The RXR Agonist MSU-42011 Reduces Tumor Burden in a Murine Preclinical NF1-Deficient Model

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Cell Culture
2.2.1. Culture of Human Immortalized NF1−/− Schwann Cells, Murine Nf1-Related MPNSTs, and Murine Lewis Lung Carcinoma Cells (LL2)
2.2.2. Culture of Primary Murine Nf1 Cdkn2a−/− and Nf1−/− DNSCs
2.2.3. Culture and Differentiation of Human THP1 Cells and BMDMs
2.3. Conditioned Medium (CM) Collection
2.4. Cytokine Production
2.5. Western Blot
2.6. Cell Viability
2.7. Real Time-PCR
2.8. In Vivo Experiments
2.9. Immunohistochemistry
2.10. Flow Cytometry
2.11. Triglyceride Assay
2.12. Statistical Analysis
3. Results
3.1. MSU-42011 Reduced Tumor Burden, pERK Levels, and Tumor-Promoting CD206+ Macrophages in a Subcutaneous Kras-Driven Lung Cancer Model
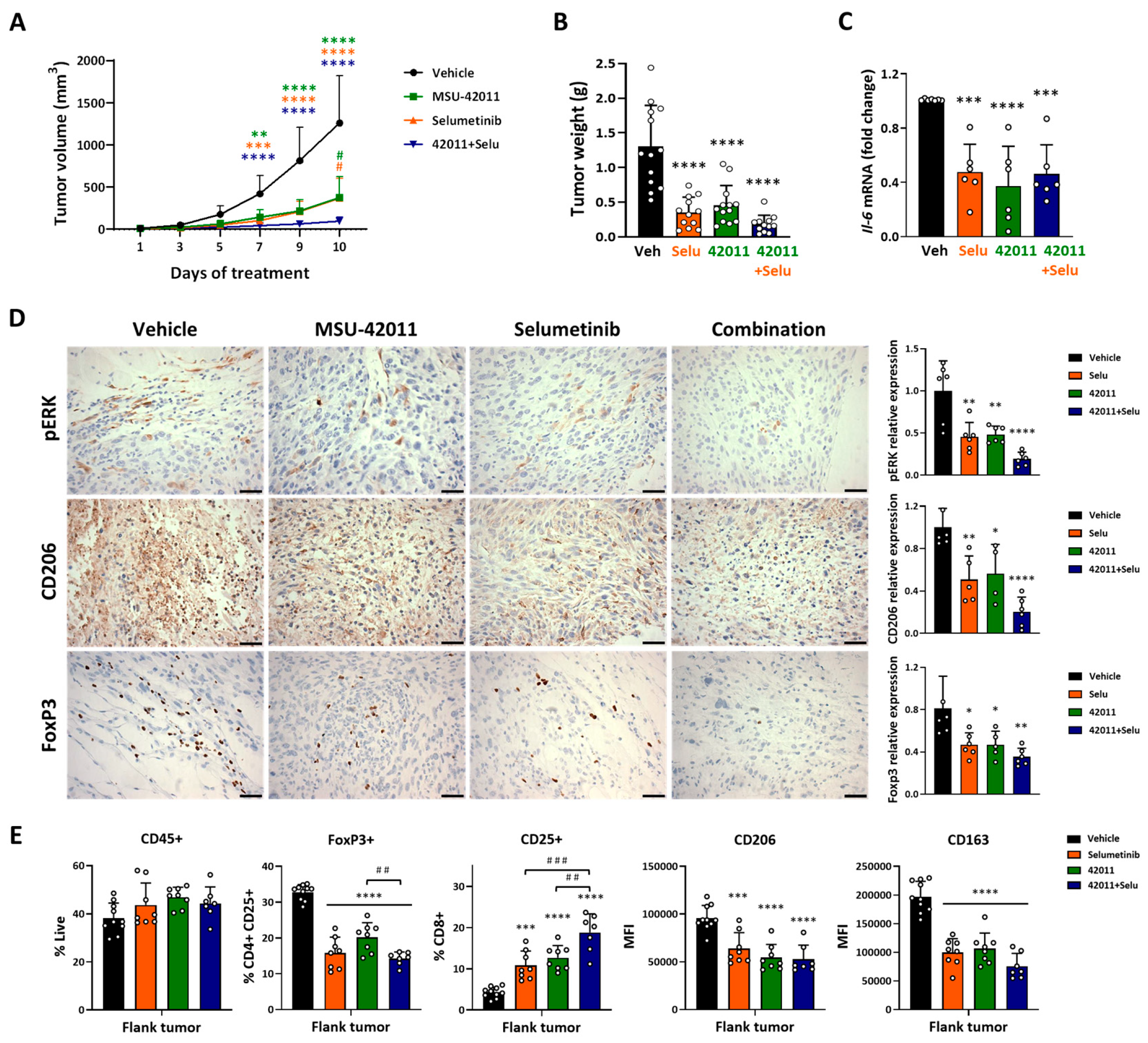
3.2. MSU-42011, Selumetinib, and the Combination Suppressed Tumor Growth, Reduced pERK Levels, and Modulated the Tumor Microenvironment in a Mouse Model of MPNST
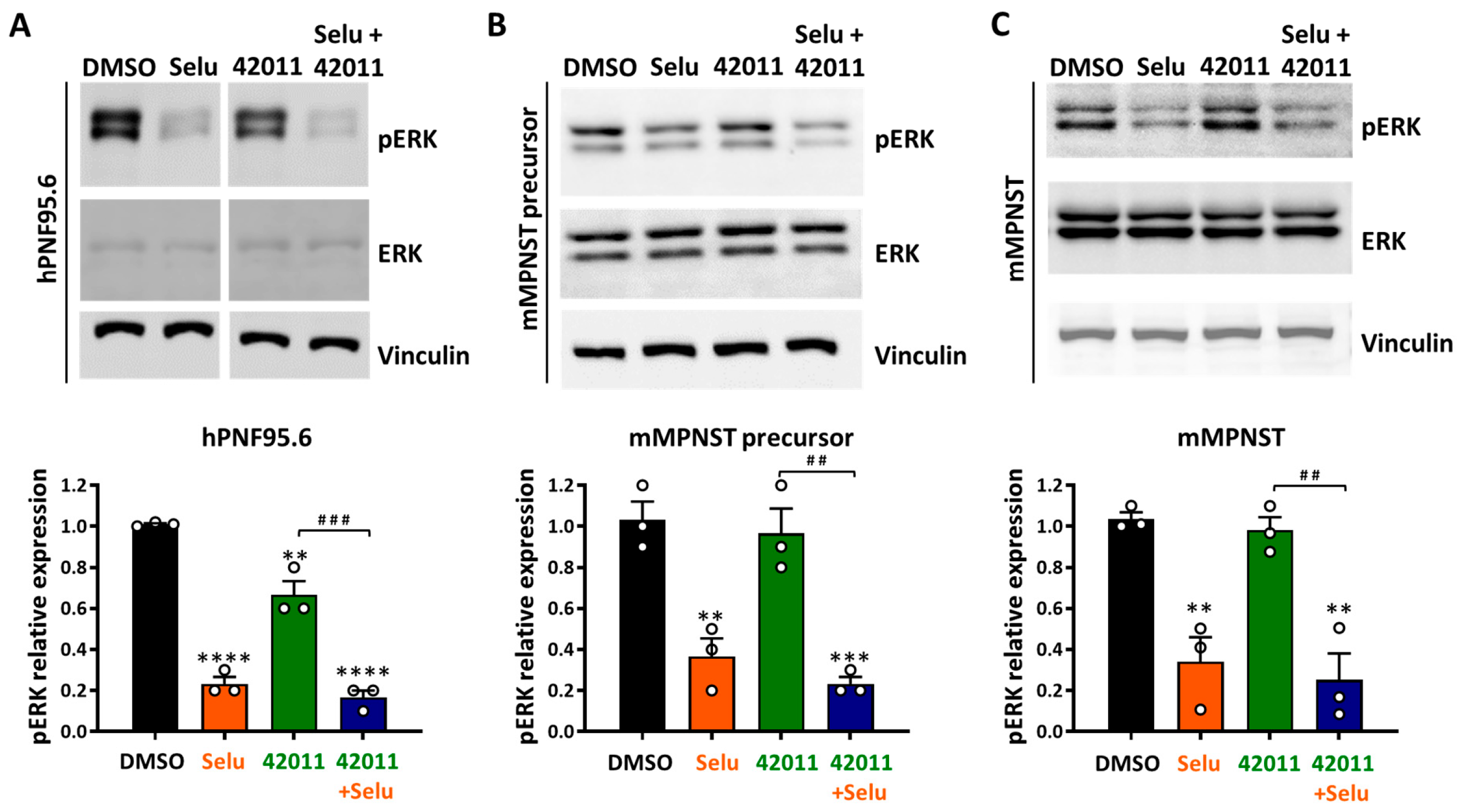
3.3. Selumetinib Reduced pERK Levels in NF1-Deficient Tumor Cells, While MSU-42011 Showed Limited Effects In Vitro
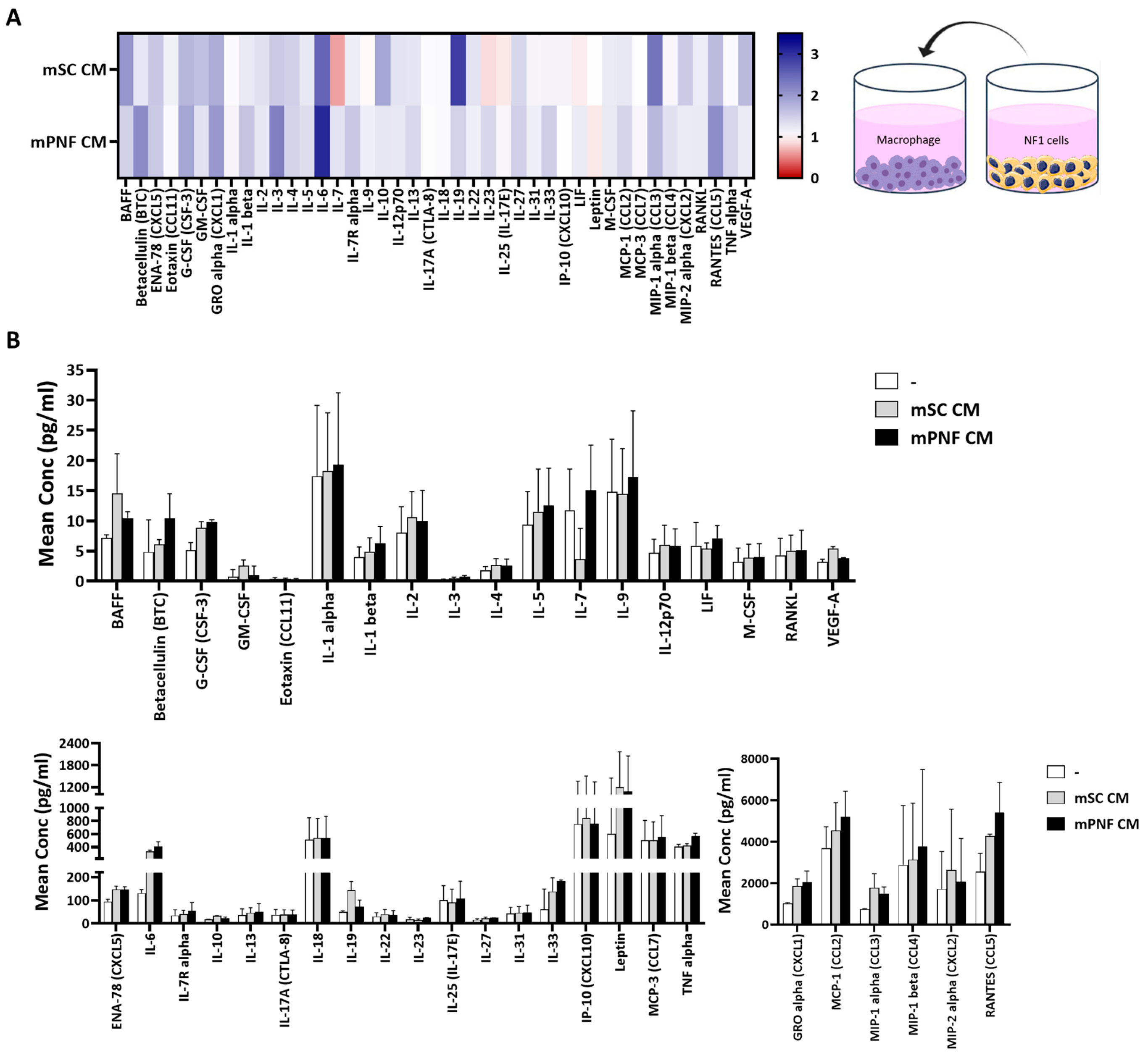
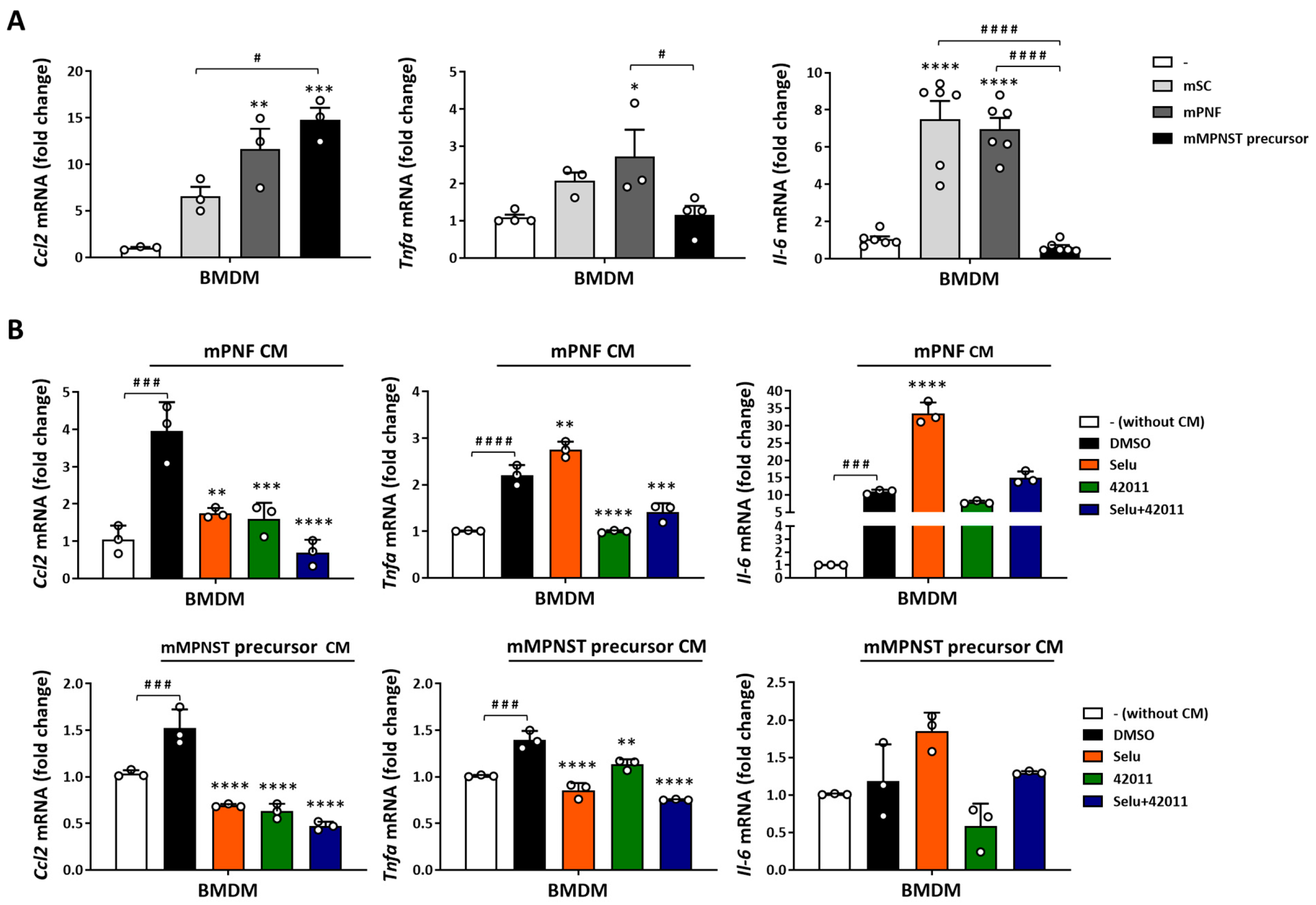
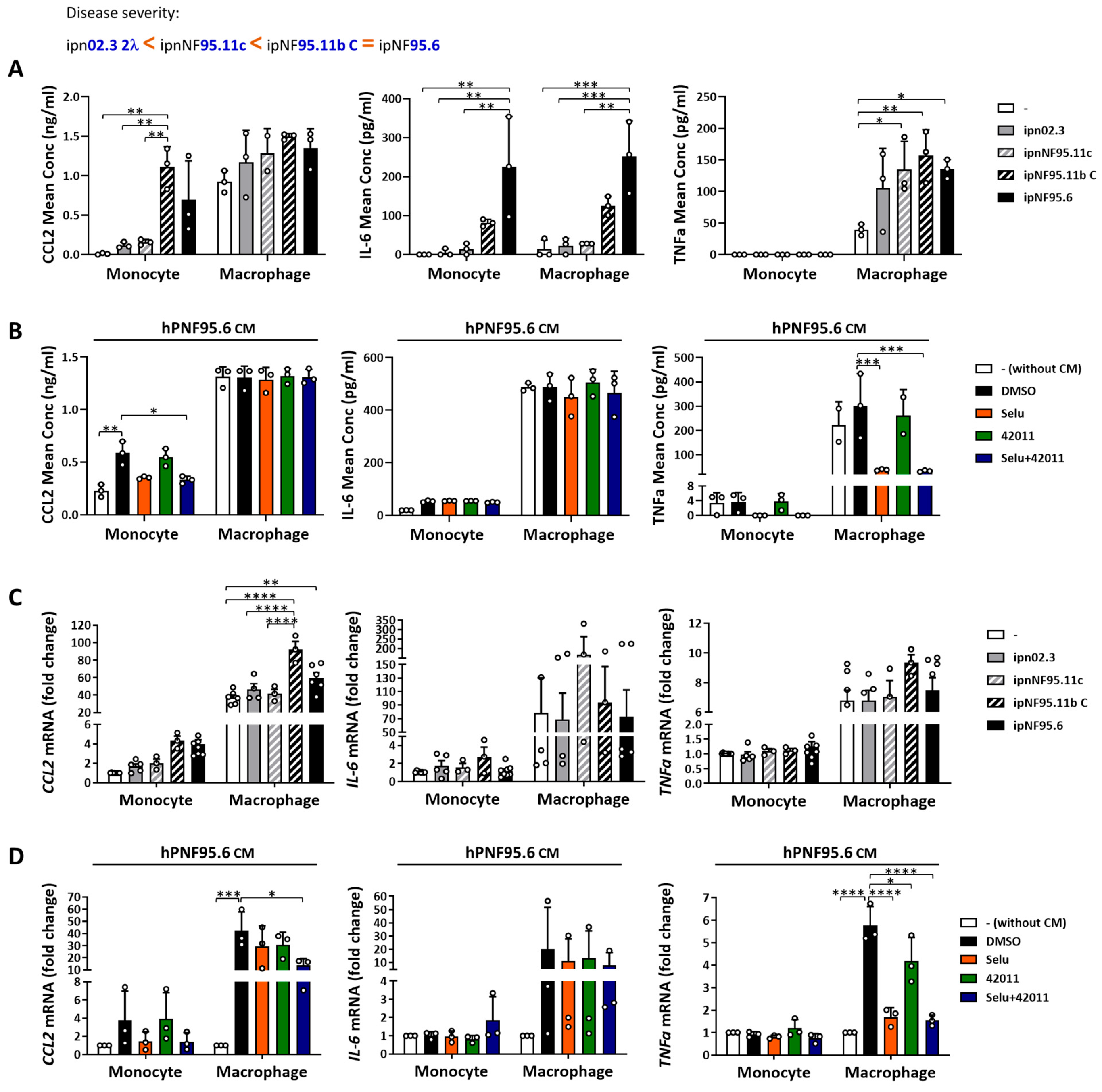
3.4. NF1-Deficient Cells Increased the Secretion and Expression of Cytokine and Chemokine in Macrophages, and Treatment with MSU-42011 and Selumetinib Partially Reduced the Increase
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lalloo, F. Birth Incidence and Prevalence of Tumor-prone Syndromes: Estimates from a UK Family Genetic Register Service. Am. J. Med. Genet. A 2010, 152, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, O.N.; Viskochil, D.H.; Couldwell, W.T. Neurofibromatosis Type 1 and Tumorigenesis: Molecular Mechanisms and Therapeutic Implications. Neurosurg. Focus 2010, 28, E8. [Google Scholar] [CrossRef] [PubMed]
- Le, L.Q.; Parada, L.F. Tumor Microenvironment and Neurofibromatosis Type I: Connecting the GAPs. Oncogene 2007, 26, 4609–4616. [Google Scholar] [CrossRef]
