
Interplay Between the Epigenome, the Microenvironment, and the Immune System in Neuroblastoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Oncogenes in Neuroblastoma
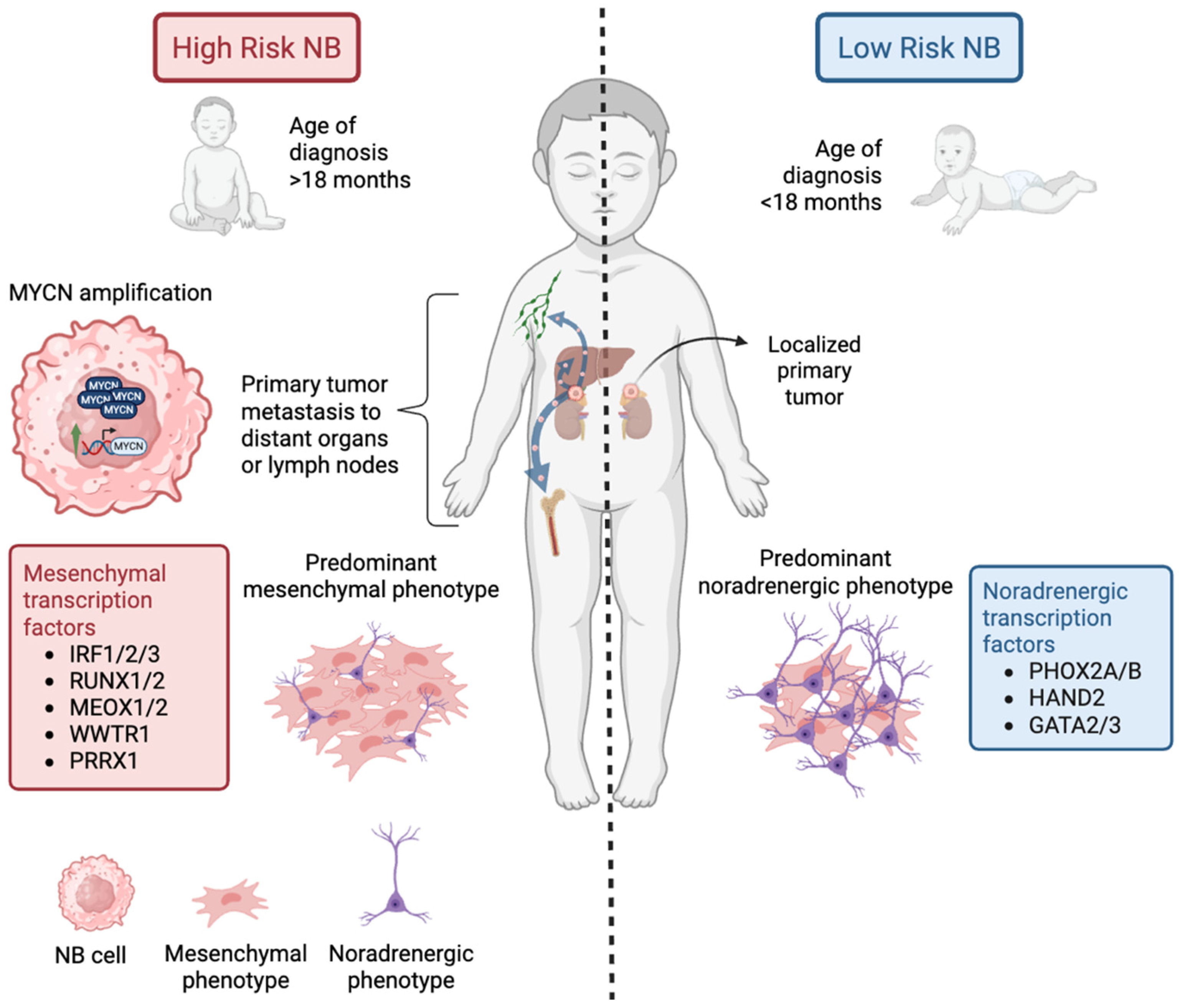
2.1. MYCN
2.2. ALK
3. Epigenetic Alterations in Neuroblastoma
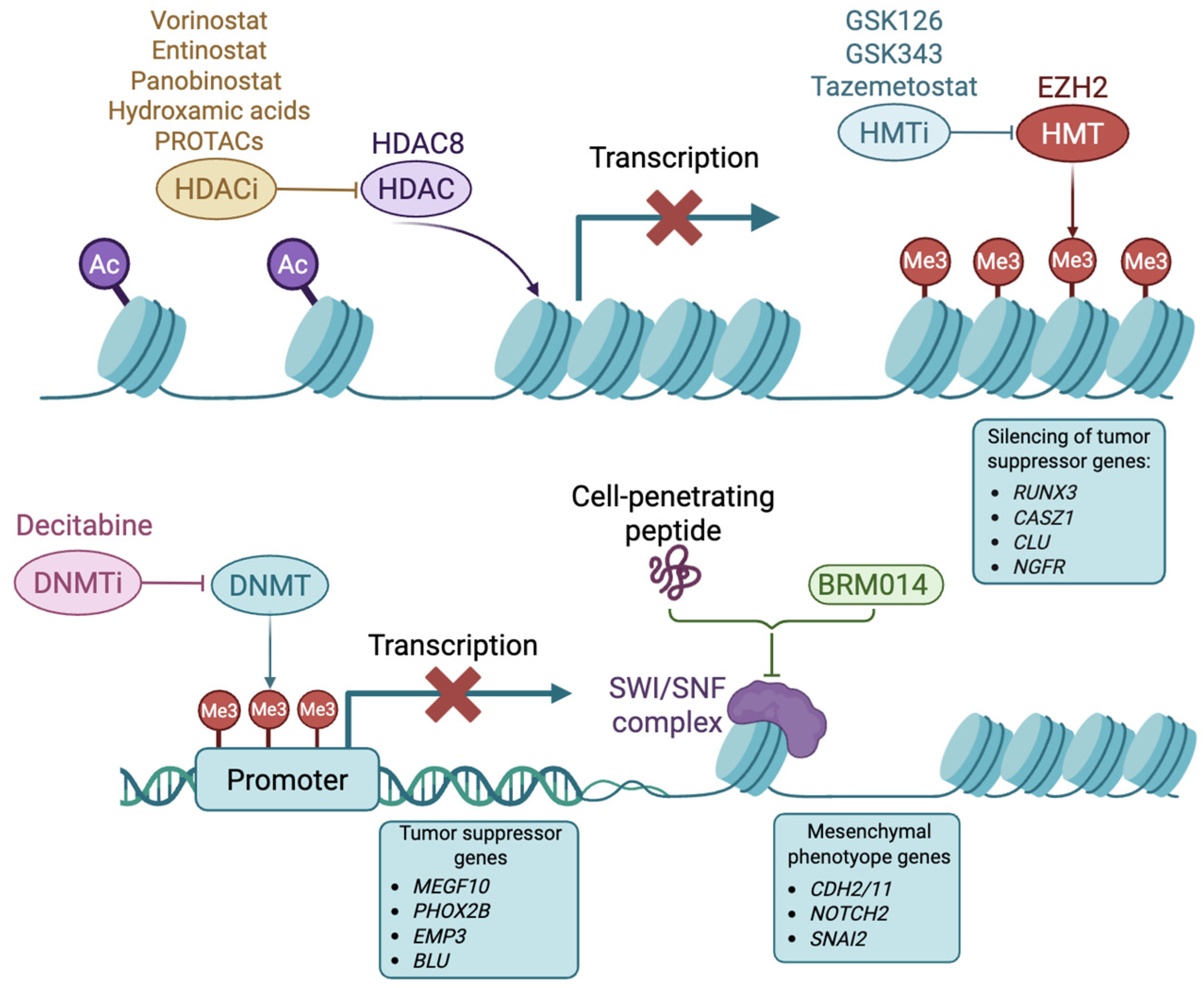
3.1. Histone Deacetylases
3.2. Histone Methyltransferases
3.3. DNA Methyltransferases
3.4. Chromatin Remodelers
3.5. Super-Enhancers
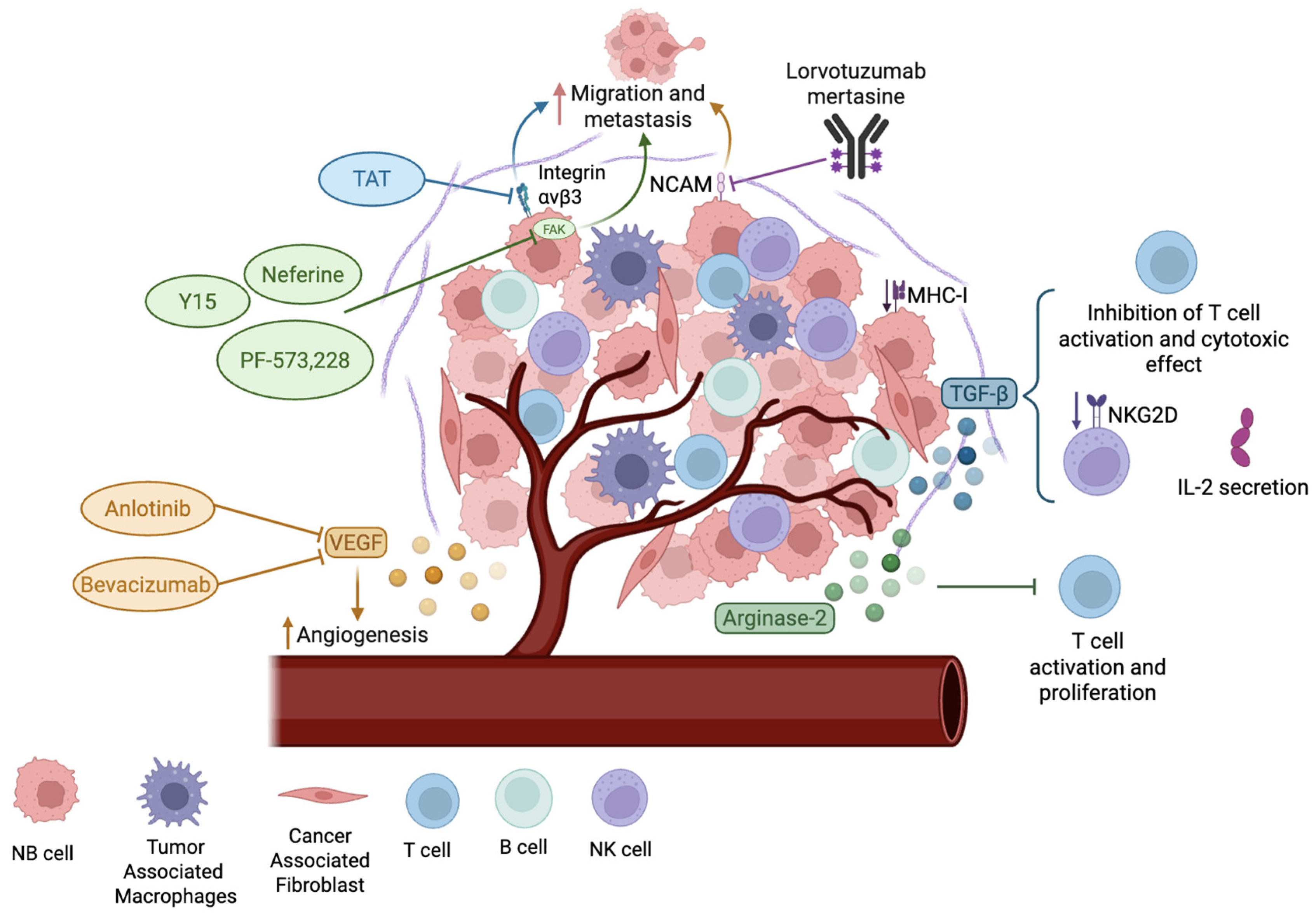
4. Microenvironment in Neuroblastoma
5. Targeting Immunological Alterations in NB
5.1. Targeting GD2 as an Approved Immunotherapeutic Treatment of NB
5.2. Immune Checkpoint Inhibitor (ICI) Therapy
5.3. T Cell and CART Approaches in NB
5.4. NK Cells and CAR NK Approaches in NB Treatment
6. Future Perspectives
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zafar, A.; Wang, W.; Liu, G.; Wang, X.; Xian, W.; McKeon, F.; Foster, J.; Zhou, J.; Zhang, R. Molecular Targeting Therapies for Neuroblastoma: Progress and Challenges. Med. Res. Rev. 2021, 41, 961–1021. [Google Scholar] [CrossRef] [PubMed]
- Jansky, S.; Sharma, A.K.; Körber, V.; Quintero, A.; Toprak, U.H.; Wecht, E.M.; Gartlgruber, M.; Greco, A.; Chomsky, E.; Grünewald, T.G.P.; et al. Single-Cell Transcriptomic Analyses Provide Insights into the Developmental Origins of Neuroblastoma. Nat. Genet. 2021, 53, 683–693. [Google Scholar] [CrossRef]
- Beaudry, A.; Jacques-Ricard, S.; Darracq, A.; Sgarioto, N.; Garcia, A.; García, T.R.; Lemieux, W.; Béland, K.; Haddad, E.; Cordeiro, P.; et al. Repurposing Disulfiram, an Alcohol-Abuse Drug, in Neuroblastoma Causes KAT2A Downregulation and in Vivo Activity with a Water/Oil Emulsion. Sci. Rep. 2023, 13, 16443. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, X.; Li, N.; Guo, Y.; Yang, X.; Lei, Y. Therapy Resistance in Neuroblastoma: Mechanisms and Reversal Strategies. Front. Pharmacol. 2023, 14, 1114295. [Google Scholar] [CrossRef] [PubMed]
- Zeineldin, M.; Patel, A.G.; Dyer, M.A. Neuroblastoma: When Differentiation Goes Awry. Neuron 2022, 110, 2916–2928. [Google Scholar] [CrossRef]
- Körber, V.; Stainczyk, S.A.; Kurilov, R.; Henrich, K.-O.; Hero, B.; Brors, B.; Westermann, F.; Höfer, T. Neuroblastoma Arises in Early Fetal Development and Its Evolutionary Duration Predicts Outcome. Nat. Genet. 2023, 55, 619–630. [Google Scholar] [CrossRef]
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2021, 39, 3229–3241. [Google Scholar] [CrossRef]
- Chung, C.; Boterberg, T.; Lucas, J.; Panoff, J.; Valteau-Couanet, D.; Hero, B.; Bagatell, R.; Hill-Kayser, C.E. Neuroblastoma. Pediatr. Blood Cancer 2021, 68, e28473. [Google Scholar] [CrossRef] [PubMed]
- Sokol, E.; Desai, A. The Evolution of Risk Classification for Neuroblastoma. Children 2019, 6, 27. [Google Scholar] [CrossRef]
- Chidiac, C.; Hu, A.; Dunn, E.; Rhee, D.S. Characteristics of Image Defined Risk Factors on Outcomes for Primary Resection of Neuroblastoma. Surg. Pract. Sci. 2023, 14, 100195. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The International Neuroblastoma Risk Group (INRG) Staging System: An INRG Task Force Report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef]
- Dong, R.; Yang, R.; Zhan, Y.; Lai, H.-D.; Ye, C.-J.; Yao, X.-Y.; Luo, W.-Q.; Cheng, X.-M.; Miao, J.-J.; Wang, J.-F.; et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell 2020, 38, 716–733.e6. [Google Scholar] [CrossRef]
- Yu, E.Y.; Cheung, N.-K.V.; Lue, N.F. Connecting Telomere Maintenance and Regulation to the Developmental Origin and Differentiation States of Neuroblastoma Tumor Cells. J. Hematol. Oncol. 2022, 15, 117. [Google Scholar] [CrossRef] [PubMed]
- Ponzoni, M.; Bachetti, T.; Corrias, M.V.; Brignole, C.; Pastorino, F.; Calarco, E.; Bensa, V.; Giusto, E.; Ceccherini, I.; Perri, P. Recent Advances in the Developmental Origin of Neuroblastoma: An Overview. J. Exp. Clin. Cancer Res. 2022, 41, 92. [Google Scholar] [CrossRef]
- Kameneva, P.; Artemov, A.V.; Kastriti, M.E.; Faure, L.; Olsen, T.K.; Otte, J.; Erickson, A.; Semsch, B.; Andersson, E.R.; Ratz, M.; et al. Single-Cell Transcriptomics of Human Embryos Identifies Multiple Sympathoblast Lineages with Potential Implications for Neuroblastoma Origin. Nat. Genet. 2021, 53, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, R.; DuBois, S.G.; Naranjo, A.; Belle, J.; Goldsmith, K.C.; Park, J.R.; Irwin, M.S. Children’s Oncology Group’s 2023 Blueprint for Research: Neuroblastoma. Pediatr. Blood Cancer 2023, 70, e30572. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, V.P.; Matthay, K.K. Neuroblastoma: Clinical and Biological Approach to Risk Stratification and Treatment. Cell Tissue Res. 2018, 372, 195–209. [Google Scholar] [CrossRef]
- Meany, H.J. Non-High-Risk Neuroblastoma: Classification and Achievements in Therapy. Children 2019, 6, 5. [Google Scholar] [CrossRef]
- Krystal, J.; Foster, J.H. Treatment of High-Risk Neuroblastoma. Children 2023, 10, 1302. [Google Scholar] [CrossRef]
- DuBois, S.G.; Macy, M.E.; Henderson, T.O. High-Risk and Relapsed Neuroblastoma: Toward More Cures and Better Outcomes. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 768–780. [Google Scholar] [CrossRef]
- Campos Cogo, S.; Gradowski Farias da Costa do Nascimento, T.; de Almeida Brehm Pinhatti, F.; de França Junior, N.; Santos Rodrigues, B.; Cavalli, L.R.; Elifio-Esposito, S. An Overview of Neuroblastoma Cell Lineage Phenotypes and in Vitro Models. Exp. Biol. Med. 2020, 245, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma Is Composed of Two Super-Enhancer-Associated Differentiation States. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of Neuroblastoma Cell Identity Defined by Transcriptional Circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef]
