
Steroid Sulfatase Regulates Metabolic Reprogramming in Advanced Prostate Cancer

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Seahorse Extracellular Flux Analyzer Experiments
2.3. Electron Transport Chain Complex I Activity
2.4. Organoids Culture
2.5. Cell Viability Assay
2.6. Colony Formation Assay
2.7. RNA-Seq
2.8. Gene Set Enrichment Analysis
2.9. Statistical Analysis
3. Results
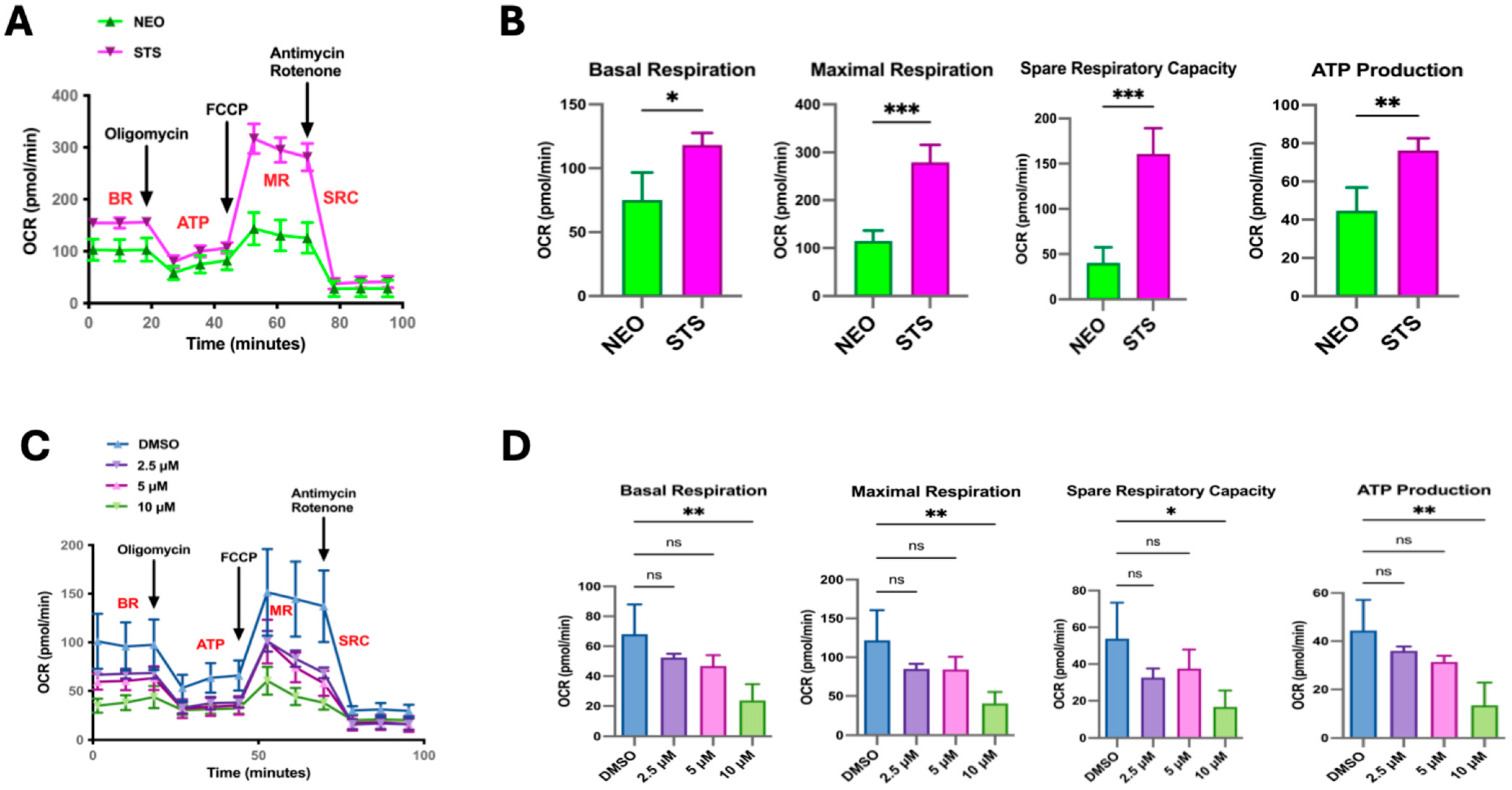
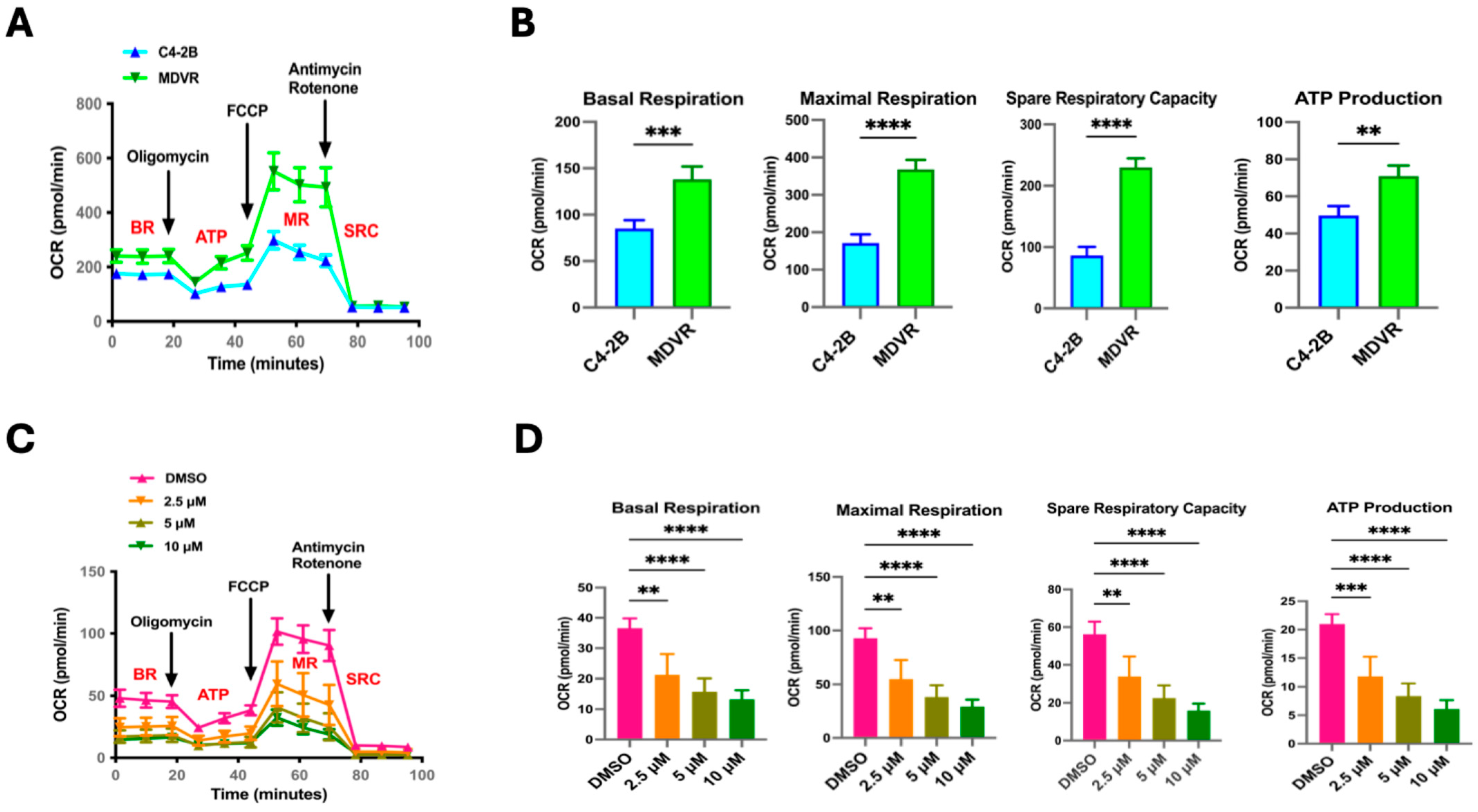
3.1. Metabolic Signaling Pathways Are Enriched in STS-Overexpressing Prostate Cancer Cells
3.2. STS Overexpression Enhances OXPHOS Electron Transport Chain Complex I Activity
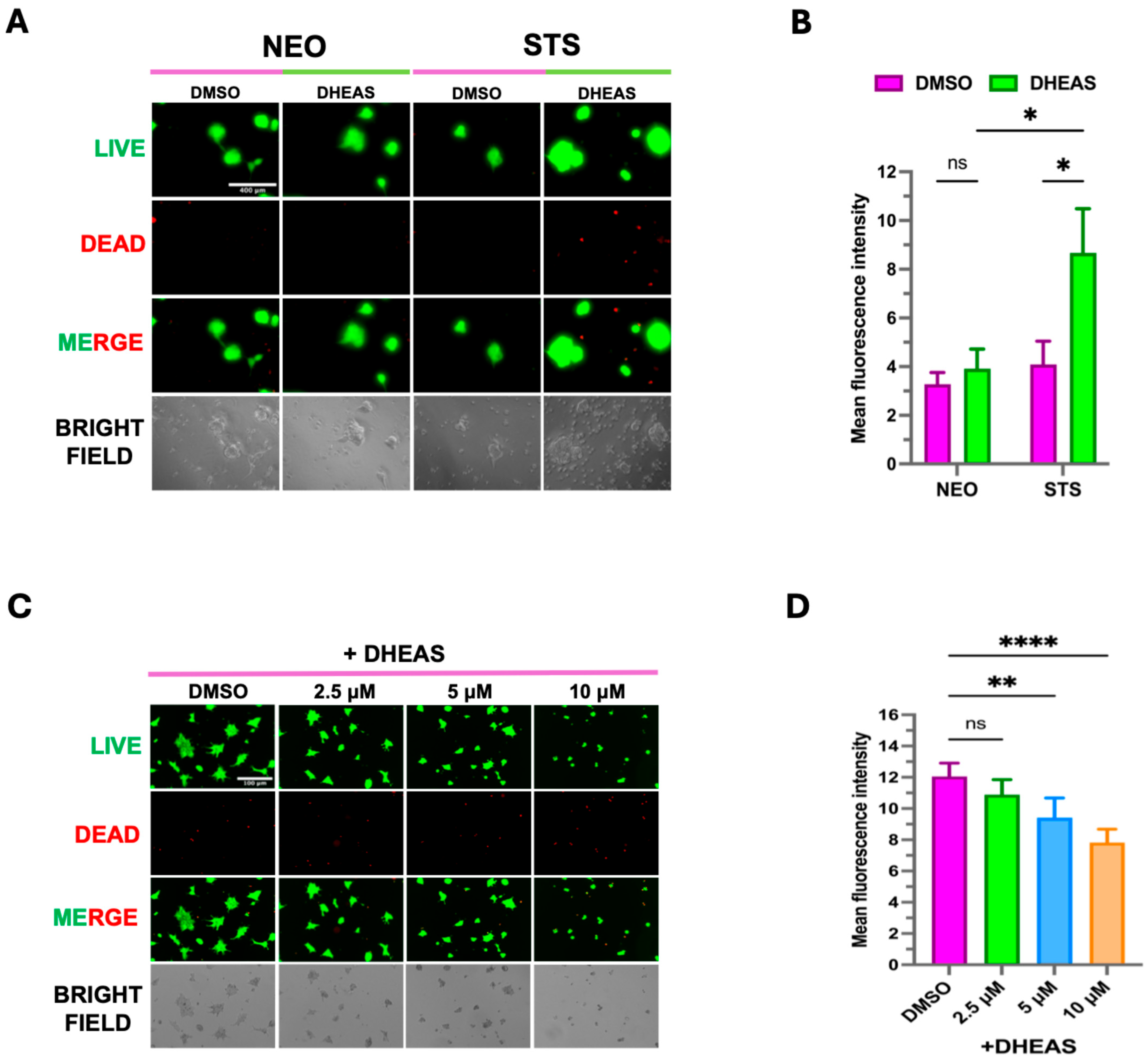
3.3. Elevated STS Induces DHEAS-Mediated Organoids Formation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PCa | Prostate Cancer |
| AR | Androgen Receptor |
| NGATs | Next-Generation Anti-Androgen Therapies |
| CRPC | Castration-Resistant Prostate Cancer |
| STS | Steroid Sulfatase |
| RNA-Seq | RNA-Sequencing |
| Enza | Enzalutamide |
| Abi | Abiraterone |
| Daro | Darolutamide |
| Apa | Apalutamide |
| OCR | Oxygen Consumption Rate |
| BR | Basal Respiration |
| SRC | Spare Respiratory Capacity |
| MR | Maximal Respiration |
| ATP | ATP Production |
| DHEA | dehydroepiandrosterone |
| DHEAS | Dehydroepiandrosterone sulfate |
| GSEA | Gene Set Enrichment Analysis |
| ETC | Electron Transport Chain |
| ROS | Reactive Oxygen Species |
References
- Chen, F.Z.; Zhao, X.K. Prostate cancer: Current treatment and prevention strategies. Iran. Red. Crescent Med. J. 2013, 15, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Pernar, C.H.; Ebot, E.M.; Wilson, K.M.; Mucci, L.A. The Epidemiology of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Sekhoacha, M.; Riet, K.; Motloung, P.; Gumenku, L.; Adegoke, A.; Mashele, S. Prostate Cancer Review: Genetics, Diagnosis, Treatment Options, and Alternative Approaches. Molecules 2022, 27, 5730. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Le, T.K.; Duong, Q.H.; Baylot, V.; Fargette, C.; Baboudjian, M.; Colleaux, L.; Taïeb, D.; Rocchi, P. Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments. Cancers 2023, 15, 5047. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, Q.; Hankey, W.; Fang, X.; Yuan, F. Second generation androgen receptor antagonists and challenges in prostate cancer treatment. Cell Death Dis. 2022, 13, 632. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- Nakazawa, M.; Paller, C.; Kyprianou, N. Mechanisms of Therapeutic Resistance in Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 13. [Google Scholar] [CrossRef]
- Reed, M.J.; Purohit, A.; Woo, L.W.; Newman, S.P.; Potter, B.V. Steroid sulfatase: Molecular biology, regulation, and inhibition. Endocr. Rev. 2005, 26, 171–202. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Liu, C.; Liu, L.; Yang, J.C.; Lou, W.; Zhao, R.; Ning, S.; Lombard, A.P.; Zhao, J.; D’Abronzo, L.S.; et al. Steroid Sulfatase Stimulates Intracrine Androgen Synthesis and is a Therapeutic Target for Advanced Prostate Cancer. Clin. Cancer Res. 2020, 26, 6064–6074. [Google Scholar] [CrossRef]
- Campbell, B. DHEAS and Human Development: An Evolutionary Perspective. Front. Endocrinol. 2020, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.A.; Katyare, S.S. Treatment with dehydroepiandrosterone (DHEA) stimulates oxidative energy metabolism in the cerebral mitochondria: A comparative study of effects in old and young adult rats. Neurosci. Lett. 2006, 402, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, K.; Sowa, P.; Rutkowska-Talipska, J.; Kuryliszyn-Moskal, A.; Rutkowski, R. Dehydroepiandrosterone (DHEA): Hypes and hopes. Drugs 2014, 74, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-J.; Lin, L.-T.; Tsui, K.-H. Dehydroepiandrosterone Shifts Energy Metabolism to Increase Mitochondrial Biogenesis in Female Fertility with Advancing Age. Nutrients 2021, 13, 2449. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Kwon, Y.J.; Ye, D.J.; Baek, H.S.; Kwon, T.U.; Kim, D.; Chun, Y.J. Human steroid sulfatase enhances aerobic glycolysis through induction of HIF1α and glycolytic enzymes. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2464–2474. [Google Scholar] [CrossRef]