- DeClue, J.E.; Papageorge, A.G.; Fletcher, J.A.; Diehl, S.R.; Ratner, N.; Vass, W.C.; Lowy, D.R. Abnormal Regulation of Mammalian P21ras Contributes to Malignant Tumor Growth in von Recklinghausen (Type 1) Neurofibromatosis. Cell 1992, 69, 265–273. [Google Scholar] [CrossRef]
- Korf, B.R. Plexiform Neurofibromas. Am. J. Med. Genet. 1999, 89, 31–37. [Google Scholar] [CrossRef]
- Prada, C.E.; Rangwala, F.A.; Martin, L.J.; Lovell, A.M.; Saal, H.M.; Schorry, E.K.; Hopkin, R.J. Pediatric Plexiform Neurofibromas: Impact on Morbidity and Mortality in Neurofibromatosis Type 1. J. Pediatr. 2012, 160, 461–467. [Google Scholar] [CrossRef]
- Choucha, A.; Troude, L.; Morin, L.; Fernandes, S.; Baucher, G.; De Simone, M.; Lihi, A.; Mazen, K.; Alseirihi, M.; Passeri, T.; et al. Management of Large Trigeminal Schwannoma: Long-Term Oncologic and Functional Outcome from a Multicentric Retrospective Cohort. Acta Neurochir 2024, 166, 440. [Google Scholar] [CrossRef]
- De Simone, M.; Choucha, A.; Ranalli, C.; Pecoraro, G.; Appay, R.; Chinot, O.L.; Dufour, H.; Iaconetta, G. Astrocytomas IDH-Mutant of Posterior Cranial Fossa, Clinical Presentation, Imaging Features and Onco-Functional Balance in Surgical Management. Neurosurg. Rev. 2025, 48, 271. [Google Scholar] [CrossRef]
- Rabab’h, O.; Gharaibeh, A.; Al-Ramadan, A.; Ismail, M.; Shah, J. Pharmacological Approaches in Neurofibromatosis Type 1-Associated Nervous System Tumors. Cancers 2021, 13, 3880. [Google Scholar] [CrossRef]
- Natalie Wu, L.M.; Lu, Q.R. Therapeutic Targets for Malignant Peripheral Nerve Sheath Tumors. Future Neurol. 2019, 14, FNL7. [Google Scholar] [CrossRef]
- Reilly, K.M.; Kim, A.; Blakely, J.; Ferner, R.E.; Gutmann, D.H.; Legius, E.; Miettinen, M.M.; Randall, R.L.; Ratner, N.; Jumbé, N.L.; et al. Neurofibromatosis Type 1–Associated MPNST State of the Science: Outlining a Research Agenda for the Future. JNCI J. Natl. Cancer Inst. 2017, 109, djx124. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Stewart, D.R.; Reilly, K.M.; Viskochil, D.; Miettinen, M.M.; Widemann, B.C. Malignant Peripheral Nerve Sheath Tumors State of the Science: Leveraging Clinical and Biological Insights into Effective Therapies. Sarcoma 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Rhodes, S.D.; He, Y.; Smith, A.; Jiang, L.; Lu, Q.; Mund, J.; Li, X.; Bessler, W.; Qian, S.; Dyer, W.; et al. Cdkn2a (Arf) Loss Drives NF1-Associated Atypical Neurofibroma and Malignant Transformation. Hum. Mol. Genet. 2019, 28, 2752–2762. [Google Scholar] [CrossRef]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic Evaluation of Atypical Neurofibromatous Tumors and Their Transformation into Malignant Peripheral Nerve Sheath Tumor in Patients with Neurofibromatosis 1—A Consensus Overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA Approves Selumetinib for Neurofibromatosis Type 1 with Symptomatic, Inoperable Plexiform Neurofibromas. 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selumetinib-neurofibromatosis-type-1-symptomatic-inoperable-plexiform-neurofibromas. (accessed on 13 March 2025).
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. KOSELUGO Safely and Effectively. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213756s000lbl.pdf. (accessed on 13 March 2025).
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Reddy, A.T.; Dombi, E.; Allen, J.; Packer, R.; Clapp, W.; Goldman, S.; Schorry, E.; Tonsgard, J.; Blakeley, J.; et al. NFB-17. Mek Inhibitor Binimetinib Shows Clinical Activity in Children with Neurofibromatosis Type 1-Associated Plexiform Neurofibromas: A Report from Pnoc and the NF Clinical Trials Consortium. Neuro Oncol. 2020, 22, iii420–iii421. [Google Scholar] [CrossRef]
- McCowage, G.B.; Mueller, S.; Pratilas, C.A.; Hargrave, D.R.; Moertel, C.L.; Whitlock, J.; Fox, E.; Hingorani, P.; Russo, M.W.; Dasgupta, K.; et al. Trametinib in Pediatric Patients with Neurofibromatosis Type 1 (NF-1)–Associated Plexiform Neurofibroma: A Phase I/IIa Study. J. Clin. Oncol. 2018, 36, 10504. [Google Scholar] [CrossRef]
