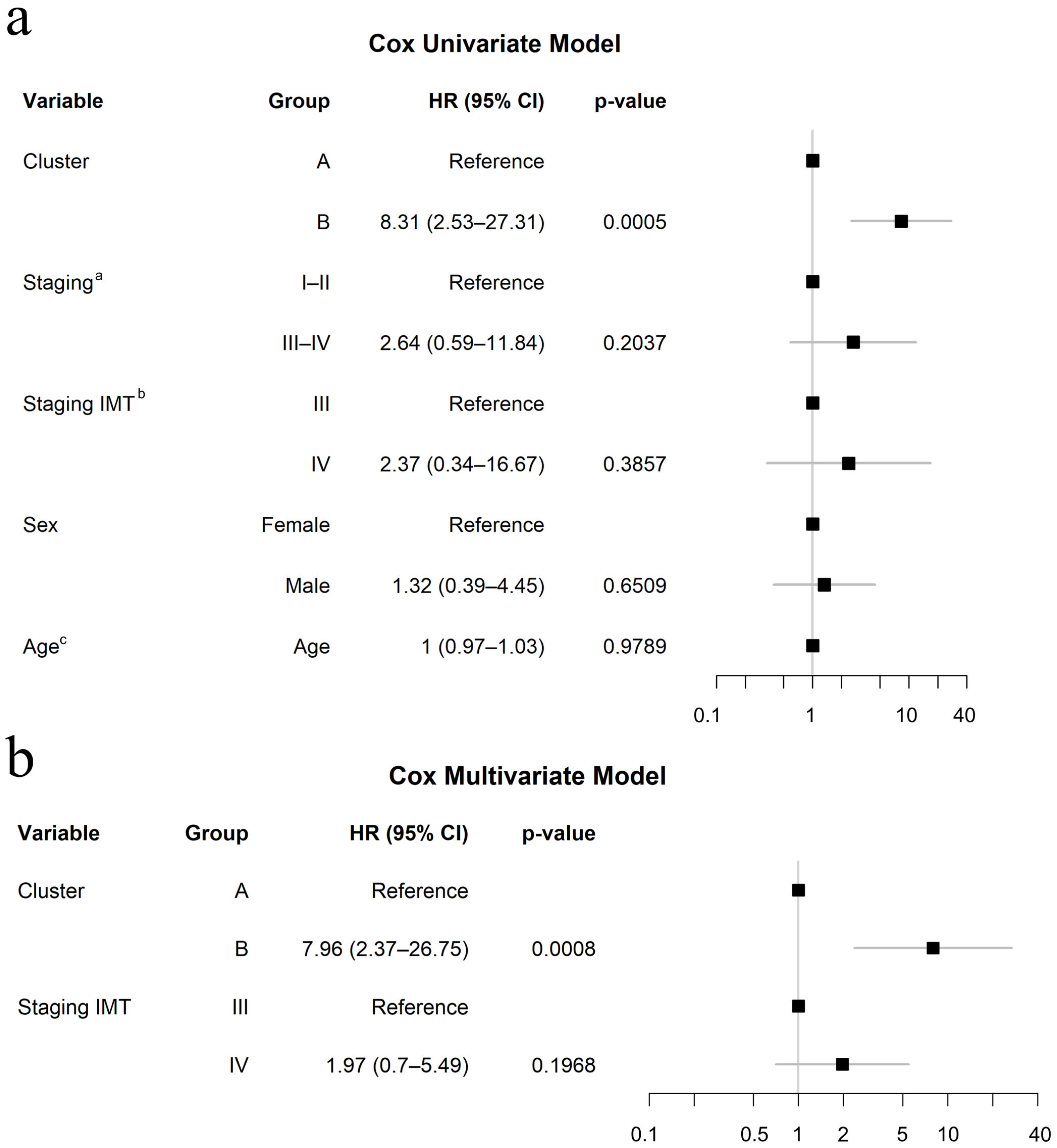
3.5. Clinical Relevance of Molecular Signature Clusters in Response to Immunotherapy
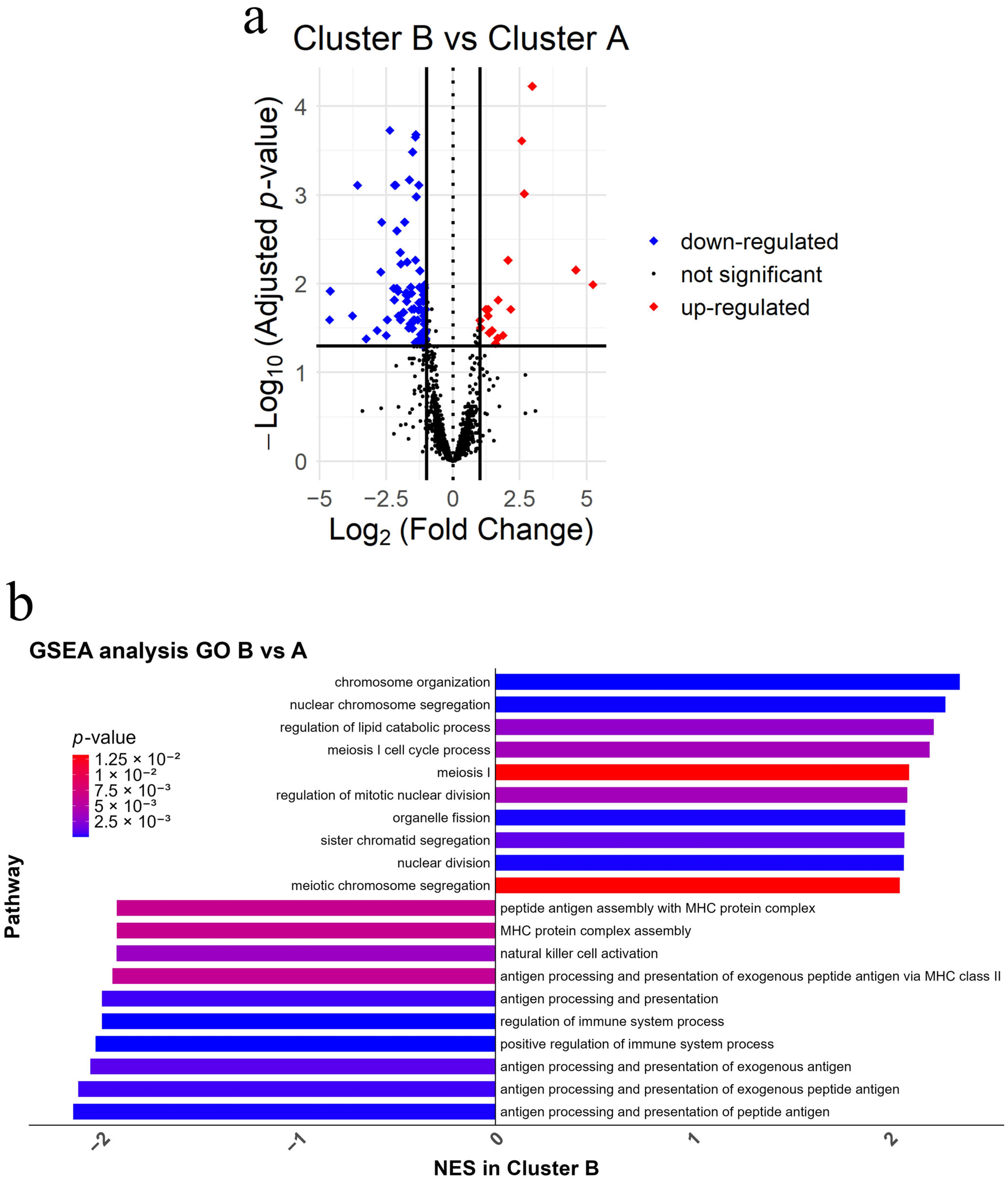
3.6. Immune Suppression and Cell Cycle Pathway Alterations Have the Capacity to Define Molecular Differences Between Clusters
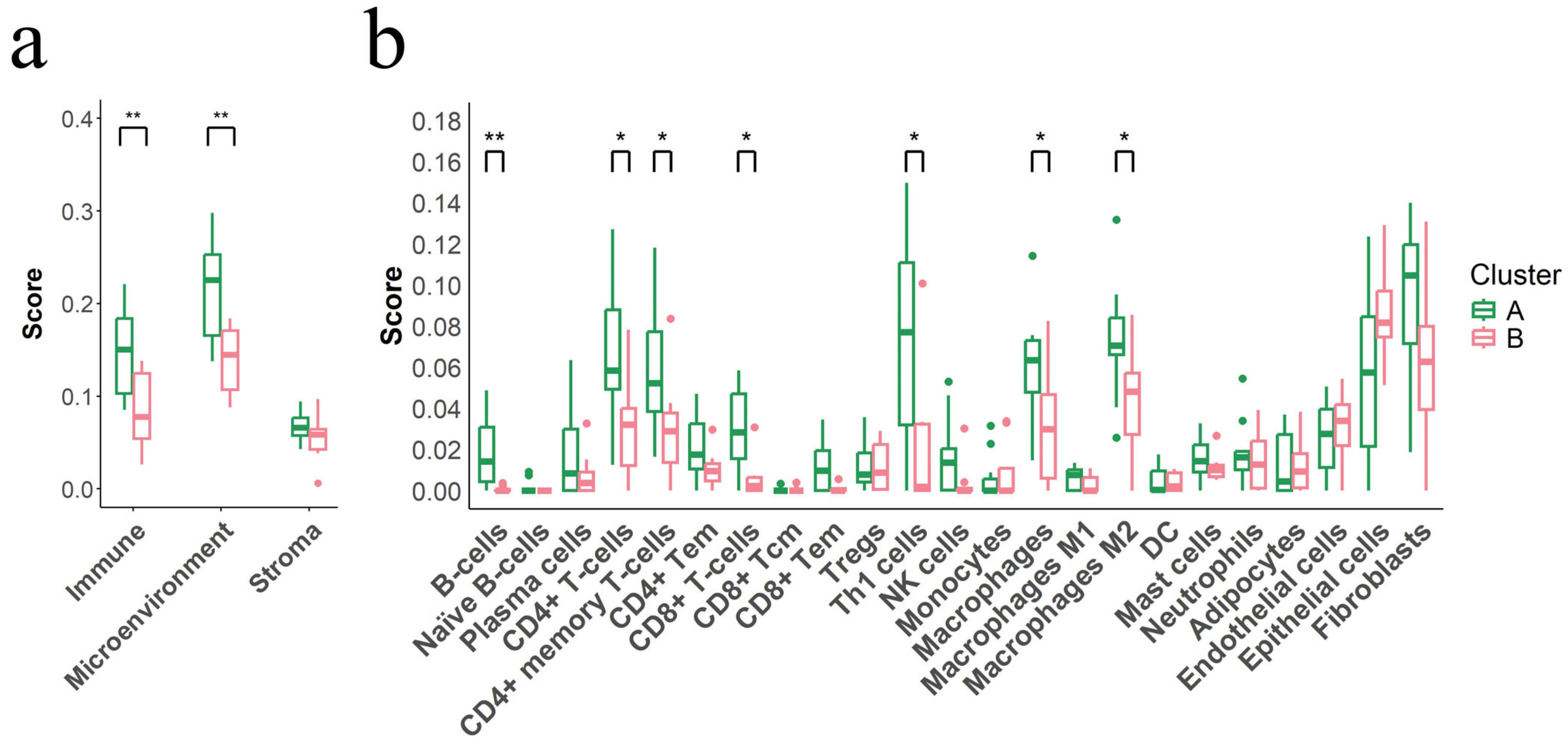
3.7. Immunosuppressive Tumor Microenvironment Defines Cluster B
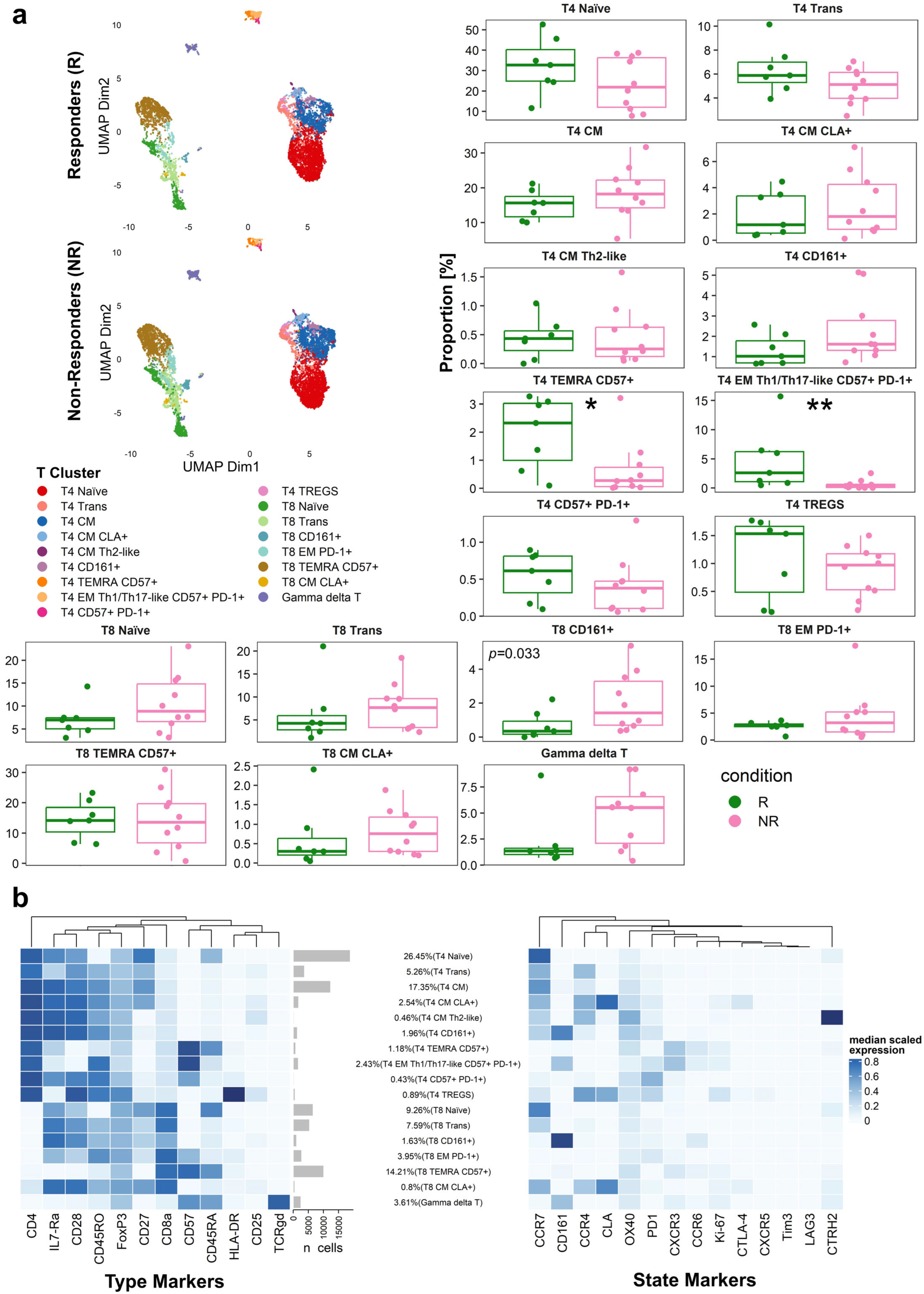
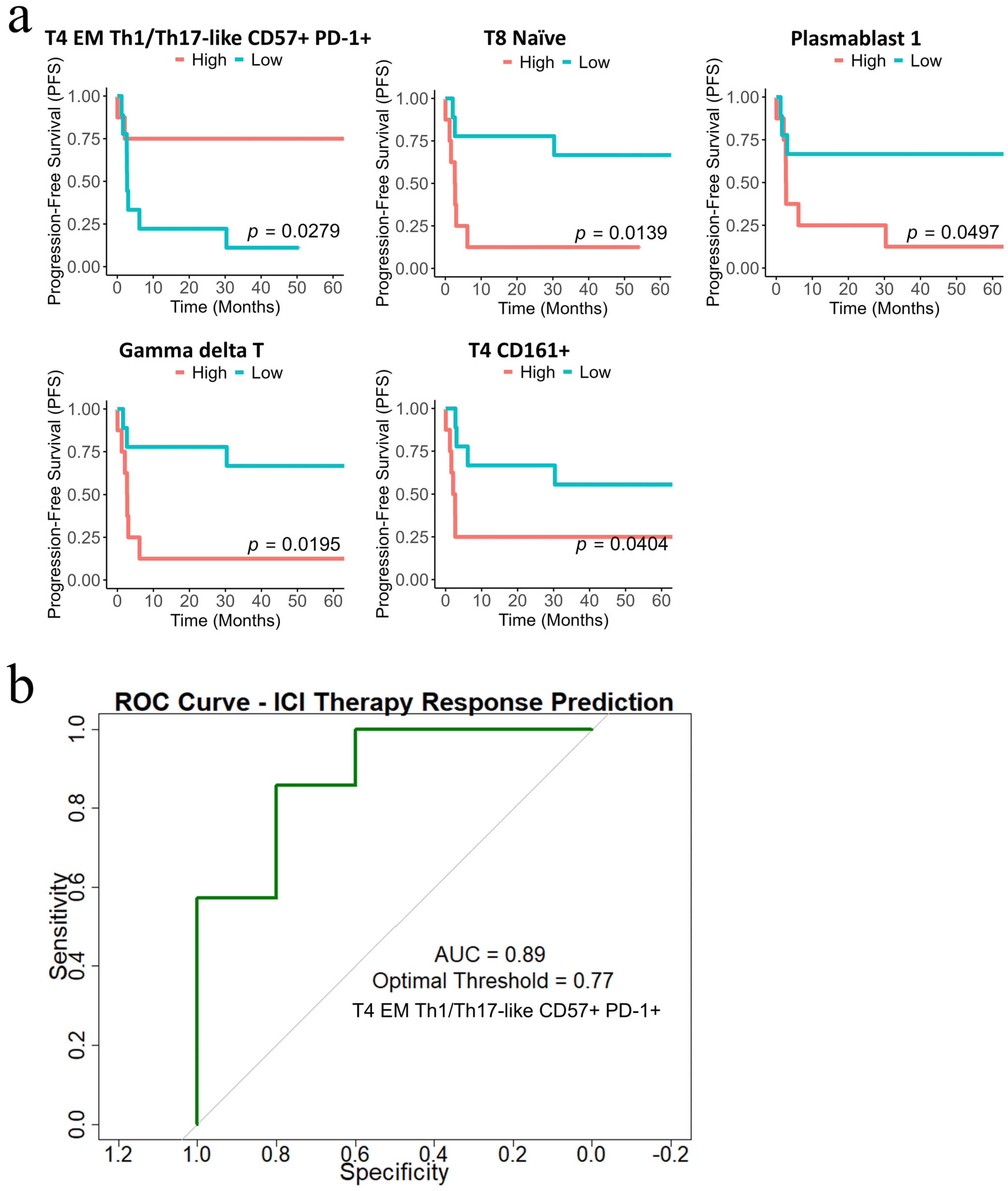
3.8. Distinct Peripheral T-Cell Subpopulations Associate with Immunotherapy Response in Patients with Advanced Melanoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACD | Acid-citrate-dextrose |
| AJCC | American Joint Committee on Cancer |
| ALM | Acral lentiginous melanoma |
| ANM | Acral nodular melanoma |
| APL | Acute promyelocytic leukemia |
| ATRA | All-trans retinoic acid |
| AUC | Area under the curve |
| BP | Biological Process |
| CI | Confidence interval |
| CPM | Counts per million |
| CR | Complete response |
| CyTOF | Cytometry by time-of-flight |
| dds | DESeqDataSet |
| DEA | Differential expression analysis |
| DEGs | Differentially expressed genes |
| EM | Effector memory |
| FCS | Flow Cytometry Standard |
| FFPE | Formalin-fixed, paraffin-embedded |
| FlowSOM | Flow Self-Organizing Map |
| GO | Gene Ontology |
| GSEA | Gene set enrichment analysis |
| H&E | Hematoxylin and eosin |
| HCA | Hierarchical clustering analysis |
| HCB | Hospital Clinic of Barcelona |
| HIV | Human immunodeficiency virus |
| HR | Hazard ratio |
| ICI | Immune-checkpoint inhibitor |
| IFN-γ | Interferon gamma |
| IMT | immunotherapy |
| IQR | Interquartile range |
| irAEs | Immune-related adverse events |
| MDIPA | Maxpar Direct Immune Profiling Assay |
| MLM | Mucosal lentiginous melanoma |
| MSigDB | Molecular signatures database |