- Patel, M.A.; Katyare, S.S. Effect of dehydroepiandrosterone (DHEA) treatment on oxidative energy metabolism in rat liver and brain mitochondria. A dose–response study. Clin. Biochem. 2007, 40, 57–65. [Google Scholar] [CrossRef]
- Ahmad, F.; Cherukuri, M.K.; Choyke, P.L. Metabolic reprogramming in prostate cancer. Br. J. Cancer 2021, 125, 1185–1196. [Google Scholar] [CrossRef]
- Beier, A.K.; Puhr, M.; Stope, M.B.; Thomas, C.; Erb, H.H.H. Metabolic changes during prostate cancer development and progression. J. Cancer Res. Clin. Oncol. 2023, 149, 2259–2270. [Google Scholar] [CrossRef]
- Pujana-Vaquerizo, M.; Bozal-Basterra, L.; Carracedo, A. Metabolic adaptations in prostate cancer. Br. J. Cancer 2024, 131, 1250–1262. [Google Scholar] [CrossRef]
- Chen, C.L.; Lin, C.Y.; Kung, H.J. Targeting Mitochondrial OXPHOS and Their Regulatory Signals in Prostate Cancers. Int. J. Mol. Sci. 2021, 22, 13435. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.T.H.; Chu, S. Measurement of Oxidative Stress: Mitochondrial Function Using the Seahorse System. Methods Mol. Biol. 2018, 1710, 285–293. [Google Scholar] [CrossRef]
- Gu, X.; Ma, Y.; Liu, Y.; Wan, Q. Measurement of mitochondrial respiration in adherent cells by Seahorse XF96 Cell Mito Stress Test. STAR Protoc. 2021, 2, 100245. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Shankar, E.; Chan, E.R.; Gupta, S. Metabolic Reprogramming and Predominance of Solute Carrier Genes during Acquired Enzalutamide Resistance in Prostate Cancer. Cells 2020, 9, 2535. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, Y.; Yuan, S.; Sun, L. Targeting Mitochondrial Metabolic Reprogramming as a Potential Approach for Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 4954. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Wolpaw, A.J.; Dang, C.V. MYC-induced metabolic stress and tumorigenesis. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2018, 1870, 43–50. [Google Scholar] [CrossRef]
