1. Introduction
Chimeric antigen receptor (CAR)-T and T-cell receptor (TCR)-engineered T-cell (TCR-T) therapies have revolutionized the treatment of hematological malignancies; however, their application to solid tumors remains a formidable challenge. The immunosuppressive tumor microenvironment, antigen heterogeneity, and manufacturing complexity limit the clinical efficacy and scalability of these treatment modalities. This review provides a comprehensive analysis of the current clinical development strategies for CAR-T and TCR-T cell therapies for solid tumors.
2. Overview of CAR-T Cell Therapy for Cancer
CAR-T cell therapy is an innovative immunotherapeutic modality in which autologous T cells are genetically engineered to target and eliminate cancer cells. This approach has demonstrated remarkable clinical efficacy, particularly for hematologic malignancies [
1,
2] and has garnered significant global attention. In Japan, CAR-T cell therapies for several indications, including relapsed/refractory CD19-positive B-cell acute lymphoblastic leukemia in patients aged ≤25 years, large B-cell lymphoma, follicular lymphoma, and multiple myeloma, have been reimbursed under the national health insurance system.
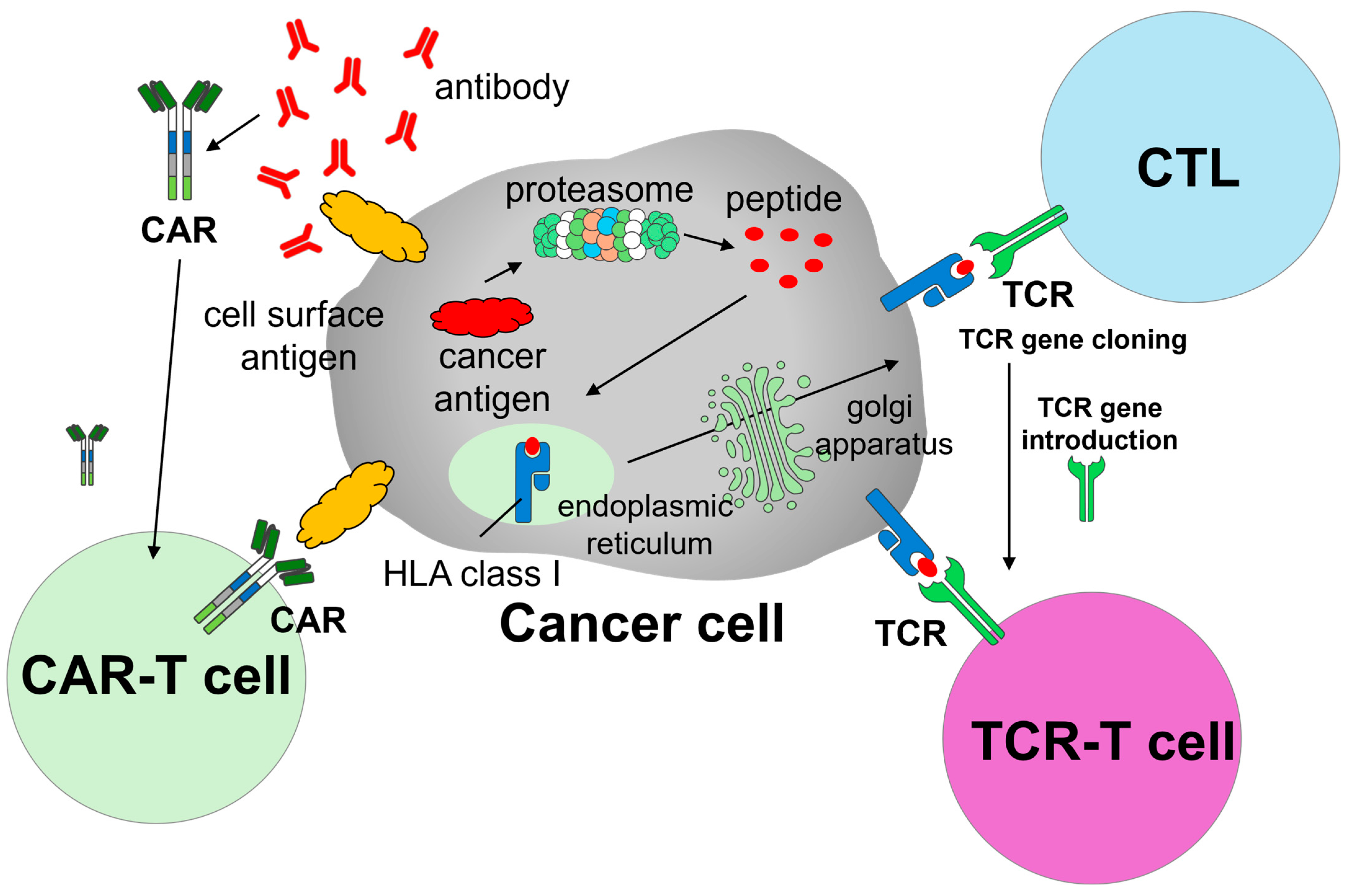
CAR-T cells are produced by introducing chimeric antigen receptors (i.e., CARs) into autologous T cells. CARs are designed to recognize tumor-associated surface antigens, enabling engineered T cells to specifically identify and eliminate malignant cells (
Figure 1). The manufacturing process consists of multiple stages: leukapheresis, transportation to a Good Manufacturing Practice (GMP)-compliant manufacturing facility, viral transduction or other gene editing to insert the CAR constructs, ex vivo expansion, and rigorous quality control, followed by reinfusion into the patient. Prior to infusion, patients typically undergo bridging therapy and lymphodepleting chemotherapy to enhance CAR-T cell engraftment and efficacy.
Although CAR-T cell therapies have yielded high response rates in hematological cancers, their clinical benefits in solid tumors remain limited. Major barriers include the immunosuppressive tumor microenvironment (TME); heterogeneous or low antigen expression; antigen loss; poor tumor infiltration; and T cell exhaustion driven by the persistent stimulation of inhibitory receptors, such as PD-1 and LAG-3 [
3]. Various strategies have been explored to overcome these limitations, including optimizing antigen selection, improving CAR design, incorporating resistance mechanisms against TME-induced suppression, and enhancing T cell trafficking to tumor sites. However, serious adverse events should be carefully managed. Prominent toxicities include cytokine release syndrome [