| NM | Nodular melanoma |
| NR | Non-responder |
| padj | Adjusted p-value |
| PBMCs | Peripheral blood mononuclear cells |
| PC1 | Principal component 1 |
| PC2 | Principal component 2 |
| PCA | Principal component analysis |
| PD | Progressive disease |
| PEI | Preliminary expanded immune signature |
| PFS | Progression-free survival |
| PR | Partial response |
| QC | Quality-control |
| R | Responder |
| RAR | Retinoic acid receptor |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| ROC | Receiver-operating characteristic |
| RXR | Retinoid X receptor |
| SSM | Superficial spreading melanoma |
| TILs | Tumor-infiltrating lymphocytes |
| TME | Tumor microenvironment |
| UMAP | Uniform manifold approximation and projection |
References
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Previously Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab for Advanced Melanoma: Final Overall Survival Results of a Multicentre, Randomised, Open-Label Phase 3 Study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.J.; Weber, J.S.; et al. Association of Pembrolizumab with Tumor Response and Survival among Patients with Advanced Melanoma. JAMA J. Am. Med. Assoc. 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Andrews, M.C.; Li, G.; Graf, R.P.; Fisher, V.A.; Mitchell, J.; Aboosaiedi, A.; O’Rourke, H.; Shackleton, M.; Iddawela, M.; Oxnard, G.R.; et al. Predictive Impact of Tumor Mutational Burden on Real-World Outcomes of First-Line Immune Checkpoint Inhibition in Metastatic Melanoma. JCO Precis. Oncol. 2024, 8, e2300640. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Wong, P.F.; Wei, W.; Smithy, J.W.; Acs, B.; Toki, M.I.; Blenman, K.R.M.; Zelterman, D.; Kluger, H.M.; Rimm, D.L. Multiplex Quantitative Analysis of Tumor-Infiltrating Lymphocytes and Immunotherapy Outcome in Metastatic Melanoma. Clin. Cancer Res. 2019, 25, 2442–2449. [Google Scholar] [CrossRef]