- Gautier, M.; Thirant, C.; Delattre, O.; Janoueix-Lerosey, I. Plasticity in Neuroblastoma Cell Identity Defines a Noradrenergic-to-Mesenchymal Transition (NMT). Cancers 2021, 13, 2904. [Google Scholar] [CrossRef]
- Van Nes, J.; Chan, A.; Van Groningen, T.; Van Sluis, P.; Koster, J.; Versteeg, R. A NOTCH3 Transcriptional Module Induces Cell Motility in Neuroblastoma. Clin. Cancer Res. 2013, 19, 3485–3494. [Google Scholar] [CrossRef]
- Durbin, A.D.; Versteeg, R. Cell State Plasticity in Neuroblastoma. EJC Paediatr. Oncol. 2024, 4, 100184. [Google Scholar] [CrossRef]
- Avitabile, M.; Bonfiglio, F.; Aievola, V.; Cantalupo, S.; Maiorino, T.; Lasorsa, V.A.; Domenicotti, C.; Marengo, B.; Heger, Z.; Adam, V.; et al. Single-Cell Transcriptomics of Neuroblastoma Identifies Chemoresistance-Associated Genes and Pathways. Comput. Struct. Biotechnol. J. 2022, 20, 4437. [Google Scholar] [CrossRef]
- Jahangiri, L. Epithelial to Mesenchymal Transition in Neuroblastoma: Mechanisms and Therapeutic Considerations. Curr. Tissue Microenviron. Rep. 2024, 5, 91–108. [Google Scholar] [CrossRef]
- Friedman, D.; Henderson, T. Late Effects and Survivorship Issues in Patients with Neuroblastoma. Children 2018, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Yeung, V.; Gabriel, M.; Padhye, B.D. Late Effects and Treatment Related Morbidity Associated with Treatment of Neuroblastoma Patients in a Tertiary Paediatric Centre. Cancer Rep. 2023, 6, e1738. [Google Scholar] [CrossRef] [PubMed]
- Lerone, M.; Ognibene, M.; Pezzolo, A.; Martucciello, G.; Zara, F.; Morini, M.; Mazzocco, K. Molecular Genetics in Neuroblastoma Prognosis. Children 2021, 8, 456. [Google Scholar] [CrossRef] [PubMed]
- Braoudaki, M.; Hatziagapiou, K.; Zaravinos, A.; Lambrou, G.I. MYCN in Neuroblastoma: “Old Wine into New Wineskins”. Diseases 2021, 9, 78. [Google Scholar] [CrossRef]
- Higashi, M.; Sakai, K.; Fumino, S.; Aoi, S.; Furukawa, T.; Tajiri, T. The Roles Played by the MYCN, Trk, and ALK Genes in Neuroblastoma and Neural Development. Surg. Today 2019, 49, 721–727. [Google Scholar] [CrossRef]
- Zhang, J.T.; Weng, Z.H.; Tsang, K.S.; Tsang, L.L.; Chan, H.C.; Jiang, X.H. MycN Is Critical for the Maintenance of Human Embryonic Stem Cell-Derived Neural Crest Stem Cells. PLoS ONE 2016, 11, e0148062. [Google Scholar] [CrossRef]
- Otte, J.; Dyberg, C.; Pepich, A.; Johnsen, J.I. MYCN Function in Neuroblastoma Development. Front. Oncol. 2021, 10, 624079. [Google Scholar] [CrossRef]
- Bartolucci, D.; Montemurro, L.; Raieli, S.; Lampis, S.; Pession, A.; Hrelia, P.; Tonelli, R. MYCN Impact on High-Risk Neuroblastoma: From Diagnosis and Prognosis to Targeted Treatment. Cancers 2022, 14, 4421. [Google Scholar] [CrossRef]
- Izumi, H.; Kaneko, Y. Evidence of Asymmetric Cell Division and Centrosome Inheritance in Human Neuroblastoma Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 18048–18053. [Google Scholar] [CrossRef]
- Bansal, M.; Gupta, A.; Ding, H.-F. MYCN and Metabolic Reprogramming in Neuroblastoma. Cancers 2022, 14, 4113. [Google Scholar] [CrossRef] [PubMed]
- Cotterman, R.; Knoepfler, P.S. N-Myc Regulates Expression of Pluripotency Genes in Neuroblastoma Including Lif, Klf2, Klf4, and Lin28b. PLoS ONE 2009, 4, e5799. [Google Scholar] [CrossRef]
- Selmi, A.; de Saint-Jean, M.; Jallas, A.-C.; Garin, E.; Hogarty, M.D.; Bénard, J.; Puisieux, A.; Marabelle, A.; Valsesia-Wittmann, S. TWIST1 Is a Direct Transcriptional Target of MYCN and MYC in Neuroblastoma. Cancer Lett. 2015, 357, 412–418. [Google Scholar] [CrossRef]
- Pastor, E.R.; Mousa, S.A. Current Management of Neuroblastoma and Future Direction. Crit. Rev. Oncol. Hematol. 2019, 138, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Fox, E.; Teachey, D.T.; Reid, J.M.; Safgren, S.L.; Carol, H.; Lock, R.B.; Houghton, P.J.; Smith, M.A.; Hall, D.; et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children’s Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin. Cancer Res. 2019, 25, 3229–3238. [Google Scholar] [CrossRef]
- DuBois, S.G.; Mosse, Y.P.; Fox, E.; Kudgus, R.A.; Reid, J.M.; McGovern, R.; Groshen, S.; Bagatell, R.; Maris, J.M.; Twist, C.J.; et al. Phase II Trial of Alisertib in Combination with Irinotecan and Temozolomide for Patients with Relapsed or Refractory Neuroblastoma. Clin. Cancer Res. 2018, 24, 6142–6149. [Google Scholar] [CrossRef]
- Boi, D.; Souvalidou, F.; Capelli, D.; Polverino, F.; Marini, G.; Montanari, R.; Pochetti, G.; Tramonti, A.; Contestabile, R.; Trisciuoglio, D.; et al. PHA-680626 Is an Effective Inhibitor of the Interaction between Aurora-A and N-Myc. Int. J. Mol. Sci. 2021, 22, 13122. [Google Scholar] [CrossRef]
- Gustafson, W.C.; Meyerowitz, J.G.; Nekritz, E.A.; Chen, J.; Benes, C.; Charron, E.; Simonds, E.F.; Seeger, R.; Matthay, K.K.; Hertz, N.T.; et al. Drugging MYCN through an Allosteric Transition in Aurora Kinase A. Cancer Cell 2014, 26, 414–427. [Google Scholar] [CrossRef]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef]
- Kurimchak, A.M.; Shelton, C.; Duncan, K.E.; Johnson, K.J.; Brown, J.; O’Brien, S.; Gabbasov, R.; Fink, L.S.; Li, Y.; Lounsbury, N.; et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Rep. 2016, 16, 1273–1286. [Google Scholar] [CrossRef]
- Lai, J.; Liu, Z.; Zhao, Y.; Ma, C.; Huang, H. Anticancer Effects of I-BET151, an Inhibitor of Bromodomain and Extra-Terminal Domain Proteins. Front. Oncol. 2021, 11, 716830. [Google Scholar] [CrossRef] [PubMed]
- Felgenhauer, J.; Tomino, L.; Selich-Anderson, J.; Bopp, E.; Shah, N. Dual BRD4 and AURKA Inhibition Is Synergistic against MYCN-Amplified and Nonamplified Neuroblastoma. Neoplasia 2018, 20, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as Emerging Anticancer Therapeutics. Oncogene 2020, 39, 4909–4924. [Google Scholar] [CrossRef]