4,
5], immune effector cell-associated neurotoxicity syndrome [
6], and hemophagocytic lymphohistiocytosis-like syndrome [
7]. These complications could be life-threatening and require vigilant monitoring.
Unlike for hematologic malignancies, CAR-T cell therapy for solid tumors faces challenges unique to the tissue context. For example, on-target/off-tumor toxicity is more common in normal tissues owing to the expression of target antigens, such as human epidermal growth factor receptor 2 and mesothelin [
3]. Dense extracellular matrices may also restrict CAR-T cell infiltration and migration. Moreover, the TME in solid tumors is enriched with immunosuppressive components, such as regulatory T cells, M2 macrophages, myeloid-derived suppressor cells, and cytokines (e.g., transforming growth factor-β and interleukin [IL]-10). This hostile environment diminishes CAR-T cell activity and efficacy. Finally, the typically high tumor burden in solid tumors can lead to chronic antigen stimulation, resulting in T cell exhaustion and diminished antitumor responses. These complex barriers underscore the pressing need for next-generation CAR-T cell therapies specifically engineered to function effectively in solid tumors.
3. Overview of TCR- T Cell Therapy for Cancer
TCR-T cell therapy represents another promising adoptive cell therapy strategy, in which autologous T cells are genetically modified to express tumor-specific TCRs. Unlike CAR-T cells, which target surface antigens in a major histocompatibility complex (MHC)-independent manner, TCR-T cells recognize intracellular tumor-derived peptides presented by MHC molecules, enabling them to target a broader range of tumor-associated antigens [
8,
9,
10] (
Figure 1). This MHC-dependent mechanism enables TCR-T cells to recognize intracellular tumor-associated or tumor-specific antigens that are processed and presented via MHC molecules, such as cancer/testis antigens (e.g., NY-ESO-1 and MAGE-A4) and neoantigens derived from somatic mutations (e.g., KRAS G12D) [
11]. This expands the range of targetable tumor antigens beyond cell surface molecules, which are the sole targets of CAR-T cells. The overall manufacturing process for TCR-T cells is similar to that for CAR-T cells: T cells are collected from the patient, transduced or edited to express tumor-specific TCRs, expanded ex vivo under GMP conditions, and reinfused following lymphodepletion.