- Fasih, S.; Suppiyah, S.; Barron, J.; Barnett-Tapia, C.; Avery, R.; Dickson, B.; Ferguson, P.; Swallow, C.; Zadeh, G.; Gupta, A.A. Malignant Transformation of Plexiform Neurofibroma to MPNST While on MEK Inhibitor. Neuro-Oncol. Adv. 2021, 3, vdab033. [Google Scholar] [CrossRef]
- Karmakar, S.; Reilly, K.M. The Role of the Immune System in Neurofibromatosis Type 1-Associated Nervous System Tumors. CNS Oncol. 2017, 6, 45–60. [Google Scholar] [CrossRef]
- Wei, C.-J.; Gu, S.-C.; Ren, J.-Y.; Gu, Y.-H.; Xu, X.-W.; Chou, X.; Lian, X.; Huang, X.; Li, H.-Z.; Gao, Y.-S.; et al. The Impact of Host Immune Cells on the Development of Neurofibromatosis Type 1: The Abnormal Immune System Provides an Immune Microenvironment for Tumorigenesis. Neuro-Oncol. Adv. 2020, 2, i33–i39. [Google Scholar] [CrossRef]
- Fletcher, J.S.; Pundavela, J.; Ratner, N. After Nf1 Loss in Schwann Cells, Inflammation Drives Neurofibroma Formation. Neuro-Oncol. Adv. 2020, 2, i23–i32. [Google Scholar] [CrossRef]
- Prada, C.E.; Jousma, E.; Rizvi, T.A.; Wu, J.; Dunn, R.S.; Mayes, D.A.; Cancelas, J.A.; Dombi, E.; Kim, M.-O.; West, B.L.; et al. Neurofibroma-Associated Macrophages Play Roles in Tumor Growth and Response to Pharmacological Inhibition. Acta Neuropathol. 2013, 125, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, P.P.; Surriga, O.; Beckman, M.J.; de Stanchina, E.; Dematteo, R.P.; Tap, W.D.; Schwartz, G.K. Sustained Inhibition of Receptor Tyrosine Kinases and Macrophage Depletion by PLX3397 and Rapamycin as a Potential New Approach for the Treatment of MPNSTs. Clin. Cancer Res. 2014, 20, 3146–3158. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.S.; Wu, J.; Jessen, W.J.; Pundavela, J.; Miller, J.A.; Dombi, E.; Kim, M.-O.; Rizvi, T.A.; Chetal, K.; Salomonis, N.; et al. Cxcr3-Expressing Leukocytes Are Necessary for Neurofibroma Formation in Mice. JCI Insight 2019, 4, e98601. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeyer, J.L.; Lingo, J.J.; Kaemmer, C.A.; Scherer, A.; Warrier, A.; Voigt, E.; Raygoza Garay, J.A.; McGivney, G.R.; Brockman, Q.R.; Tang, A.; et al. CDK4/6-MEK Inhibition in MPNSTs Causes Plasma Cell Infiltration, Sensitization to PD-L1 Blockade, and Tumor Regression. Clin. Cancer Res. 2023, 29, 3484–3497. [Google Scholar] [CrossRef]