- Jia, S.-Q.; Zhuo, R.; Zhang, Z.-M.; Yang, Y.; Tao, Y.-F.; Wang, J.-W.; Li, X.-L.; Xie, Y.; Li, G.; Wu, D.; et al. The BRD4 Inhibitor DBET57 Exerts Anticancer Effects by Targeting Superenhancer-Related Genes in Neuroblastoma. J. Immunol. Res. 2022, 2022, 7945884. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, X.; Zhuo, R.; Tao, Y.; Liang, W.; Yang, R.; Chen, Y.; Cao, H.; Jia, S.; Yu, J.; et al. BRD4 Inhibitor MZ1 Exerts Anti-Cancer Effects by Targeting MYCN and MAPK Signaling in Neuroblastoma. Biochem. Biophys. Res. Commun. 2022, 604, 63–69. [Google Scholar] [CrossRef]
- Hogarty, M.D.; Norris, M.D.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1 Is a Critical Determinant of MYCN Oncogenesis and a Therapeutic Target in Neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef]
- Jiang, J.; Yu, Y. Eflornithine for Treatment of High-Risk Neuroblastoma. Trends Pharmacol. Sci. 2024, 45, 577–578. [Google Scholar] [CrossRef]
- Alborzinia, H.; Flórez, A.F.; Kreth, S.; Brückner, L.M.; Yildiz, U.; Gartlgruber, M.; Odoni, D.I.; Poschet, G.; Garbowicz, K.; Shao, C.; et al. MYCN Mediates Cysteine Addiction and Sensitizes Neuroblastoma to Ferroptosis. Nat. Cancer 2022, 3, 471–485. [Google Scholar] [CrossRef]
- Tao, L.; Mohammad, M.A.; Milazzo, G.; Moreno-Smith, M.; Patel, T.D.; Zorman, B.; Badachhape, A.; Hernandez, B.E.; Wolf, A.B.; Zeng, Z.; et al. MYCN-Driven Fatty Acid Uptake Is a Metabolic Vulnerability in Neuroblastoma. Nat. Commun. 2022, 13, 3728. [Google Scholar] [CrossRef]
- Yoda, H.; Inoue, T.; Shinozaki, Y.; Lin, J.; Watanabe, T.; Koshikawa, N.; Takatori, A.; Nagase, H. Direct Targeting of MYCN Gene Amplification by Site-Specific DNA Alkylation in Neuroblastoma. Cancer Res. 2019, 79, 830–840. [Google Scholar] [CrossRef]
- Obata, H.; Tsuji, A.B.; Sudo, H.; Sugyo, A.; Hashiya, K.; Ikeda, H.; Itoh, M.; Minegishi, K.; Nagatsu, K.; Ogawa, M.; et al. Novel Auger-Electron-Emitting 191Pt-Labeled Pyrrole–Imidazole Polyamide Targeting MYCN Increases Cytotoxicity and Cytosolic DsDNA Granules in MYCN-Amplified Neuroblastoma. Pharmaceuticals 2023, 16, 1526. [Google Scholar] [CrossRef]
- Huang, H. Anaplastic Lymphoma Kinase (ALK) Receptor Tyrosine Kinase: A Catalytic Receptor with Many Faces. Int. J. Mol. Sci. 2018, 19, 3448. [Google Scholar] [CrossRef]
- Wulf, A.M.; Moreno, M.M.; Paka, C.; Rampasekova, A.; Liu, K.J. Defining Pathological Activities of ALK in Neuroblastoma, a Neural Crest-Derived Cancer. Int. J. Mol. Sci. 2021, 22, 11718. [Google Scholar] [CrossRef]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Javanmardi, N.; Bernard, V.; Leroy, Q.; Cappo, J.; Rio Frio, T.; Pierron, G.; Lapouble, E.; Combaret, V.; Speleman, F.; et al. Emergence of New ALK Mutations at Relapse of Neuroblastoma. J. Clin. Oncol. 2014, 32, 2727–2734. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a Major Familial Neuroblastoma Predisposition Gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef]
- Bresler, S.C.; Wood, A.C.; Haglund, E.A.; Courtright, J.; Belcastro, L.T.; Plegaria, J.S.; Cole, K.; Toporovskaya, Y.; Zhao, H.; Carpenter, E.L.; et al. Differential Inhibitor Sensitivity of Anaplastic Lymphoma Kinase Variants Found in Neuroblastoma. Sci. Transl. Med. 2011, 3, 108ra114. [Google Scholar] [CrossRef]
- Foster, J.H.; Voss, S.D.; Hall, D.C.; Minard, C.G.; Balis, F.M.; Wilner, K.; Berg, S.L.; Fox, E.; Adamson, P.C.; Blaney, S.M.; et al. Activity of Crizotinib in Patients with ALK-Aberrant Relapsed/Refractory Neuroblastoma: A Children’s Oncology Group Study (ADVL0912). Clin. Cancer Res. 2021, 27, 3543–3548. [Google Scholar] [CrossRef]
- Infarinato, N.R.; Park, J.H.; Krytska, K.; Ryles, H.T.; Sano, R.; Szigety, K.M.; Li, Y.; Zou, H.Y.; Lee, N.V.; Smeal, T.; et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016, 6, 96–107. [Google Scholar] [CrossRef]
- Goldsmith, K.C.; Park, J.R.; Kayser, K.; Malvar, J.; Chi, Y.-Y.; Groshen, S.G.; Villablanca, J.G.; Krytska, K.; Lai, L.M.; Acharya, P.T.; et al. Lorlatinib with or without Chemotherapy in ALK-Driven Refractory/Relapsed Neuroblastoma: Phase 1 Trial Results. Nat. Med. 2023, 29, 1092–1102. [Google Scholar] [CrossRef]
- Ota, Y.; Yoda, H.; Inoue, T.; Watanabe, T.; Shinozaki, Y.; Takatori, A.; Nagase, H. Targeting Anaplastic Lymphoma Kinase (ALK) Gene Alterations in Neuroblastoma by Using Alkylating Pyrrole-Imidazole Polyamides. PLoS ONE 2021, 16, e0257718. [Google Scholar] [CrossRef]
- Pucci, P.; Lee, L.; Turner, S.; Jahangiri, L.; Matthews, J.; Burke, A.; Reynolds, P.; Kenner, L.; Merkel, O.; Rifatbegovic, F.; et al. Abstract 5344: High-Throughput CRISPR-Cas9 Knockout Screens Identify Loss of MiRNA1304-5p Targeting the RAS/MAPK Pathway as a Modulator of Resistance to ALK Inhibitors in High-Risk Neuroblastoma. Cancer Res. 2022, 82, 5344. [Google Scholar] [CrossRef]
- Lee, H.M.; Wright, W.C.; Pan, M.; Low, J.; Currier, D.; Fang, J.; Singh, S.; Nance, S.; Delahunty, I.; Kim, Y.; et al. A CRISPR-Drug Perturbational Map for Identifying Compounds to Combine with Commonly Used Chemotherapeutics. Nat. Commun. 2023, 14, 7332. [Google Scholar] [CrossRef]
- Fetahu, I.S.; Taschner-Mandl, S. Neuroblastoma and the Epigenome. Cancer Metastasis Rev. 2021, 40, 173–189. [Google Scholar] [CrossRef]
- Durinck, K.; Speleman, F. Epigenetic Regulation of Neuroblastoma Development. Cell Tissue Res. 2018, 372, 309–324. [Google Scholar] [CrossRef]
- Epp, S.; Chuah, S.M.; Halasz, M. Epigenetic Dysregulation in MYCN-Amplified Neuroblastoma. Int. J. Mol. Sci. 2023, 24, 17085. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, X.; Sun, H.; Zhao, D.; Liu, H.; Huang, B.; Li, X.; Gu, Y. Epigenome-Wide Association Study Reveals CpG Sites Related to COG of Neuroblastoma. Biosci. Rep. 2020, 40, BSR20200826. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. Pathological Role of HDAC8: Cancer and Beyond. Cells 2022, 11, 3161. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef]