Table 1 outlines the key differences between CAR-T and TCR-T cell therapies.
In August 2024, the United States (US) Food and Drug Administration (FDA) granted accelerated approval for afamitresgene autoleucel, a TCR-T cell therapy targeting the MAGE-A4 antigen presented by a specific human leukocyte antigen (HLA) type, for unresectable or metastatic synovial sarcoma. This approval was based on the SPEARHEAD-1 trial, which demonstrated an overall response rate of 39% among 44 patients [
12]. This marked a milestone for TCR-T cell therapy in solid tumors and initiated further clinical development across a range of malignancies. However, despite its potential, TCR-T cell therapy faces several challenges. Many of these issues overlap with those encountered by CAR-T cells, such as limited tumor infiltration owing to physical barriers, immunosuppressive TMEs, T cell exhaustion, and the risk of on-target/off-tumor toxicity when targeting shared antigens. However, TCR-T cells rely on MHC expression; therefore, their therapeutic efficacy may be impaired by tumor-driven MHC down-regulation, a common immune evasion mechanism in advanced cancers.
Moreover, the HLA system is highly polymorphic, and each TCR construct must be tailored to a specific HLA type, posing a significant challenge for broad clinical application [
9]. These factors highlight the need for individualized approaches to TCR-T cell therapy and represent key bottlenecks in its development and scalability. However, ongoing research to overcome these limitations, including improvements in TCR affinity, persistence, and safety, continues to drive progress toward optimized TCR-T cell therapies for solid tumors.
4. Strategies to Enhance the Efficacy of CAR-T Cell Therapy Against Refractory Solid Tumors
4.1. CAR-T Cell Approval Strategy for Solid Tumor Treatment
There has been notable progress in applying TCR-T cell therapy for refractory solid tumors, with one product, afamitresgene autoleucel, receiving FDA approval for unresectable or metastatic synovial sarcoma. However, CAR-T cells have not been approved for solid tumors. The major barriers include tumor antigen escape, lack of identifying effective tumor-specific surface antigens, the immunosuppressive tumor microenvironment, and difficulties in T cell infiltration [
13,
14,
15]. Overcoming these challenges is crucial to achieving success in CAR-T cell therapy for solid tumors. Although numerous clinical trials are ongoing worldwide, this review does not attempt to summarize them comprehensively. Instead, we recommend recent high-quality reviews focusing on CAR-T cell combination strategies, such as those by Uslu et al. [
16].
4.2. In Vivo CAR-T Cell Therapy
Gene therapies can be broadly categorized into ex vivo and in vivo modalities (
Table 2). Currently approved CAR-T cell therapies for hematologic malignancies are ex vivo gene therapies that require leukapheresis, T-cell modification and expansion, and reinfusion. This process demands significant infrastructure, personnel, and financial resources and limits accessibility. In contrast, in vivo gene therapy involves the direct delivery of gene-editing components (e.g., viral or non-viral vectors) to patients, potentially bypassing the need for leukapheresis and complex manufacturing [
17]. Efforts to generate CAR-T cells in vivo have demonstrated a proof-of-concept in murine tumor models using injectable viral vectors or nanoparticle platforms [
18]. However, although these approaches are promising, the long-term safety concerns remain unresolved. Potential risks include persistent antigen stimulation, unexpected recognition of normal cells, off-target effects due to nonspecific interactions, and unintended immune activation. Integrating vectors, such as lentiviruses, may result in insertional mutagenesis [
19], and individual differences in immune cell composition and thymic selection add further uncertainty. To mitigate these risks, research is shifting toward non-integrating vector systems (e.g., adeno-associated virus [AAV] vectors [
18,
20]) or transient gene expression platforms (e.g., messenger RNA [mRNA] delivery via lipid nanoparticles [LNPs]) [
21,
22]. These strategies aim to enable short-term CAR expression while reducing genotoxic risk.