- O’Donnell, K.A.; Yu, D.; Zeller, K.I.; Kim, J.-w.; Racke, F.; Thomas-Tikhonenko, A.; Dang, C.V. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol. Cell. Biol. 2006, 26, 2373–2386. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Warburg, O.H. Über den Stoffwechsel der Tumoren: Arbeiten aus dem Kaiser Wilhelm Institut für Biologie, Berlin-Dahlem; Springer: Berlin/Heidelberg, Germany, 1926. [Google Scholar]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231. [Google Scholar] [CrossRef]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic investigations of the mitochondrial complex I inhibitor rotenone in the context of pharmacological and safety evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Gandhi, J.; Joshi, G.; Smith, N.L.; Khan, S.A. The anticancer potential of metformin on prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, H.; Jang, H.; Woo, S.M.; Park, J.B.; Lee, S.H.; Kang, J.H.; Kim, H.Y.; Song, J.; Kim, S.Y. Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling. Cells 2020, 9, 2013. [Google Scholar] [CrossRef]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Basu, H.S.; Wilganowski, N.; Robertson, S.; Reuben, J.M.; Cohen, E.N.; Zurita, A.; Ramachandran, S.; Xiao, L.C.; Titus, M.; Wilding, G. Prostate cancer cells survive anti-androgen and mitochondrial metabolic inhibitors by modulating glycolysis and mitochondrial metabolic activities. Prostate 2021, 81, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Simpson, K.; Allison, D.B.; He, D.; Liu, J.; Wang, C.; Liu, X. Metformin in overcoming enzalutamide resistance in castration-resistant prostate cancer. J. Pharmacol. Exp. Ther. 2025, 392, 100034. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Qi, S.; Zhu, Z. Advances in mitochondria-centered mechanism behind the roles of androgens and androgen receptor in the regulation of glucose and lipid metabolism. Front. Endocrinol. 2023, 14, 1267170. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, W.; Wang, B.; Lin, G.; Wei, Y.; Abudurexiti, M.; Zhu, W.; Liu, C.; Qin, X.; Dai, B.; et al. GLUT1 is an AR target contributing to tumor growth and glycolysis in castration-resistant and enzalutamide-resistant prostate cancers. Cancer Lett. 2020, 485, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Newell-Fugate, A.E. Role of androgens and androgen receptor in control of mitochondrial function. Am. J. Physiol.—Cell Physiol. 2022, 323, C835–C846. [Google Scholar] [CrossRef] [PubMed]