- Keshelava, N.; Houghton, P.J.; Morton, C.L.; Lock, R.B.; Carol, H.; Keir, S.T.; Maris, J.M.; Reynolds, C.P.; Gorlick, R.; Kolb, E.A.; et al. Initial Testing (Stage 1) of Vorinostat (SAHA) by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2009, 53, 505–508. [Google Scholar] [CrossRef]
- Cheung, B.B.; Kleynhans, A.; Mittra, R.; Kim, P.Y.; Holien, J.K.; Nagy, Z.; Ciampa, O.C.; Seneviratne, J.A.; Mayoh, C.; Raipuria, M.; et al. A Novel Combination Therapy Targeting Ubiquitin-Specific Protease 5 in MYCN-Driven Neuroblastoma. Oncogene 2021, 40, 2367–2381. [Google Scholar] [CrossRef]
- Seneviratne, J.A.; Carter, D.R.; Mittra, R.; Gifford, A.; Kim, P.Y.; Luo, J.; Mayoh, C.; Salib, A.; Rahmanto, A.S.; Murray, J.; et al. Inhibition of Mitochondrial Translocase SLC25A5 and Histone Deacetylation Is an Effective Combination Therapy in Neuroblastoma. Int. J. Cancer 2023, 152, 1399–1413. [Google Scholar] [CrossRef]
- Xiao, L.; Somers, K.; Murray, J.; Pandher, R.; Karsa, M.; Ronca, E.; Bongers, A.; Terry, R.; Ehteda, A.; Gamble, L.D.; et al. Dual Targeting of Chromatin Stability By The Curaxin CBL0137 and Histone Deacetylase Inhibitor Panobinostat Shows Significant Preclinical Efficacy in Neuroblastoma. Clin. Cancer Res. 2021, 27, 4338–4352. [Google Scholar] [CrossRef]
- Lock, R.; Carol, H.; Maris, J.M.; Kolb, E.A.; Gorlick, R.; Reynolds, C.P.; Kang, M.H.; Keir, S.T.; Wu, J.; Purmal, A.; et al. Initial Testing (Stage 1) of the Curaxin CBL0137 by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2017, 64, e26263. [Google Scholar] [CrossRef]
- Carter, D.R.; Murray, J.; Cheung, B.B.; Gamble, L.; Koach, J.; Tsang, J.; Sutton, S.; Kalla, H.; Syed, S.; Gifford, A.J.; et al. Therapeutic Targeting of the MYC Signal by Inhibition of Histone Chaperone FACT in Neuroblastoma. Sci. Transl. Med. 2015, 7, 312ra176. [Google Scholar] [CrossRef]
- Shah, R.R. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective Inhibition of HDAC8 Decreases Neuroblastoma Growth in Vitro and in Vivo and Enhances Retinoic Acid-Mediated Differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef]
- Heimburg, T.; Kolbinger, F.R.; Zeyen, P.; Ghazy, E.; Herp, D.; Schmidtkunz, K.; Melesina, J.; Shaik, T.B.; Erdmann, F.; Schmidt, M.; et al. Structure-Based Design and Biological Characterization of Selective Histone Deacetylase 8 (HDAC8) Inhibitors with Anti-Neuroblastoma Activity. J. Med. Chem. 2017, 60, 10188–10204. [Google Scholar] [CrossRef]
- Morgen, M.; Steimbach, R.R.; Géraldy, M.; Hellweg, L.; Sehr, P.; Ridinger, J.; Witt, O.; Oehme, I.; Herbst-Gervasoni, C.J.; Osko, J.D.; et al. Design and Synthesis of Dihydroxamic Acids as HDAC6/8/10 Inhibitors. ChemMedChem 2020, 15, 1163–1174. [Google Scholar] [CrossRef]
- Kolbinger, F.R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Müller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A.K.; et al. The HDAC6/8/10 Inhibitor TH34 Induces DNA Damage-Mediated Cell Death in Human High-Grade Neuroblastoma Cell Lines. Arch. Toxicol. 2018, 92, 2649–2664. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Darwish, S.; Ghazy, E.; Heimburg, T.; Herp, D.; Zeyen, P.; Salem-Altintas, R.; Ridinger, J.; Robaa, D.; Schmidtkunz, K.; Erdmann, F.; et al. Design, Synthesis and Biological Characterization of Histone Deacetylase 8 (HDAC8) Proteolysis Targeting Chimeras (PROTACs) with Anti-Neuroblastoma Activity. Int. J. Mol. Sci. 2022, 23, 7535. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Das, S.; Crespo, A.C.; Cornel, A.M.; Patel, A.G.; Mahadevan, N.R.; Campisi, M.; Ali, A.K.; Sharma, B.; Rowe, J.H.; et al. Mesenchymal and Adrenergic Cell Lineage States in Neuroblastoma Possess Distinct Immunogenic Phenotypes. Nat. Cancer 2022, 3, 1228–1246. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Z.; Woo, C.-W.; Li, Z.; Wang, L.; Wei, J.S.; Marquez, V.E.; Bates, S.E.; Jin, Q.; Khan, J.; et al. EZH2 Mediates Epigenetic Silencing of Neuroblastoma Suppressor Genes CASZ1, CLU, RUNX3, and NGFR. Cancer Res. 2012, 72, 315–324. [Google Scholar] [CrossRef]
- Mellini, P.; Marrocco, B.; Borovika, D.; Polletta, L.; Carnevale, I.; Saladini, S.; Stazi, G.; Zwergel, C.; Trapencieris, P.; Ferretti, E.; et al. Pyrazole-Based Inhibitors of Enhancer of Zeste Homologue 2 Induce Apoptosis and Autophagy in Cancer Cells. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170150. [Google Scholar] [CrossRef]
- Bownes, L.V.; Williams, A.P.; Marayati, R.; Stafman, L.L.; Markert, H.; Quinn, C.H.; Wadhwani, N.; Aye, J.M.; Stewart, J.E.; Yoon, K.J.; et al. EZH2 Inhibition Decreases Neuroblastoma Proliferation and in Vivo Tumor Growth. PLoS ONE 2021, 16, e0246244. [Google Scholar] [CrossRef]
- Abe, M.; Ohira, M.; Kaneda, A.; Yagi, Y.; Yamamoto, S.; Kitano, Y.; Takato, T.; Nakagawara, A.; Ushijima, T. CpG Island Methylator Phenotype Is a Strong Determinant of Poor Prognosis in Neuroblastomas. Cancer Res. 2005, 65, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Ram Kumar, R.M.; Schor, N.F. Methylation of DNA and Chromatin as a Mechanism of Oncogenesis and Therapeutic Target in Neuroblastoma. Oncotarget 2018, 9, 22184–22193. [Google Scholar] [CrossRef]
- Olsson, M.; Beck, S.; Kogner, P.; Martinsson, T.; Carén, H. Genome-Wide Methylation Profiling Identifies Novel Methylated Genes in Neuroblastoma Tumors. Epigenetics 2016, 11, 74–84. [Google Scholar] [CrossRef]
- Bartolucci, S.; Rossi, M.; Longo, A.; Rossi, M.; Estenoz, M.; Momparler, R.L.; Santoro, B.; Augusti-Tocco, G. 5-Aza-2′-Deoxycytidine as Inducer of Differentiation and Growth Inhibition in Mouse Neuroblastoma Cells. Cell Differ. Dev. 1989, 27, 47–55. [Google Scholar] [CrossRef]
- George, R.E.; Lahti, J.M.; Adamson, P.C.; Zhu, K.; Finkelstein, D.; Ingle, A.M.; Reid, J.M.; Krailo, M.; Neuberg, D.; Blaney, S.M.; et al. Phase I Study of Decitabine with Doxorubicin and Cyclophosphamide in Children with Neuroblastoma and Other Solid Tumors: A Children’s Oncology Group Study. Pediatr. Blood Cancer 2010, 55, 629–638. [Google Scholar] [CrossRef]