4.3. Allogeneic iPSC-Derived CAR-T Cell Therapy
Allogeneic CAR-T cell therapy, especially induced pluripotent stem cell (iPSC)-derived CAR-T cells, is gaining attention as a scalable off-the-shelf option. These therapies use T cells derived from healthy donors, modified to inactivate TCRs (to reduce graft-versus-host disease [GVHD] risk), and stored as ready-to-use frozen products. Although this approach circumvents the logistical challenges of autologous therapy and reduces production time and cost, significant concerns regarding long-term persistence and immune rejection, including long-term GVHD over the long term, remain [
23].
4.4. Developing Strategies for CAR-T Cell Therapy
Several approaches that can be applied to both autologous and allogeneic CAR-T cell platforms have been developed. Armored CAR-T cells or T cells redirected for universal cytokine-mediated killing are engineered to secrete cytokines [
24] or express costimulatory ligands (e.g., CD28L and 4-1BBL), enhancing survival in the hostile TME. These cytokines can also recruit innate immune cells, such as natural killer (NK) cells and macrophages, amplifying antitumor responses. Dual- or multitargeting-CAR-T cells are currently being developed to address antigen escape. For example, relapse following anti-CD19 CAR-T cell therapy occurs in 30–60% of cases [
25], and is one of the main reasons associated with CD19 loss. To overcome this problem, dual-targeting CAR-T cells (e.g., CD19/CD22) and multiple-targeting CARs are under investigation [
26].
Combination therapies are also being investigated. These include immune checkpoint inhibitors (e.g., anti–PD-1, anti–CTLA-4) that restore T cell activity by blocking inhibitory pathways; chemotherapy and radiotherapy that may enhance CAR-T cell infiltration and promote immunogenic tumor cell death; molecularly targeted therapies that modulate tumor signaling pathways; and other immunotherapies, such as cancer vaccines and onco-lytic viruses. However, although combination approaches hold promise, they introduce new challenges, such as optimal sequencing, dosing strategies, and cost-effectiveness.
4.5. Enhancing Long-Term Safety
Long-term safety is an increasingly important concern for CAR-T cell therapy. In 2023, the FDA reported 22 cases of secondary T-cell cancers in more than 27,000 CAR-T dose [
27]. Although causality remains uncertain, two main considerations are worth noting: (1) secondary malignancies can also arise from prior therapies, including chemotherapy, radiotherapy, and stem cell transplantation, with reported prevalence rates of one in six patients [
28], and (2) current data suggest that T-cell lymphoma post-CAR-T cell therapy occurs in 0.22% of cases [
29]. Although further research is required to clarify whether these cases are causally associated with CAR-T cell therapy, they have raised concerns regarding integration-based CAR technologies. Transient expression strategies using AAV vectors or mRNA, or LNPs may offer safer alternatives.
4.6. Beyond T Cells: CAR-Modified Myeloid and Innate Immune Cells
Although T cells remain the cornerstone of adoptive cell therapy, other immune cell types, such as macrophages, neutrophils, and NK cells, are being investigated for CAR engineering. These cells lack TCRs and therefore carry a lower risk of side effects. However, their use is limited by poor tumor tropism, susceptibility to TME-mediated suppression, functional exhaustion.
To address these issues, emerging strategies include the introduction of tumor-homing receptors (e.g., CCR2 and CXCR2); optimization of intracellular signaling domains; synthetic receptors (e.g., PD-1-CD28); cytokine co-expression (e.g., IL-7, IL-12, IL-15, and IL-18); use of rejuvenated or stem-like immune cells (e.g., iPSC-derived or stem cell-like memory T cells); and bispecific or dual-antigen recognition systems. Such developments may enhance the applicability and efficacy of cellular immunotherapies.