- Leal, A.S.; Moerland, J.A.; Zhang, D.; Carapellucci, S.; Lockwood, B.; Krieger-Burke, T.; Aleiwi, B.; Ellsworth, E.; Liby, K.T. The RXR Agonist MSU42011 Is Effective for the Treatment of Preclinical HER2+ Breast Cancer and Kras-Driven Lung Cancer. Cancers 2021, 13, 5004. [Google Scholar] [CrossRef]
- Moerland, J.A.; Zhang, D.; Reich, L.A.; Carapellucci, S.; Lockwood, B.; Leal, A.S.; Krieger-Burke, T.; Aleiwi, B.; Ellsworth, E.; Liby, K.T. The Novel Rexinoid MSU-42011 Is Effective for the Treatment of Preclinical Kras-Driven Lung Cancer. Sci. Rep. 2020, 10, 22244. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune Modulatory Effects of Oncogenic KRAS in Cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Pereira, F.; Ferreira, A.; Reis, C.A.; Sousa, M.J.; Oliveira, M.J.; Preto, A. KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications. Cells 2022, 11, 398. [Google Scholar] [CrossRef]
- Liby, K.T.; Sporn, M.B. Rexinoids for Prevention and Treatment of Cancer: Opportunities and Challenges. Curr. Top. Med. Chem. 2016. Epub ahead of print. [Google Scholar]
- Leal, A.S.; Hung, P.-Y.; Chowdhury, A.S.; Liby, K.T. Retinoid X Receptor Agonists as Selective Modulators of the Immune System for the Treatment of Cancer. Pharmacol. Ther. 2023, 252, 108561. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.S.; Zydeck, K.; Carapellucci, S.; Reich, L.A.; Zhang, D.; Moerland, J.A.; Sporn, M.B.; Liby, K.T. Retinoid X Receptor Agonist LG100268 Modulates the Immune Microenvironment in Preclinical Breast Cancer Models. NPJ Breast Cancer 2019, 5, 39. [Google Scholar] [CrossRef]
- He, Q.; Sun, C.; Pan, Y. Whole-exome Sequencing Reveals Lewis Lung Carcinoma Is a Hypermutated Kras/Nras–Mutant Cancer with Extensive Regional Mutation Clusters in Its Genome. Sci. Rep. 2024, 14, 100. [Google Scholar] [CrossRef]
- Li, H.; Chang, L.-J.; Neubauer, D.R.; Muir, D.F.; Wallace, M.R. Immortalization of Human Normal and NF1 Neurofibroma Schwann Cells. Lab. Investig. 2016, 96, 1105–1115. [Google Scholar] [CrossRef]