- Shui, I.M.; Liu, X.Q.; Zhao, Q.; Kim, S.T.; Sun, Y.; Yearley, J.H.; Choudhury, T.; Webber, A.L.; Krepler, C.; Cristescu, R.; et al. Baseline and Post-Treatment Biomarkers of Resistance to Anti-PD-1 Therapy in Acral and Mucosal Melanoma: An Observational Study. J. Immunother. Cancer 2022, 10, e004879. [Google Scholar] [CrossRef] [PubMed]
- Madore, J.; Strbenac, D.; Vilain, R.; Menzies, A.M.; Yang, J.Y.H.; Thompson, J.F.; Long, G.V.; Mann, G.J.; Scolyer, R.A.; Wilmott, J.S. PD-L1 Negative Status Is Associated with Lower Mutation Burden, Differential Expression of Immune-Related Genes, and Worse Survival in Stage III Melanoma. Clin. Cancer Res. 2016, 22, 3915–3923. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Larkin, J.; Ascierto, P.A.; Hodi, F.S.; Rutkowski, P.; Sileni, V.; Hassel, J.; Lebbe, C.; Pavlick, A.C.; Wagstaff, J.; et al. PD-L1 Expression as a Biomarker for Nivolumab (NIVO) plus Ipilimumab (IPI) and NIVO Alone in Advanced Melanoma (MEL): A Pooled Analysis. Ann. Oncol. 2016, 27, vi381. [Google Scholar] [CrossRef]
- Daud, A.I.; Loo, K.; Pauli, M.L.; Sanchez-Rodriguez, R.; Sandoval, P.M.; Taravati, K.; Tsai, K.; Nosrati, A.; Nardo, L.; Alvarado, M.D.; et al. Tumor Immune Profiling Predicts Response to Anti-PD-1 Therapy in Human Melanoma. J. Clin. Investig. 2016, 126, 3447–3452. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ—Related MRNA Profile Predicts Clinical Response to PD-1 Blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Karachaliou, N.; Gonzalez-cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Gimenez-capitan, A.; Teixido, C.; Molina-vila, M.A.; Viteri, S.; Gil, M.D.L.L.; et al. Interferon Gamma, an Important Marker of Response to Immune Checkpoint Blockade in Non-Small Cell Lung Cancer and Melanoma Patients. Ther. Adv. Med. Oncol. 