5. Global Development of CAR-T and TCR-T Cell Therapies for Solid Tumors: An Intellectual Property Perspective
5.1. Trends in Patent Filings
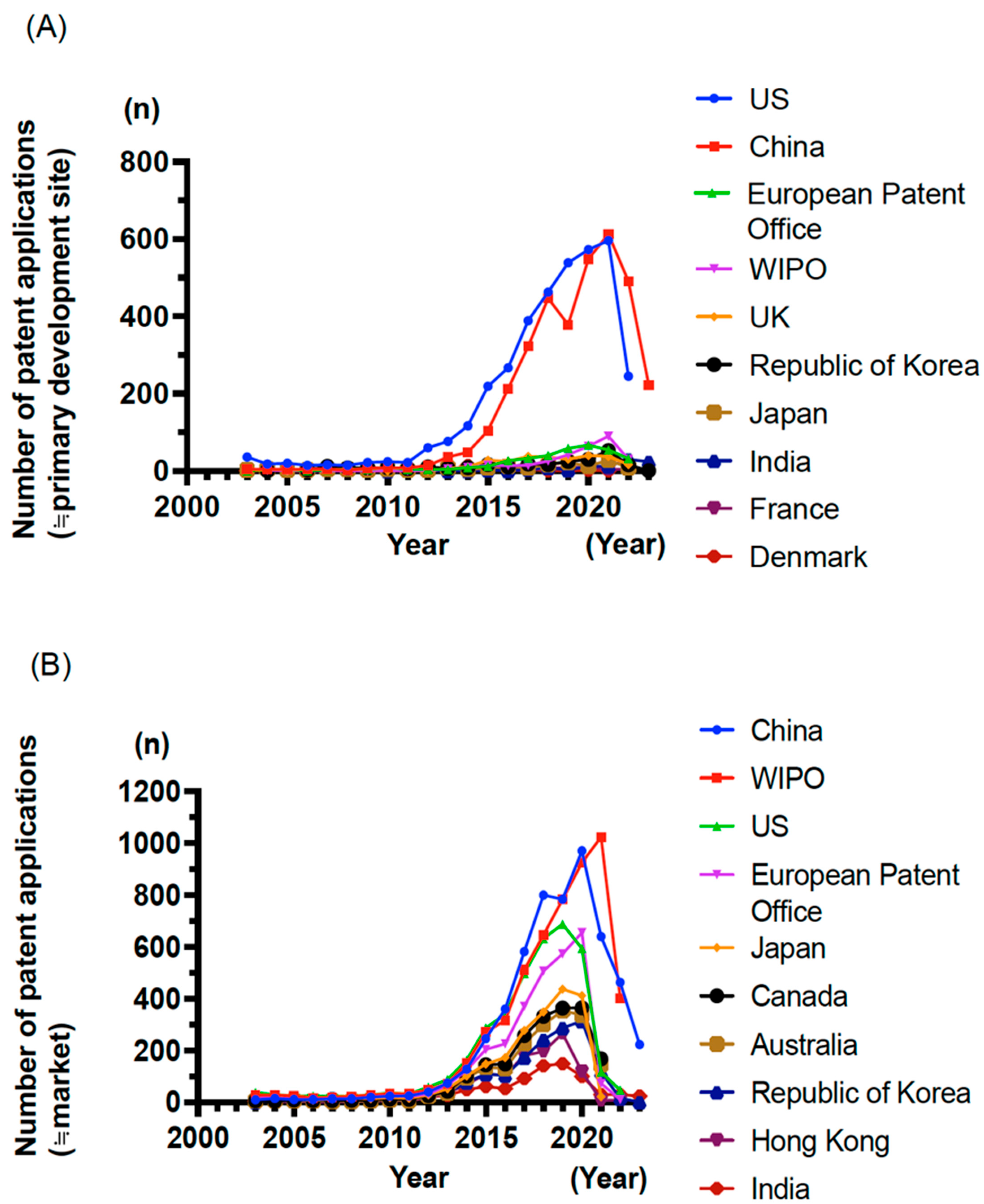
Building upon the preceding sections outlining CAR-T and TCR-T cell therapies, this section provides an advanced analysis of global development trends in these therapies for solid tumors based on a comprehensive patent landscape analysis conducted in collaboration with Clarivate (London, United Kingdom [UK], and Philadelphia, PA, US). Patent data were retrieved from the Derwent Innovation-DWPI and Cortellis Competitive Intelligence (CCI)-Patents databases, covering a broad range of intellectual properties related to CAR-T and TCR-T technologies. Between 2003 and 2011, the number of patent filings remained modest at approximately 50 per year, but it started to increase around 2012. This upward trend continues, reflecting a growing global interest in adoptive cell therapies.