- Crowell, P.D.; Giafaglione, J.M.; Jones, A.E.; Nunley, N.M.; Hashimoto, T.; Delcourt, A.M.L.; Petcherski, A.; Bernard, M.J.; Huang, R.R.; Low, J.-Y.; et al. Androgen receptor inhibition induces metabolic reprogramming and increased reliance on oxidative mitochondrial metabolism in prostate cancer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Li, C.J.; Chen, S.N.; Lin, L.T.; Chern, C.U.; Wang, P.H.; Wen, Z.H.; Tsui, K.H. Dehydroepiandrosterone Ameliorates Abnormal Mitochondrial Dynamics and Mitophagy of Cumulus Cells in Poor Ovarian Responders. J. Clin. Med. 2018, 7, 293. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Yuan, L.L.; Wang, Z.C.; Zhuang, W.B.; Zhang, W.J.; Liu, H.T.; Li, M.; Fan, L.H. DHEA down-regulates mitochondrial dynamics and promotes apoptosis of lung adenocarcinoma cells through FASTKD2. J. Cancer 2024, 15, 2110–2122. [Google Scholar] [CrossRef]
- Hou, Y.-L.; Li, C.-J.; Lin, L.-T.; Chen, S.-N.; Wen, Z.-H.; Tsui, K.-H. DHEA restores mitochondrial dynamics of cumulus cells by regulating PGAM5 expression in poor ovarian responders. Taiwan J. Obstet. Gynecol. 2022, 61, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wang, D.; Li, L.; Ma, H. Dehydroepiandrosterone ameliorates H2O2-induced Leydig cells oxidation damage and apoptosis through inhibition of ROS production and activation of PI3K/Akt pathways. Int. J. Biochem. Cell Biol. 2016, 70, 126–139. [Google Scholar] [CrossRef]
- Nakashima, N.; Haji, M.; Sakai, Y.; Ono, Y.; Umeda, F.; Nawata, H. Effect of dehydroepiandrosterone on glucose uptake in cultured human fibroblasts. Metabolism 1995, 44, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Vieira-Marques, C.; Arbo, B.D.; Ruiz-Palmero, I.; Ortiz-Rodriguez, A.; Ghorbanpoor, S.; Kucharski, L.C.; Arevalo, M.A.; Garcia-Segura, L.M.; Ribeiro, M.F.M. Dehydroepiandrosterone protects male and female hippocampal neurons and neuroblastoma cells from glucose deprivation. Brain Res. 2016, 1644, 176–182. [Google Scholar] [CrossRef]