2018, 10, 1758834017749748. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G. V Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti-PD-1 Therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in Tumor Progression and Regression: A Review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Han, J.; Wu, M.; Liu, Z. Dysregulation in IFN-γ Signaling and Response: The Barricade to Tumor Immunotherapy. Front. Immunol. 2023, 14, 1190333. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T Cells in Cancer and Cancer Immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Macian, F. Mechanisms of Self-Inactivation in Anergic T Cells. Inmunologia 2010, 29, 20–33. [Google Scholar] [CrossRef]
- Nüssing, S.; Trapani, J.A.; Parish, I.A. Revisiting T Cell Tolerance as a Checkpoint Target for Cancer Immunotherapy. Front. Immunol. 2020, 11, 589641. [Google Scholar] [CrossRef]
- Zhou, X.; Ni, Y.; Liang, X.; Lin, Y.; An, B.; He, X.; Zhao, X. Mechanisms of Tumor Resistance to Immune Checkpoint Blockade and Combination Strategies to Overcome Resistance. Front. Immunol. 2022, 13, 915094. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Weiping, Z. T Cell Anergy, Exhaustion, Senescence, and Stemness in the Tumor Microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef]
- Reschke, R.; Gussek, P.; Boldt, A.; Sack, U.; Köhl, U.; Lordick, F.; Gora, T.; Kreuz, M.; Reiche, K.; Simon, J.C.; et al. Distinct Immune Signatures Indicative of Treatment Response and Immune-related Adverse Events in Melanoma Patients under Immune Checkpoint Inhibitor Therapy. Int. J. Mol. Sci. 2021, 22, 8017. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-Cell Invigoration to Tumour Burden Ratio Associated with Anti-PD-1 Response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef]