5.2. Leading Countries and Filing Strategies
An analysis of priority-filing countries (≒primary development site) revealed that the US and China dominate CAR-T/TCR-T-related patent applications (
Figure 2A), indicating their central role in driving innovation. The European Patent Office ranked third, followed by the World Intellectual Property Organization (WIPO), the UK, Republic of Korea, and Japan. Among priority countries, the WIPO has emerged as the frequent destination (≒market) for patent filings (
Figure 2B), suggesting a strong intent to pursue global protection. However, the filing patterns differ by country. While more than 50% of applications filed via the WIPO are from the US, Europe, Japan, and Republic of Korea, the corresponding proportions for China and India are lower at 20.9% and 16.7%, respectively (
Table 3). These data suggest that although most Western countries aim for international commercialization, China and India prioritize domestic protection and deployment.
5.3. Top Assignees and Research Sectors
Among the top 20 patent assignees in the CAR-T/TCR-T domain, 11 are academic institutions (including universities, hospitals, and government/research institutes), and 9 are pharmaceutical or biotechnology companies (
Table 4). This distribution reflects the translational nature of the field, in which academic discovery is rapidly moving toward clinical implementation and commercialization.
5.4. Technological Hotspots and Emerging Themes
The most frequently patented technological areas include immune cell engineering (e.g., T cells, NK cells, and stem cells), CAR structural components (e.g., intracellular signaling domains, transmembrane and extracellular domains, single-domain antibodies, and amino acid sequences), capable of binding antibody, immune response, and gene transfer technologies (e.g., lentiviral vectors and circular RNAs). These trends underscore the diverse innovations in the CAR-T/TCR-T cell therapeutic field.
5.5. Disease Focus and Clinical Translation Potential
Oncology remains the dominant therapeutic area in CAR-T and TCR-T intellectual properties. Initially, most filings focused on hematologic malignancies, such as leukemia, lymphoma, and multiple myeloma. In recent years, research and development of CAR-T cell therapy have been increasingly conducted for various solid tumors, including breast, ovarian, lung, prostate, liver, kidney, and pancreatic cancers. These indications remain largely experimental owing to challenges such as antigen specificity and tumor penetration; however, they represent high-priority targets for future expansion. Beyond specific diseases, active patenting is also observed in broader categories, such as adoptive cell therapies, immunotherapy and immune modulation, gene delivery vectors, effector and antibody fragments. These trends reflect the multidisciplinary nature of the field, which includes immunology, cell biology, genetic engineering, and biomanufacturing.
6. Conclusions and Future Directions
Despite significant scientific and clinical advances, the development of CAR-T and TCR-T cell therapies for solid tumors remains a formidable challenge. These modalities face multifaceted barriers, including antigen heterogeneity, an immunosuppressive TME, and complex manufacturing and delivery logistics. Nevertheless, the number of global clinical trials targeting solid tumors has markedly increased in the past decade, reflecting the growing momentum and investment in this field. Encouraging progress has been made in technological innovation and translational research. TCR-T cell therapy has achieved a historic milestone with the FDA approval of afamitresgene autoleucel for unresectable or metastatic synovial sarcoma. Although no CAR-T product has been approved for solid tumors, advances in engineering strategies, including in vivo programming, armored and dual-target CAR designs, and integration with checkpoint blockade, are rapidly pushing the boundaries of clinically achievable.