- Tsui, K.H.; Wang, P.H.; Lin, L.T.; Li, C.J. DHEA protects mitochondria against dual modes of apoptosis and necroptosis in human granulosa HO23 cells. Reproduction 2017, 154, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.A.; Katyare, S.S. Dehydroepiandrosterone (DHEA) treatment stimulates oxidative energy metabolism in the cerebral mitochondria from developing rats. Int. J. Dev. Neurosci. 2006, 24, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.-S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef]
- Soga, T. Cancer metabolism: Key players in metabolic reprogramming. Cancer Sci. 2013, 104, 275–281. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef]
- Kim, J.-w.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, Y.; Xu, J.; Zhang, T.; Wang, H.; Wang, Z.; Jiang, Y.; Zhou, L.; Yang, M.; Hua, Y.; et al. Her4 promotes cancer metabolic reprogramming via the c-Myc-dependent signaling axis. Cancer Lett. 2021, 496, 57–71. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharifi, M.; Armstrong, C.M.; Ning, S.; Leslie, A.R.; Schaaf, Z.A.; Maine, J.P.; Lou, W.; Li, P.-K.; Xu, H.; Liu, C.; et al. Steroid Sulfatase Regulates Metabolic Reprogramming in Advanced Prostate Cancer. Cancers 2025, 17, 1959. https://doi.org/10.3390/cancers17121959
Sharifi M, Armstrong CM, Ning S, Leslie AR, Schaaf ZA, Maine JP, Lou W, Li P-K, Xu H, Liu C, et al. Steroid Sulfatase Regulates Metabolic Reprogramming in Advanced Prostate Cancer. Cancers. 2025; 17(12):1959. https://doi.org/10.3390/cancers17121959
Chicago/Turabian StyleSharifi, Masuda, Cameron M. Armstrong, Shu Ning, Amy R. Leslie, Zachary A. Schaaf, James P. Maine, Wei Lou, Pui-Kai Li, Hongyu Xu, Chengfei Liu, and et al. 2025. "Steroid Sulfatase Regulates Metabolic Reprogramming in Advanced Prostate Cancer" Cancers 17, no. 12: 1959. https://doi.org/10.3390/cancers17121959
APA StyleSharifi, M., Armstrong, C. M., Ning, S., Leslie, A. R., Schaaf, Z. A., Maine, J. P., Lou, W., Li, P.-K., Xu, H., Liu, C., & Gao, A. C. (2025). Steroid Sulfatase Regulates Metabolic Reprogramming in Advanced Prostate Cancer. Cancers, 17(12), 1959. https://doi.org/10.3390/cancers17121959