- Pico de Coaña, Y.; Wolodarski, M.; van der Haar Àvila, I.; Nakajima, T.; Rentouli, S.; Lundqvist, A.; Masucci, G.; Hansson, J.; Kiessling, R. PD-1 Checkpoint Blockade in Advanced Melanoma Patients: NK Cells, Monocytic Subsets and Host PD-L1 Expression as Predictive Biomarker Candidates. Oncoimmunology 2020, 9, 1786888. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Amin, M.; Edge, S.; Greene, F.; Byrd, D.; Brookland, R.; Washington, M.; Gershenwald, J.; Compton, C.; Hess, K.; Sullivan, D.; et al. (Eds.) AJCC Cancer Staging Manual, 8th ed.; Springer: Chicago, IL, USA, 2017; Volume 7, ISBN 9788578110796. [Google Scholar]
- Aran, D.; Hu, Z.; Butte, A.J. XCell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Bagwell, C.B.; Inokuma, M.; Hunsberger, B.; Herbert, D.; Bray, C.; Hill, B.; Stelzer, G.; Li, S.; Kollipara, A.; Ornatsky, O.; et al. Automated Data Cleanup for Mass Cytometry. Cytom. Part A 2020, 97, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, M.; Krieg, C.; Crowell, H.L.; Weber, L.M.; Hartmann, F.J.; Guglietta, S.; Becher, B.; Levesque, M.P.; Robinson, M.D. CyTOF Workflow: Differential Discovery in High-Throughput High-Dimensional Cytometry Datasets. F1000Research 2017, 6, 748. [Google Scholar] [CrossRef]
- Poletto, S.; Paruzzo, L.; Nepote, A.; Caravelli, D.; Sangiolo, D.; Carnevale-Schianca, F. Predictive Factors in Metastatic Melanoma Treated with Immune Checkpoint Inhibitors: From Clinical Practice to Future Perspective. Cancers 2023, 16, 101. [Google Scholar] [CrossRef]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug Signatures Database for Gene Set Analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Mittra, A.; Naqash, A.R.; Takebe, N. A Review of Mechanisms of Resistance to Immune Checkpoint Inhibitors and Potential Strategies for Therapy. Cancer Drug Resist. 2020, 3, 252–275. [Google Scholar] [CrossRef]