- Tian, Z.; You, Y.; Xiao, M.; Liu, J.; Xu, G.; Ma, C.; Du, Z.; Wang, Y. Inhibition of YAP Sensitizes the Selumetinib Treatment for Neurofibromatosis Type 1 Related Plexiform Neurofibroma. Int. J. Med. Sci. 2023, 20, 125–135. [Google Scholar] [CrossRef]
- Staedtke, V.; Gray-Bethke, T.; Riggins, G.J.; Bai, R.-Y. Preventative Effect of Mebendazole against Malignancies in Neurofibromatosis 1. Genes 2020, 11, 762. [Google Scholar] [CrossRef]
- Flint, A.C.; Mitchell, D.K.; Angus, S.P.; Smith, A.E.; Bessler, W.; Jiang, L.; Mang, H.; Li, X.; Lu, Q.; Rodriguez, B.; et al. Combined CDK4/6 and ERK1/2 Inhibition Enhances Antitumor Activity in NF1-Associated Plexiform Neurofibroma. Clin. Cancer Res. 2023, 29, 3438–3456. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, C.; Patel, A.J.; Liao, C.-P.; Wang, Y.; Le, L.Q. Cells of Origin in the Embryonic Nerve Roots for NF1-Associated Plexiform Neurofibroma. Cancer Cell 2014, 26, 695–706. [Google Scholar] [CrossRef]
- Jessen, W.J.; Miller, S.J.; Jousma, E.; Wu, J.; Rizvi, T.A.; Brundage, M.E.; Eaves, D.; Widemann, B.; Kim, M.-O.; Dombi, E.; et al. MEK Inhibition Exhibits Efficacy in Human and Mouse Neurofibromatosis Tumors. J. Clin. Investig. 2013, 123, 340–347. [Google Scholar] [CrossRef]
- Qu, L.; Tang, X. Bexarotene: A Promising Anticancer Agent. Cancer Chemother. Pharmacol. 2010, 65, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.S.; Reich, L.A.; Moerland, J.A.; Zhang, D.; Liby, K.T. Potential Therapeutic Uses of Rexinoids. Adv. Pharmacol. 2021, 91, 141–183. [Google Scholar] [CrossRef]
- Millesi, E.; Rechberger, J.S.; Wang, H.; Mardini, S.; Spinner, R.J.; Daniels, D.J. Advancements in Therapeutic Approaches for Malignant Peripheral Nerve Sheath Tumor. Ther. Deliv. 2023, 14, 385–389. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Multi-Disciplinary Review and Evaluation NDA 213756 Koselugo (Selumetinib). 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213756Orig1s000MultidisciplineR.pdf. (accessed on 13 March 2025).