- Hattori, N.; Asada, K.; Miyajima, N.; Mori, A.; Nakanishi, Y.; Kimura, K.; Wakabayashi, M.; Takeshima, H.; Nitani, C.; Hara, J.; et al. Combination of a Synthetic Retinoid and a DNA Demethylating Agent Induced Differentiation of Neuroblastoma through Retinoic Acid Signal Reprogramming. Br. J. Cancer 2021, 125, 1647–1656. [Google Scholar] [CrossRef]
- Rouse, D.C.; Agarwal, S. Abstract 7038: Direct Targeting of DNMT1 Inhibits High-Risk Neuroblastoma Growth through Epigenetic Reprogramming. Cancer Res. 2025, 85, 7038. [Google Scholar] [CrossRef]
- Jiménez, C.; Antonelli, R.; Nadal-Ribelles, M.; Devis-Jauregui, L.; Latorre, P.; Solé, C.; Masanas, M.; Molero-Valenzuela, A.; Soriano, A.; Sánchez de Toledo, J.; et al. Structural Disruption of BAF Chromatin Remodeller Impairs Neuroblastoma Metastasis by Reverting an Invasiveness Epigenomic Program. Mol. Cancer 2022, 21, 175. [Google Scholar] [CrossRef]
- Hu, A.; Chen, G.; Bao, B.; Guo, Y.; Li, D.; Wang, X.; Wang, J.; Li, Q.; Zhou, Y.; Gao, H.; et al. Therapeutic Targeting of CNBP Phase Separation Inhibits Ribosome Biogenesis and Neuroblastoma Progression via Modulating SWI/SNF Complex Activity. Clin. Transl. Med. 2023, 13, e1235. [Google Scholar] [CrossRef]
- Cermakova, K.; Tao, L.; Dejmek, M.; Sala, M.; Montierth, M.D.; Chan, Y.S.; Patel, I.; Chambers, C.; Loeza Cabrera, M.; Hoffman, D.; et al. Reactivation of the G1 Enhancer Landscape Underlies Core Circuitry Addiction to SWI/SNF. Nucleic Acids Res. 2024, 52, 4–21. [Google Scholar] [CrossRef]
- Gomez, R.L.; Woods, L.M.; Ramachandran, R.; Abou Tayoun, A.N.; Philpott, A.; Ali, F.R. Super-Enhancer Associated Core Regulatory Circuits Mediate Susceptibility to Retinoic Acid in Neuroblastoma Cells. Front. Cell Dev. Biol. 2022, 10, 943924. [Google Scholar] [CrossRef]
- Banerjee, D.; Bagchi, S.; Liu, Z.; Chou, H.C.; Xu, M.; Sun, M.; Aloisi, S.; Vaksman, Z.; Diskin, S.J.; Zimmerman, M.; et al. Lineage Specific Transcription Factor Waves Reprogram Neuroblastoma from Self-Renewal to Differentiation. Nat. Commun. 2024, 15, 1–18. [Google Scholar] [CrossRef]
- Zhang, D.; Gong, B.; Zhao, Q.; Li, Z.; Tan, X.; Hua, Z. SOX4 Mediates ATRA-Induced Differentiation in Neuroblastoma Cells. Cancers 2022, 14, 5642. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Volegova, M.; Nasholm, N.; Das, S.; Kwiatkowski, N.; Abraham, B.J.; Zhang, T.; Gray, N.S.; Gustafson, C.; Krajewska, M.; et al. Synergistic Anti-Tumor Effect of Combining Selective CDK7 and BRD4 Inhibition in Neuroblastoma. Front. Oncol. 2022, 11, 773186. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S. Targeting the Tumor Microenvironment in Neuroblastoma: Recent Advances and Future Directions. Cancers 2020, 12, 2057. [Google Scholar] [CrossRef] [PubMed]
- Gavin, C.; Geerts, N.; Cavanagh, B.; Haynes, M.; Reynolds, C.P.; Loessner, D.; Ewald, A.J.; Piskareva, O. Neuroblastoma Invasion Strategies Are Regulated by the Extracellular Matrix. Cancers 2021, 13, 736. [Google Scholar] [CrossRef] [PubMed]
- Horwacik, I. The Extracellular Matrix and Neuroblastoma Cell Communication—A Complex Interplay and Its Therapeutic Implications. Cells 2022, 11, 3172. [Google Scholar] [CrossRef]
- Lam, W.A.; Cao, L.; Umesh, V.; Keung, A.J.; Sen, S.; Kumar, S. Extracellular Matrix Rigidity Modulates Neuroblastoma Cell Differentiation and N-Myc Expression. Mol. Cancer 2010, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Chen, Y.; Liu, J.-T.; Bao, H.; Wang, W.-B.; Qi, Y.-X.; Lv, F. Extracellular Matrix Stiffness Controls VEGF165 Secretion and Neuroblastoma Angiogenesis via the YAP/RUNX2/SRSF1 Axis. Angiogenesis 2022, 25, 71–86. [Google Scholar] [CrossRef]
- López-Carrasco, A.; Martín-Vañó, S.; Burgos-Panadero, R.; Monferrer, E.; Berbegall, A.P.; Fernández-Blanco, B.; Navarro, S.; Noguera, R. Impact of Extracellular Matrix Stiffness on Genomic Heterogeneity in MYCN-Amplified Neuroblastoma Cell Line. J. Exp. Clin. Cancer Res. 2020, 39, 226. [Google Scholar] [CrossRef]
- Burgos-Panadero, R.; Noguera, I.; Cañete, A.; Navarro, S.; Noguera, R. Vitronectin as a Molecular Player of the Tumor Microenvironment in Neuroblastoma. BMC Cancer 2019, 19, 479. [Google Scholar] [CrossRef]
- Tadeo, I.; Berbegall, A.P.; Navarro, S.; Castel, V.; Noguera, R. A Stiff Extracellular Matrix Is Associated with Malignancy in Peripheral Neuroblastic Tumors. Pediatr. Blood Cancer 2017, 64, e26449. [Google Scholar] [CrossRef]
- Darvishi, B.; Eisavand, M.R.; Majidzadeh-A, K.; Farahmand, L. Matrix Stiffening and Acquired Resistance to Chemotherapy: Concepts and Clinical Significance. Br. J. Cancer 2022, 126, 1253–1263. [Google Scholar] [CrossRef]
- Mai, Z.; Lin, Y.; Lin, P.; Zhao, X.; Cui, L. Modulating Extracellular Matrix Stiffness: A Strategic Approach to Boost Cancer Immunotherapy. Cell Death Dis. 2024, 15, 307. [Google Scholar] [CrossRef]
- Beierle, E.A.; Trujillo, A.; Nagaram, A.; Kurenova, E.V.; Finch, R.; Ma, X.; Vella, J.; Cance, W.G.; Golubovskaya, V.M. N-MYC Regulates Focal Adhesion Kinase Expression in Human Neuroblastoma. J. Biol. Chem. 2007, 282, 12503–12516. [Google Scholar] [CrossRef]
- Megison, M.L.; Stewart, J.E.; Nabers, H.C.; Gillory, L.A.; Beierle, E.A. FAK Inhibition Decreases Cell Invasion, Migration and Metastasis in MYCN Amplified Neuroblastoma. Clin. Exp. Metastasis 2013, 30, 555–568. [Google Scholar] [CrossRef]
- Pham, D.-C.; Chang, Y.-C.; Lin, S.-R.; Fuh, Y.-M.; Tsai, M.-J.; Weng, C.-F. FAK and S6K1 Inhibitor, Neferine, Dually Induces Autophagy and Apoptosis in Human Neuroblastoma Cells. Molecules 2018, 23, 3110. [Google Scholar] [CrossRef]