- Saito, A.; Saito, N.; Mol, W.; Furukawa, H.; Tsutsumida, A.; Oyama, A.; Sekido, M.; Sasaki, S.; Yamamoto, Y. Simvastatin Inhibits Growth via Apoptosis and the Induction of Cell Cycle Arrest in Human Melanoma Cells. Melanoma Res. 2008, 18, 85–94. [Google Scholar] [CrossRef]
- Guterres, F.A.D.L.B.; Martinez, G.R.; Rocha, M.E.M.; Winnischofer, S.M.B. Simvastatin Rises Reactive Oxygen Species Levels and Induces Senescence in Human Melanoma Cells by Activation of P53/P21 Pathway. Exp. Cell Res. 2013, 319, 2977–2988. [Google Scholar] [CrossRef]
- Duarte, J.A.; de Barros, A.L.B.; Leite, E.A. The Potential Use of Simvastatin for Cancer Treatment: A Review. Biomed. Pharmacother. 2021, 141, 111858. [Google Scholar] [CrossRef]
- Favero, G.M.; Otuki, M.F.; Oliveira, K.A.; Bohatch, M.S.; Borelli, P.; Barros, F.E.; Maria, D.A.; Fernandes, D.; Bydlowski, S.P. Simvastatin Impairs Murine Melanoma Growth. Lipids Health Dis. 2010, 9, 142. [Google Scholar] [CrossRef]
- Alupei, M.C.; Licarete, E.; Patras, L.; Banciu, M. Liposomal Simvastatin Inhibits Tumor Growth via Targeting Tumor-Associated Macrophages-Mediated Oxidative Stress. Cancer Lett. 2015, 356, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Negrea, G.; Rauca, V.F.; Meszaros, M.S.; Patras, L.; Luput, L.; Licarete, E.; Toma, V.A.; Porfire, A.; Muntean, D.; Sesarman, A.; et al. Active Tumor-Targeting Nano-Formulations Containing Simvastatin and Doxorubicin Inhibit Melanoma Growth and Angiogenesis. Front. Pharmacol. 2022, 13, 870347. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.; Lo-Coco, F.; Sanz, M.A.; Di Bona, E.; Coutre, S.; Altman, J.K.; Wetzler, M.; Allen, S.L.; Ravandi, F.; Kantarjian, H.; et al. Tamibarotene in Patients with Acute Promyelocytic Leukaemia Relapsing after Treatment with All-Trans Retinoic Acid and Arsenic Trioxide. Br. J. Haematol. 2015, 171, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-Dimensional Single-Cell Analysis Predicts Response to Anti-PD-1 Immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef]