- MacCollin, M.; Chiocca, E.A.; Evans, D.G.; Friedman, J.M.; Horvitz, R.; Jaramillo, D.; Lev, M.; Mautner, V.F.; Niimura, M.; Plotkin, S.R.; et al. Diagnostic Criteria for Schwannomatosis. Neurology 2005, 64, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Merker, V.L.; Esparza, S.; Smith, M.J.; Stemmer-Rachamimov, A.; Plotkin, S.R. Clinical Features of Schwannomatosis: A Retrospective Analysis of 87 Patients. Oncologist 2012, 17, 1317–1322. [Google Scholar] [CrossRef]
- Yin, Z.; Wu, L.; Zhang, Y.; Sun, Y.; Chen, J.W.; Subudhi, S.; Ho, W.; Lee, G.Y.; Wang, A.; Gao, X.; et al. Co-Targeting IL-6 and EGFR Signaling for the Treatment of Schwannomatosis and Associated Pain. bioRxiv 2023. [Google Scholar] [CrossRef]
- Shou, J.; Zhang, Z.; Lai, Y.; Chen, Z.; Huang, J. Worse Outcome in Breast Cancer with Higher Tumor-Infiltrating FOXP3+ Tregs: A Systematic Review and Meta-Analysis. BMC Cancer 2016, 16, 687. [Google Scholar] [CrossRef]
- Pinto, S.M.; Kim, H.; Subbannayya, Y.; Giambelluca, M.S.; Bösl, K.; Ryan, L.; Sharma, A.; Kandasamy, R.K. Comparative Proteomic Analysis Reveals Varying Impact on Immune Responses in Phorbol 12-Myristate-13-Acetate-Mediated THP-1 Monocyte-to-Macrophage Differentiation. Front. Immunol. 2021, 12, 679458. [Google Scholar] [CrossRef]
- Kundu, P.; Shankar, B.S. Macrophage Induced ERK-TGF-Β1 Signaling in MCF7 Breast Cancer Cells Result in Reversible Cancer Stem Cell Plasticity and Epithelial Mesenchymal Transition. Biochim. Biophys. Acta (BBA) General. Subj. 2022, 1866, 130215. [Google Scholar] [CrossRef]
- Kumar, S.; Principe, D.R.; Singh, S.K.; Viswakarma, N.; Sondarva, G.; Rana, B.; Rana, A. Mitogen-Activated Protein Kinase Inhibitors and T-Cell-Dependent Immunotherapy in Cancer. Pharmaceuticals 2020, 13, 9. [Google Scholar] [CrossRef]
- Fletcher, J.S.; Springer, M.G.; Choi, K.; Jousma, E.; Rizvi, T.A.; Dombi, E.; Kim, M.-O.; Wu, J.; Ratner, N. STAT3 Inhibition Reduces Macrophage Number and Tumor Growth in Neurofibroma. Oncogene 2019, 38, 2876–2884. [Google Scholar] [CrossRef]
- Mendoza, R.; Banerjee, I.; Manna, D.; Reghupaty, S.C.; Yetirajam, R.; Sarkar, D. Mouse Bone Marrow Cell Isolation and Macrophage Differentiation; Humana: New York, NY, USA, 2022; Volume 2455, pp. 85–91. [Google Scholar] [CrossRef]
- Martinez, F.O. Macrophage Activation and Polarization. Front. Biosci. 2008, 13, 453. [Google Scholar] [CrossRef] [PubMed]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 Polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of Tumor-Associated Macrophages and Cancer Chemotherapy. Oncoimmunology 2019, 8, e1596004. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Knelson, E.H.; Sequist, L.V. A Bright Future for KRAS Inhibitors. Nat. Cancer 2020, 1, 25–27. [Google Scholar] [CrossRef]
- Kitajima, S.; Thummalapalli, R.; Barbie, D.A. Inflammation as a Driver and Vulnerability of KRAS Mediated Oncogenesis. Semin. Cell Dev. Biol. 2016, 58, 127–135. [Google Scholar] [CrossRef]
- Farschtschi, S.; Park, S.-J.; Sawitzki, B.; Oh, S.-J.; Kluwe, L.; Mautner, V.F.; Kurtz, A. Effector T Cell Subclasses Associate with Tumor Burden in Neurofibromatosis Type 1 Patients. Cancer Immunol. Immunother. 2016, 65, 1113–1121. [Google Scholar] [CrossRef]
- Mitchell, D.K.; Burgess, B.; White, E.E.; Smith, A.E.; Sierra Potchanant, E.A.; Mang, H.; Hickey, B.E.; Lu, Q.; Qian, S.; Bessler, W.; et al. Spatial Gene-Expression Profiling Unveils Immuno-Oncogenic Programs of NF1-Associated Peripheral Nerve Sheath Tumor Progression. Clin. Cancer Res. 2024, 30, 1038–1053. [Google Scholar] [CrossRef] [PubMed]
- Shurell, E.; Singh, A.S.; Crompton, J.G.; Jensen, S.; Li, Y.; Dry, S.; Nelson, S.; Chmielowski, B.; Bernthal, N.; Federman, N.; et al. Characterizing the Immune Microenvironment of Malignant Peripheral Nerve Sheath Tumor by PD-L1 Expression and Presence of CD8+ Tumor Infiltrating Lymphocytes. Oncotarget 2016, 7, 64300–64308. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Komurov, K.; Fletcher, J.S.; Jousma, E.; Cancelas, J.A.; Wu, J.; Ratner, N. An Inflammatory Gene Signature Distinguishes Neurofibroma Schwann Cells and Macrophages from Cells in the Normal Peripheral Nervous System. Sci. Rep. 2017, 7, 43315. [Google Scholar] [CrossRef]