- Stafman, L.L.; Williams, A.P.; Marayati, R.; Aye, J.M.; Markert, H.R.; Garner, E.F.; Quinn, C.H.; Lallani, S.B.; Stewart, J.E.; Yoon, K.J.; et al. Focal Adhesion Kinase Inhibition Contributes to Tumor Cell Survival and Motility in Neuroblastoma Patient-Derived Xenografts. Sci. Rep. 2019, 9, 13259. [Google Scholar] [CrossRef]
- Bui, C.-B.; To, K.D.; Vu, D.M.; Nguyen, Q.-G.; Nguyen, H.T.; Nguyen, S.-B. Denatured Collagen Inhibits Neuroblastoma Tumor-Sphere Migration and Growth via the LOX/LOXL2—FAK Signaling Pathway. J. Therm. Biol. 2023, 115, 103624. [Google Scholar] [CrossRef]
- Meyer, A.; van Golen, C.M.; Kim, B.; van Golen, K.L.; Feldman, E.L. Integrin Expression Regulates Neuroblastoma Attachment and Migration. Neoplasia 2004, 6, 332–342. [Google Scholar] [CrossRef]
- Ozen Karakus, O.; Godugu, K.; Mousa, S.A. Discovery of Dual Targeting PEGylated BG-P1600-TAT to Norepinephrine Transporter (NET) and Thyrointegrin Avβ3 in the Treatment of Neuroblastoma. Bioorg Med. Chem. 2021, 43, 116278. [Google Scholar] [CrossRef]
- Karakus, O.O.; Godugu, K.; Rajabi, M.; Mousa, S.A. Dual Targeting of Norepinephrine Transporter (NET) Function and Thyrointegrin Avβ3 Receptors in the Treatment of Neuroblastoma. J. Med. Chem. 2020, 63, 7653–7662. [Google Scholar] [CrossRef]
- Wachowiak, R.; Rawnaq, T.; Metzger, R.; Quaas, A.; Fiegel, H.; Kähler, N.; Rolle, U.; Izbicki, J.R.; Kaifi, J.; Till, H. Universal Expression of Cell Adhesion Molecule NCAM in Neuroblastoma in Contrast to L1: Implications for Different Roles in Tumor Biology of Neuroblastoma? Pediatr. Surg. Int. 2008, 24, 1361–1364. [Google Scholar] [CrossRef]
- Winter, C.; Pawel, B.; Seiser, E.; Zhao, H.; Raabe, E.; Wang, Q.; Judkins, A.R.; Attiyeh, E.; Maris, J.M. Neural Cell Adhesion Molecule (NCAM) Isoform Expression Is Associated with Neuroblastoma Differentiation Status. Pediatr. Blood Cancer 2008, 51, 10–16. [Google Scholar] [CrossRef]
- Geller, J.I.; Pressey, J.G.; Smith, M.A.; Kudgus, R.A.; Cajaiba, M.; Reid, J.M.; Hall, D.; Barkauskas, D.A.; Voss, S.D.; Cho, S.Y.; et al. ADVL1522: A Phase 2 Study of Lorvotuzumab Mertansine (IMGN901) in Children with Relapsed or Refractory Wilms Tumor, Rhabdomyosarcoma, Neuroblastoma, Pleuropulmonary Blastoma, Malignant Peripheral Nerve Sheath Tumor, or Synovial Sarcoma—A Children’s Oncology Group Study. Cancer 2020, 126, 5303–5310. [Google Scholar] [CrossRef]
- Su, Y.; Luo, B.; Lu, Y.; Wang, D.; Yan, J.; Zheng, J.; Xiao, J.; Wang, Y.; Xue, Z.; Yin, J.; et al. Anlotinib Induces a T Cell–Inflamed Tumor Microenvironment by Facilitating Vessel Normalization and Enhances the Efficacy of PD-1 Checkpoint Blockade in Neuroblastoma. Clin. Cancer Res. 2022, 28, 793–809. [Google Scholar] [CrossRef]
- Moreno, L.; Weston, R.; Owens, C.; Valteau-Couanet, D.; Gambart, M.; Castel, V.; Zwaan, C.M.; Nysom, K.; Gerber, N.; Castellano, A.; et al. Bevacizumab, Irinotecan, or Topotecan Added to Temozolomide for Children With Relapsed and Refractory Neuroblastoma: Results of the ITCC-SIOPEN BEACON-Neuroblastoma Trial. J. Clin. Oncol. 2024, 42, 1135–1145. [Google Scholar] [CrossRef]
- Rivera, Z.; Escutia, C.; Madonna, M.B.; Gupta, K.H. Biological Insight and Recent Advancement in the Treatment of Neuroblastoma. Int. J. Mol. Sci. 2023, 24, 8470. [Google Scholar] [CrossRef]
- Pathania, A.S.; Prathipati, P.; Murakonda, S.P.; Murakonda, A.B.; Srivastava, A.; Avadhesh; Byrareddy, S.N.; Coulter, D.W.; Gupta, S.C.; Challagundla, K.B. Immune Checkpoint Molecules in Neuroblastoma: A Clinical Perspective. Semin. Cancer Biol. 2022, 86, 247–258. [Google Scholar] [CrossRef]
- Layer, J.P.; Kronmüller, M.T.; Quast, T.; van den Boorn-Konijnenberg, D.; Effern, M.; Hinze, D.; Althoff, K.; Schramm, A.; Westermann, F.; Peifer, M.; et al. Amplification of N-Myc Is Associated with a T-Cell-Poor Microenvironment in Metastatic Neuroblastoma Restraining Interferon Pathway Activity and Chemokine Expression. Oncoimmunology 2017, 6, e1320626. [Google Scholar] [CrossRef]
- Raieli, S.; Di Renzo, D.; Lampis, S.; Amadesi, C.; Montemurro, L.; Pession, A.; Hrelia, P.; Fischer, M.; Tonelli, R. MYCN Drives a Tumor Immunosuppressive Environment Which Impacts Survival in Neuroblastoma. Front. Oncol. 2021, 11, 625207. [Google Scholar] [CrossRef]
- Lundberg, K.I.; Treis, D.; Johnsen, J.I. Neuroblastoma Heterogeneity, Plasticity, and Emerging Therapies. Curr. Oncol. Rep. 2022, 24, 1053–1062. [Google Scholar] [CrossRef]
- Brandetti, E.; Veneziani, I.; Melaiu, O.; Pezzolo, A.; Castellano, A.; Boldrini, R.; Ferretti, E.; Fruci, D.; Moretta, L.; Pistoia, V.; et al. MYCN Is an Immunosuppressive Oncogene Dampening the Expression of Ligands for NK-Cell-Activating Receptors in Human High-Risk Neuroblastoma. Oncoimmunology 2017, 6, e1316439. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, M.; Mehrazma, M.; Karimian, M. Tumor Infiltrating Cytotoxic CD8 T-Cells Predict Clinical Outcome of Neuroblastoma in Children. Indian J. Med. Paediatr. Oncol. 2018, 39, 159–164. [Google Scholar] [CrossRef]
- Mina, M.; Boldrini, R.; Citti, A.; Romania, P.; D’Alicandro, V.; De Ioris, M.; Castellano, A.; Furlanello, C.; Locatelli, F.; Fruci, D. Tumor-Infiltrating T Lymphocytes Improve Clinical Outcome of Therapy-Resistant Neuroblastoma. Oncoimmunology 2015, 4, e1019981. [Google Scholar] [CrossRef] [PubMed]
- Kacher, J.; Manches, O.; Aspord, C.; Sartelet, H.; Chaperot, L. Impaired Antitumor Immune Response in MYCN-Amplified Neuroblastoma Is Associated with Lack of CCL2 Secretion and Poor Dendritic Cell Recruitment. Cancer Res. Commun. 2022, 2, 577–589. [Google Scholar] [CrossRef]
- Qin, X.; Lam, A.; Zhang, X.; Sengupta, S.; Iorgulescu, J.B.; Ni, H.; Das, S.; Rager, M.; Zhou, Z.; Zuo, T.; et al. CKLF Instigates a “Cold” Microenvironment to Promote MYCN-Mediated Tumor Aggressiveness. Sci. Adv. 2024, 10, 9547. [Google Scholar] [CrossRef]