- Ratnayake, G.; Reinwald, S.; Edwards, J.; Wong, N.; Yu, D.; Ward, R.; Smith, R.; Haydon, A.; Au, P.M.; van Zelm, M.C.; et al. Blood T-Cell Profiling in Metastatic Melanoma Patients as a Marker for Response to Immune Checkpoint Inhibitors Combined with Radiotherapy. Radiother. Oncol. 2022, 173, 299–305. [Google Scholar] [CrossRef]
- Iliopoulou, E.G.; Karamouzis, M.V.; Missitzis, I.; Ardavanis, A.; Sotiriadou, N.N.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. Increased Frequency of CD4+ Cells Expressing CD161 in Cancer Patients. Clin. Cancer Res. 2006, 12, 6901–6909. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Cluster A (n = 11) | Cluster B (n = 8) | p-Value |
|---|---|---|---|
| Sex, n (%) | 1 | ||
| Females | 5 (45.5%) | 3 (37.5%) | |
| Males | 6 (54.5%) | 5 (62.5%) | |
| Age at diagnosis (years), mean (SD) a | 60.2 (17.2) | 73.6 (10.5) | 0.068 |
| Age at start of IMT b (years), mean (SD) | 62.5 (17.4) | 75.6 (9.1) | 0.072 |
| Staging c at diagnosis | 0.898 | ||
| I–II | 5 (45.5%) | 1 (12.5%) | |
| III–IV | 6 (54.5%) | 7 (87.5%) | |
| Staging at start of IMT | 0.307 | ||
| III | 2 (18.2%) | 1 (12.5%) | |
| IV | 9 (81.8%) | 7 (87.5%) | |
| Mitotic Index d, median (IQR) e | 7 (6) | 6.5 (4.75) | 1 |
| PFS f, median (IQR) | 59.4 (42.2) | 2.4 (7.7) | 0.002 |
| Response g, n (%) | 0.003 | ||
| Responder | 8 (72.7%) | 0 (0%) | |
| Non-responder | 3 (27.3%) | 8 (100%) | |
| irAEs h, n (%) | 0.058 | ||
| Yes | 9 (81.8%) | 6 (75%) | |