- Ramlau, R.; Zatloukal, P.; Jassem, J.; Schwarzenberger, P.; Orlov, S.V.; Gottfried, M.; Pereira, J.R.; Temperley, G.; Negro-Vilar, R.; Rahal, S.; et al. Randomized Phase III Trial Comparing Bexarotene (L1069-49)/Cisplatin/Vinorelbine With Cisplatin/Vinorelbine in Chemotherapy-Naïve Patients With Advanced or Metastatic Non–Small-Cell Lung Cancer: SPIRIT I. J. Clin. Oncol. 2008, 26, 1886–1892. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Khuri, F.R.; von Pawel, J.; Gatzemeier, U.; Miller, W.H.; Jotte, R.M.; Le Treut, J.; Sun, S.-L.; Zhang, J.K.; Dziewanowska, Z.E.; et al. Phase III Trial Comparing Carboplatin, Paclitaxel, and Bexarotene With Carboplatin and Paclitaxel in Chemotherapy-Naïve Patients With Advanced or Metastatic Non–Small-Cell Lung Cancer: SPIRIT II. J. Clin. Oncol. 2008, 26, 1879–1885. [Google Scholar] [CrossRef]
- Dragnev, K.H.; Ma, T.; Cyrus, J.; Galimberti, F.; Memoli, V.; Busch, A.M.; Tsongalis, G.J.; Seltzer, M.; Johnstone, D.; Erkmen, C.P.; et al. Bexarotene Plus Erlotinib Suppress Lung Carcinogenesis Independent of KRAS Mutations in Two Clinical Trials and Transgenic Models. Cancer Prev. Res. 2011, 4, 818–828. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Hill, J.; Kim, H.-T.; Shen, Q.; Bissonnette, R.P.; Lamph, W.W.; Brown, P.H. The Rexinoid, Bexarotene, Prevents the Development of Premalignant Lesions in MMTV-ErbB2 Mice. Br. J. Cancer 2008, 98, 1380–1388. [Google Scholar] [CrossRef]
- Li, Y.; Shen, Q.; Kim, H.-T.; Bissonnette, R.P.; Lamph, W.W.; Yan, B.; Brown, P.H. The Rexinoid Bexarotene Represses Cyclin D1 Transcription by Inducing the DEC2 Transcriptional Repressor. Breast Cancer Res. Treat. 2011, 128, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T.; Menéndez-Gutiérrez, M.P.; Cedenilla, M.; Ricote, M. Retinoid X Receptors in Macrophage Biology. Trends Endocrinol. Metab. 2013, 24, 460–468. [Google Scholar] [CrossRef]
- Kiss, M.; Czimmerer, Z.; Nagy, G.; Bieniasz-Krzywiec, P.; Ehling, M.; Pap, A.; Poliska, S.; Boto, P.; Tzerpos, P.; Horvath, A.; et al. Retinoid X Receptor Suppresses a Metastasis-Promoting Transcriptional Program in Myeloid Cells via a Ligand-Insensitive Mechanism. Proc. Natl. Acad. Sci. USA 2017, 114, 10725–10730. [Google Scholar] [CrossRef]
- Ma, F.; Liu, S.-Y.; Razani, B.; Arora, N.; Li, B.; Kagechika, H.; Tontonoz, P.; Núñez, V.; Ricote, M.; Cheng, G. Retinoid X Receptor α Attenuates Host Antiviral Response by Suppressing Type I Interferon. Nat. Commun. 2014, 5, 5494. [Google Scholar] [CrossRef] [PubMed]
- Standeven, A.M.; Thacher, S.M.; Yuan, Y.-D.; Escobar, M.; Vuligonda, V.; Beard, R.L.; Chandraratna, R.A.S. Retinoid X Receptor Agonist Elevation of Serum Triglycerides in Rats by Potentiation of Retinoic Acid Receptor Agonist Induction or by Action as Single Agents 2 2Abbreviations: RARs, Retinoic Acid Receptors; and RXRs, Retinoid X Receptors. Biochem. Pharmacol. 2001, 62, 1501–1509. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, P.-Y.; Moerland, J.A.; Leal, A.S.; Aleiwi, B.; Ellsworth, E.; Clapp, D.W.; Staedtke, V.; Bai, R.; Liby, K.T. The RXR Agonist MSU-42011 Reduces Tumor Burden in a Murine Preclinical NF1-Deficient Model. Cancers 2025, 17, 1920. https://doi.org/10.3390/cancers17121920
Hung P-Y, Moerland JA, Leal AS, Aleiwi B, Ellsworth E, Clapp DW, Staedtke V, Bai R, Liby KT. The RXR Agonist MSU-42011 Reduces Tumor Burden in a Murine Preclinical NF1-Deficient Model. Cancers. 2025; 17(12):1920. https://doi.org/10.3390/cancers17121920
Chicago/Turabian StyleHung, Pei-Yu, Jessica A. Moerland, Ana S. Leal, Bilal Aleiwi, Edmund Ellsworth, D. Wade Clapp, Verena Staedtke, Renyuan Bai, and Karen T. Liby. 2025. "The RXR Agonist MSU-42011 Reduces Tumor Burden in a Murine Preclinical NF1-Deficient Model" Cancers 17, no. 12: 1920. https://doi.org/10.3390/cancers17121920
APA StyleHung, P.-Y., Moerland, J. A., Leal, A. S., Aleiwi, B., Ellsworth, E., Clapp, D. W., Staedtke, V., Bai, R., & Liby, K. T. (2025). The RXR Agonist MSU-42011 Reduces Tumor Burden in a Murine Preclinical NF1-Deficient Model. Cancers, 17(12), 1920. https://doi.org/10.3390/cancers17121920