- Hashimoto, O.; Yoshida, M.; Koma, Y.I.; Yanai, T.; Hasegawa, D.; Kosaka, Y.; Nishimura, N.; Yokozaki, H. Collaboration of Cancer-Associated Fibroblasts and Tumour-Associated Macrophages for Neuroblastoma Development. J. Pathol. 2016, 240, 211–223. [Google Scholar] [CrossRef]
- Tucker, E.R.; Poon, E.; Chesler, L. Targeting MYCN and ALK in Resistant and Relapsing Neuroblastoma. Cancer Drug Resist. 2019, 2, 803. [Google Scholar] [CrossRef]
- Lombardo, S.D.; Presti, M.; Mangano, K.; Petralia, M.C.; Basile, M.S.; Libra, M.; Candido, S.; Fagone, P.; Mazzon, E.; Nicoletti, F.; et al. Prediction of PD-L1 Expression in Neuroblastoma via Computational Modeling. Brain Sci. 2019, 9, 221. [Google Scholar] [CrossRef]
- Dahmani, A.; Delisle, J.-S. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-β Directly Targets Cytotoxic T Cell Functions during Tumor Evasion of Immune Surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF- Downregulates the Activating Receptor NKG2D on NK Cells and CD8+ T Cells in Glioma Patients. Neuro Oncol. 2010, 12, 7–13. [Google Scholar] [CrossRef]
- Louault, K.; Porras, T.; Lee, M.-H.; Muthugounder, S.; Kennedy, R.J.; Blavier, L.; Sarte, E.; Fernandez, G.E.; Yang, F.; Pawel, B.R.; et al. Fibroblasts and Macrophages Cooperate to Create a Pro-Tumorigenic and Immune Resistant Environment via Activation of TGF-β/IL-6 Pathway in Neuroblastoma. Oncoimmunology 2022, 11, 2146860. [Google Scholar] [CrossRef]
- Pei, L.; Liu, Y.; Liu, L.; Gao, S.; Gao, X.; Feng, Y.; Sun, Z.; Zhang, Y.; Wang, C. Roles of Cancer-Associated Fibroblasts (CAFs) in Anti- PD-1/PD-L1 Immunotherapy for Solid Cancers. Mol. Cancer 2023, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.X.; Joshi, S. “Re-Educating” Tumor Associated Macrophages as a Novel Immunotherapy Strategy for Neuroblastoma. Front. Immunol. 2020, 11, 1947. [Google Scholar] [CrossRef]
- Mussai, F.; Egan, S.; Hunter, S.; Webber, H.; Fisher, J.; Wheat, R.; McConville, C.; Sbirkov, Y.; Wheeler, K.; Bendle, G.; et al. Neuroblastoma Arginase Activity Creates an Immunosuppressive Microenvironment That Impairs Autologous and Engineered Immunity. Cancer Res. 2015, 75, 3043–3053. [Google Scholar] [CrossRef]
- Kudva, A.; Modak, S. Immunotherapy for Neuroblastoma. In Neuroblastoma; Elsevier: Amsterdam, The Netherlands, 2019; pp. 147–173. [Google Scholar]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Wienke, J.; Dierselhuis, M.P.; Tytgat, G.A.M.; Künkele, A.; Nierkens, S.; Molenaar, J.J. The Immune Landscape of Neuroblastoma: Challenges and Opportunities for Novel Therapeutic Strategies in Pediatric Oncology. Eur. J. Cancer 2021, 144, 123–150. [Google Scholar] [CrossRef]
- Kennedy, P.T.; Zannoupa, D.; Son, M.H.; Dahal, L.N.; Woolley, J.F. Neuroblastoma: An Ongoing Cold Front for Cancer Immunotherapy. J. Immunother. Cancer 2023, 11, e007798. [Google Scholar] [CrossRef] [PubMed]
- Kushner, B.; Cheung, N. GM-CSF Enhances 3F8 Monoclonal Antibody-Dependent Cellular Cytotoxicity against Human Melanoma and Neuroblastoma. Blood 1989, 73, 1936–1941. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Kushner, B.H.; Yeh, S.D.; Larson, S.M. 3F8 Monoclonal Antibody Treatment of Patients with Stage 4 Neuroblastoma: A Phase II Study. Int. J. Oncol. 1998, 12, 1299–1306. [Google Scholar] [CrossRef]
- Cheung, N.K.; Lazarus, H.; Miraldi, F.D.; Abramowsky, C.R.; Kallick, S.; Saarinen, U.M.; Spitzer, T.; Strandjord, S.E.; Coccia, P.F.; Berger, N.A. Ganglioside GD2 Specific Monoclonal Antibody 3F8: A Phase I Study in Patients with Neuroblastoma and Malignant Melanoma. J. Clin. Oncol. 1987, 5, 1430–1440. [Google Scholar] [CrossRef]
- Cheung, N.-K.V.; Guo, H.; Hu, J.; Tassev, D.V.; Cheung, I.Y. Humanizing Murine IgG3 Anti-GD2 Antibody M3F8 Substantially Improves Antibody-Dependent Cell-Mediated Cytotoxicity While Retaining Targeting in Vivo. Oncoimmunology 2012, 1, 477–486. [Google Scholar] [CrossRef]
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.-K. Humanized 3F8 Anti-G D2 Monoclonal Antibody Dosing with Granulocyte-Macrophage Colony-Stimulating Factor in Patients with Resistant Neuroblastoma. JAMA Oncol. 2018, 4, 1729. [Google Scholar] [CrossRef]
- Kushner, B.H.; Morgenstern, D.A.; Nysom, K.; Bear, M.K.; Tornøe, K.; Losic, N.; Mora, J. Efficacy of Naxitamab in Patients with Refractory/Relapse (R/R) High-Risk Neuroblastoma (HR-NB) by Bone/Bone Marrow (BM) Evaluation, Potential Sites of Residual Disease. J. Clin. Oncol. 2021, 39, 10022. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; Naranjo, A.; Diccianni, M.B.; Gan, J.; Hank, J.A.; Batova, A.; London, W.B.; Tenney, S.C.; et al. Long-Term Follow-up of a Phase III Study of Ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin. Cancer Res. 2021, 27, 2179–2189. [Google Scholar] [CrossRef]
- Mohd, A.B.; Mohd, O.B.; Alabdallat, Y.J.; Al Dwairy, S.Y.; Ghannam, R.A.; Hanaqtah, B.M.; Albakri, K.A. Safety and Efficacy of Dinutuximab in the Treatment of Neuroblastoma: A Review. J. Res. Med. Sci. 2023, 28, 71. [Google Scholar] [CrossRef] [PubMed]
- Ladenstein, R.; Weixler, S.; Baykan, B.; Bleeke, M.; Kunert, R.; Katinger, D.; Pribill, I.; Glander, P.; Bauer, S.; Pistoia, V.; et al. Ch14.18 Antibody Produced in CHO Cells in Relapsed or Refractory Stage 4 Neuroblastoma Patients. MAbs 2013, 5, 801–809. [Google Scholar] [CrossRef]
- Gray, J.; Moreno, L.; Weston, R.; Barone, G.; Rubio, A.; Makin, G.; Vaidya, S.; Ng, A.; Castel, V.; Nysom, K.; et al. BEACON-Immuno: Results of the Dinutuximab Beta (DB) Randomization of the BEACON-Neuroblastoma Phase 2 Trial—A European Innovative Therapies for Children with Cancer (ITCC–International Society of Paediatric Oncology Europe Neuroblastoma Group (SIOPEN) Trial. J. Clin. Oncol. 2022, 40, 10002. [Google Scholar] [CrossRef]
- Mabe, N.W.; Huang, M.; Dalton, G.N.; Alexe, G.; Schaefer, D.A.; Geraghty, A.C.; Robichaud, A.L.; Conway, A.S.; Khalid, D.; Mader, M.M.; et al. Transition to a Mesenchymal State in Neuroblastoma Confers Resistance to Anti-GD2 Antibody via Reduced Expression of ST8SIA1. Nat. Cancer 2022, 3, 976–993. [Google Scholar] [CrossRef]