| No | 2 (18.2%) | 2 (25%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molina-García, M.; Rojas-Lechuga, M.J.; Torres Moral, T.; Crespí-Payeras, F.; Bagué, J.; Mateu, J.; Paschalidis, N.; de Souza, V.G.; Podlipnik, S.; Carrera, C.; et al. Integrative Molecular and Immune Profiling in Advanced Unresectable Melanoma: Tumor Microenvironment and Peripheral PD-1+ CD4+ Effector Memory T-Cells as Potential Markers of Response to Immune Checkpoint Inhibitor Therapy. Cancers 2025, 17, 2022. https://doi.org/10.3390/cancers17122022
Molina-García M, Rojas-Lechuga MJ, Torres Moral T, Crespí-Payeras F, Bagué J, Mateu J, Paschalidis N, de Souza VG, Podlipnik S, Carrera C, et al. Integrative Molecular and Immune Profiling in Advanced Unresectable Melanoma: Tumor Microenvironment and Peripheral PD-1+ CD4+ Effector Memory T-Cells as Potential Markers of Response to Immune Checkpoint Inhibitor Therapy. Cancers. 2025; 17(12):2022. https://doi.org/10.3390/cancers17122022
Chicago/Turabian StyleMolina-García, Manuel, María Jesús Rojas-Lechuga, Teresa Torres Moral, Francesca Crespí-Payeras, Jaume Bagué, Judit Mateu, Nikolaos Paschalidis, Vinícius Gonçalves de Souza, Sebastian Podlipnik, Cristina Carrera, and et al. 2025. "Integrative Molecular and Immune Profiling in Advanced Unresectable Melanoma: Tumor Microenvironment and Peripheral PD-1+ CD4+ Effector Memory T-Cells as Potential Markers of Response to Immune Checkpoint Inhibitor Therapy" Cancers 17, no. 12: 2022. https://doi.org/10.3390/cancers17122022
APA StyleMolina-García, M., Rojas-Lechuga, M. J., Torres Moral, T., Crespí-Payeras, F., Bagué, J., Mateu, J., Paschalidis, N., de Souza, V. G., Podlipnik, S., Carrera, C., Malvehy, J., da Silva-Júnior, R. M. P., & Puig, S. (2025). Integrative Molecular and Immune Profiling in Advanced Unresectable Melanoma: Tumor Microenvironment and Peripheral PD-1+ CD4+ Effector Memory T-Cells as Potential Markers of Response to Immune Checkpoint Inhibitor Therapy. Cancers, 17(12), 2022. https://doi.org/10.3390/cancers17122022