Moreover, insights from global patent activity indicate a vibrant and expanding ecosystem of innovation, driven by both academic institutions and biotechnology companies. The emergence of novel platforms such as mRNA-based or iPSC-derived CAR-T cell therapies points toward a future in which scalable, off-the-shelf, and safer cell therapies may become a clinical reality. Overcoming the unique challenges in solid tumors requires a synergistic approach that combines deep biological understanding, precise genetic engineering, and strategic clinical trial design. The next generation of adoptive cell therapies is likely to be characterized by enhanced specificity, persistence, safety, and accessibility.
In conclusion, although substantial hurdles remain, the collective efforts of global research and clinical communities are paving the way for the successful implementation of CAR-T and TCR-T cell therapies for solid tumors. Continued innovation with thoughtful integration into multidisciplinary cancer care holds great promise for transforming the treatment landscape of refractory solid tumors.
Author Contributions
T.S., Y.K. and T.K. contributed to the conception and design of the manuscript. T.S. and T.N. contributed to data analysis and interpretation. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by AMED under Grant Number JP23bm1123042.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Dataset available on appropriate and reasonable request from the authors.
Acknowledgments
We acknowledge the valuable contributions of Go Hasegawa (Patent Search & Analytics, Analytics-LS, IP Services Japan, Clarivate) for his expert support in this study.
Conflicts of Interest
T.S. and Y.K. have nothing to declare. T.K. has received honoraria from Chugai Pharma, Sysmex, and AstraZeneca and served in a consulting or advisory role for Amgen. Research funding has been received by the author’s institution from the following companies: PACT Pharma, Novartis, Lilly, Takeda, Daiichi Sankyo RD Novare, Chugai Pharma, Zymeworks, Pfizer, Janssen Pharma, Boehringer Ingelheim, and AstraZeneca. T.N. holds patents with OncoTherapy Science, Inc., Noile-Immune Biotech Inc., Thyas Co., Ltd., Resonac Corporation, MEDINET Co., Ltd., and Takara Bio Inc., holds stock in Noile-Immune Biotech Inc., holds stock options and has received grants or contracts from Logomix Inc. and Optieum Biotechnologies Inc., has received grants or contracts from BrightPath Biotherapeutics Co., Ltd., Thyas Co., Ltd., ONO PHARMACEUTICAL CO., LTD., Resonac Corporation, MEDINET Co., Ltd., NapaJen Pharma Inc., Heartseed Inc., Takara Bio Inc., DAICEL CORPORATION, NA Vaccine Institute CO., LTD., and MaxCyte, Inc.
Abbreviations
The following abbreviations are used in this manuscript:
| CAR | chimeric antigen receptor |
| FDA | Food and Drug Administration |
| GMP | good manufacturing practice |
| HLA | human leukocyte antigen |
| IL | interleukin |
| iPSC | induced pluripotent stem cell |
| MHC | major histocompatibility complex |
| NK | natural killer |
| TCR | T-cell receptor |
| TME | tumor microenvironment |
| WIPO | World Intellectual Property Organization |
References
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, M.; Chen, Y.; Liu, Y. Current challenges and therapeutic advances of CAR-T cell therapy for solid tumors. Cancer Cell Int. 2024, 24, 133. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018, 15, 31–46. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Grant, S.J.; Grimshaw, A.A.; Silberstein, J.; Murdaugh, D.; Wildes, T.M.; Rosko, A.E.; Giri, S. Clinical Presentation, Risk Factors, and Outcomes of Immune Effector Cell-Associated Neurotoxicity Syndrome Following Chimeric Antigen Receptor T Cell Therapy: A Systematic Review. Transplant. Cell Ther. 2022, 28, 294–302. [Google Scholar] [CrossRef]
- Hines, M.R.; Knight, T.E.; McNerney, K.O.; Leick, M.B.; Jain, T.; Ahmed, S.; Frigault, M.J.; Hill, J.A.; Jain, M.D.; Johnson, W.T.; et al. Immune Effector Cell-Associated Hemophagocytic Lymphohistiocytosis-Like Syndrome. Transplant. Cell Ther. 2023, 29, e431–e438. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef]
- Baulu, E.; Gardet, C.; Chuvin, N.; Depil, S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci. Adv. 2023, 9, eadf3700. [Google Scholar] [CrossRef]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy With TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Araujo, D.M.; Abdul Razak, A.R.; Agulnik, M.; Attia, S.; Blay, J.Y.; Carrasco Garcia, I.; Charlson, J.A.; Choy, E.; Demetri, G.D.; et al. Afamitresgene autoleucel for advanced synovial sarcoma and myxoid round cell liposarcoma (SPEARHEAD-1): An international, open-label, phase 2 trial. Lancet 2024, 403, 1460–1471. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550. [Google Scholar] [CrossRef]
- Uslu, U.; Castelli, S.; June, C.H. CAR T cell combination therapies to treat cancer. Cancer Cell. 2024, 42, 1319–1325. [Google Scholar] [CrossRef]
- Xin, T.; Cheng, L.; Zhou, C.; Zhao, Y.; Hu, Z.; Wu, X. In-Vivo Induced CAR-T Cell for the Potential Breakthrough to Overcome the Barriers of Current CAR-T Cell Therapy. Front. Oncol. 2022, 12, 809754. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.; Ho, N.; Buchholz, C.J. Precision medicine: In vivo CAR therapy as a showcase for receptor-targeted vector platforms. Mol. Ther. 2022, 30, 2401–2415. [Google Scholar] [CrossRef]
- Rossi, M.; Breman, E. Engineering strategies to safely drive CAR T-cells into the future. Front. Immunol. 2024, 15, 1411393. [Google Scholar] [CrossRef]
- Theuerkauf, S.A.; Herrera-Carrillo, E.; John, F.; Zinser, L.J.; Molina, M.A.; Riechert, V.; Thalheimer, F.B.; Börner, K.; Grimm, D.; Chlanda, P.; et al. AAV vectors displaying bispecific DARPins enable dual-control targeted gene delivery. Biomaterials 2023, 303, 122399. [Google Scholar] [CrossRef]
- Parayath, N.N.; Stephan, S.B.; Koehne, A.L.; Nelson, P.S.; Stephan, M.T. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 2020, 11, 6080. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, M.M.; Gong, N.; Mukalel, A.J.; Thatte, A.S.; El-Mayta, R.; Patel, S.K.; Metzloff, A.E.; Swingle, K.L.; Han, X.; Xue, L.; et al. In Vivo mRNA CAR T Cell Engineering via Targeted Ionizable Lipid Nanoparticles with Extrahepatic Tropism. Small 2024, 20, e2304378. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef]
- Ghorashian, S.; Lucchini, G.; Richardson, R.; Nguyen, K.; Terris, C.; Guvenel, A.; Oporto-Espuelas, M.; Yeung, J.; Pinner, D.; Chu, J.; et al. CD19/CD22 targeting with cotransduced CAR T cells to prevent antigen-negative relapse after CAR T-cell therapy for B-cell ALL. Blood 2024, 143, 118–123. [Google Scholar] [CrossRef]
- Verdun, N.; Marks, P. Secondary Cancers after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2024, 390, 584–586. [Google Scholar] [CrossRef]
- Wood, M.E.; Vogel, V.; Ng, A.; Foxhall, L.; Goodwin, P.; Travis, L.B. Second malignant neoplasms: Assessment and strategies for risk reduction. J. Clin. Oncol. 2012, 30, 3734–3745. [Google Scholar] [CrossRef]
- Ghilardi, G.; Fraietta, J.A.; Gerson, J.N.; Van Deerlin, V.M.; Morrissette, J.J.D.; Caponetti, G.C.; Paruzzo, L.; Harris, J.C.; Chong, E.A.; Susanibar Adaniya, S.P.; et al. T cell lymphoma and secondary primary malignancy risk after commercial CAR T cell therapy. Nat. Med. 2024, 30, 984–989. [Google Scholar] [CrossRef]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).





{kind=link}
{kind=link}