- Zuo, S.; Sho, M.; Sawai, T.; Kanehiro, H.; Maeda, K.; Yoshida, M.; Tsukada, R.; Nomura, M.; Okuyama, H. Potential Role of the PD-L1 Expression and Tumor-Infiltrating Lymphocytes on Neuroblastoma. Pediatr. Surg. Int. 2020, 36, 137–143. [Google Scholar] [CrossRef]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of Programmed Death-ligand 1 Expression and Tumor-associated Immune Cells in Pediatric Cancer Tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef]
- Saletta, F.; Vilain, R.E.; Gupta, A.K.; Nagabushan, S.; Yuksel, A.; Catchpoole, D.; Scolyer, R.A.; Byrne, J.A.; McCowage, G. Programmed Death-Ligand 1 Expression in a Large Cohort of Pediatric Patients with Solid Tumor and Association with Clinicopathologic Features in Neuroblastoma. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in Children and Young Adults with Relapsed or Refractory Solid Tumours or Lymphoma (ADVL1412): A Multicentre, Open-Label, Single-Arm, Phase 1–2 Trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Zwaan, C.M.; Marshall, L.V.; Michon, J.; Bourdeaut, F.; Casanova, M.; Corradini, N.; Rossato, G.; Farid-Kapadia, M.; Shemesh, C.S.; et al. Atezolizumab for Children and Young Adults with Previously Treated Solid Tumours, Non-Hodgkin Lymphoma, and Hodgkin Lymphoma (IMATRIX): A Multicentre Phase 1–2 Study. Lancet Oncol. 2020, 21, 134–144. [Google Scholar] [CrossRef]
- Principe, N.; Aston, W.J.; Hope, D.E.; Tilsed, C.M.; Fisher, S.A.; Boon, L.; Dick, I.M.; Chin, W.L.; McDonnell, A.M.; Nowak, A.K.; et al. Comprehensive Testing of Chemotherapy and Immune Checkpoint Blockade in Preclinical Cancer Models Identifies Additive Combinations. Front. Immunol. 2022, 13, 872295. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-Line Nivolumab plus Ipilimumab Combined with Two Cycles of Chemotherapy in Patients with Non-Small-Cell Lung Cancer (CheckMate 9LA): An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus Platinum–Etoposide versus Platinum–Etoposide in First-Line Treatment of Extensive-Stage Small-Cell Lung Cancer (CASPIAN): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, C.; Rubino, J.; Brard, C.; Cassard, L.; André, N.; Rondof, W.; Scoazec, J.-Y.; Marchais, A.; Nebchi, S.; Boselli, L.; et al. Phase II and Biomarker Study of Programmed Cell Death Protein 1 Inhibitor Nivolumab and Metronomic Cyclophosphamide in Paediatric Relapsed/Refractory Solid Tumours: Arm G of AcSé-ESMART, a Trial of the European Innovative Therapies for Children With Cancer Consortium. Eur. J. Cancer 2021, 150, 53–62. [Google Scholar] [CrossRef]
- Ehlert, K.; Hansjuergens, I.; Zinke, A.; Otto, S.; Siebert, N.; Henze, G.; Lode, H. Nivolumab and Dinutuximab Beta in Two Patients with Refractory Neuroblastoma. J. Immunother. Cancer 2020, 8, e000540. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, Y.; Kong, L.; Shi, F.; Zhu, H.; Yu, J. Abscopal Effect of Radiotherapy Combined with Immune Checkpoint Inhibitors. J. Hematol. Oncol. 2018, 11, 104. [Google Scholar] [CrossRef]
- Lorusso, D.; Xiang, Y.; Hasegawa, K.; Scambia, G.; Leiva, M.; Ramos-Elias, P.; Acevedo, A.; Sukhin, V.; Cloven, N.; Pereira de Santana Gomes, A.J.; et al. Pembrolizumab or Placebo with Chemoradiotherapy Followed by Pembrolizumab or Placebo for Newly Diagnosed, High-Risk, Locally Advanced Cervical Cancer (ENGOT-Cx11/GOG-3047/KEYNOTE-A18): A Randomised, Double-Blind, Phase 3 Clinical Trial. Lancet 2024, 403, 1341–1350. [Google Scholar] [CrossRef]
- Tempora, P.; D’Amico, S.; Gragera, P.; Damiani, V.; Krol, K.; Scaldaferri, V.; Pandey, K.; Chung, S.; Lucarini, V.; Giorda, E.; et al. Combining ERAP1 Silencing and Entinostat Therapy to Overcome Resistance to Cancer Immunotherapy in Neuroblastoma. J. Exp. Clin. Cancer Res. 2024, 43, 292. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M.; Khorramian-Ghahfarokhi, M.; Shafieizadeh, M.; Mahmoudi, E.; Eskandari, F.; Rashidi, M.; Arshi, A.; Mokhtari-Farsani, A. Comprehensive Review of CRISPR-Based Gene Editing: Mechanisms, Challenges, and Applications in Cancer Therapy. Mol. Cancer 2024, 23, 9. [Google Scholar] [CrossRef] [PubMed]
- Leitner, J.; Klauser, C.; Pickl, W.F.; Stöckl, J.; Majdic, O.; Bardet, A.F.; Kreil, D.P.; Dong, C.; Yamazaki, T.; Zlabinger, G.; et al. B7-H3 Is a Potent Inhibitor of Human T-cell Activation: No Evidence for B7-H3 and TREML2 Interaction. Eur. J. Immunol. 2009, 39, 1754–1764. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Augugliaro, R.; Cantoni, C.; Carnemolla, B.; Sementa, A.R.; Negri, F.; Conte, R.; Corrias, M.V.; Moretta, L.; et al. Identification of 4Ig-B7-H3 as a Neuroblastoma-Associated Molecule That Exerts a Protective Role from an NK Cell-Mediated Lysis. Proc. Natl. Acad. Sci. USA 2004, 101, 12640–12645. [Google Scholar] [CrossRef]
- Rados, M.; Landegger, A.; Schmutzler, L.; Rabidou, K.; Taschner-Mandl, S.; Fetahu, I.S. Natural Killer Cells in Neuroblastoma: Immunological Insights and Therapeutic Perspectives. Cancer Metastasis Rev. 2024, 43, 1401–1417. [Google Scholar] [CrossRef]
- Gregorio, A.; Corrias, M.V.; Castriconi, R.; Dondero, A.; Mosconi, M.; Gambini, C.; Moretta, A.; Moretta, L.; Bottino, C. Small Round Blue Cell Tumours: Diagnostic and Prognostic Usefulness of the Expression of B7-H3 Surface Molecule. Histopathology 2008, 53, 73–80. [Google Scholar] [CrossRef]
- Kendsersky, N.M.; Lindsay, J.; Kolb, E.A.; Smith, M.A.; Teicher, B.A.; Erickson, S.W.; Earley, E.J.; Mosse, Y.P.; Martinez, D.; Pogoriler, J.; et al. The B7-H3–Targeting Antibody–Drug Conjugate M276-SL-PBD Is Potently Effective Against Pediatric Cancer Preclinical Solid Tumor Models. Clin. Cancer Res. 2021, 27, 2938–2946. [Google Scholar] [CrossRef]
- Kurmasheva, R.; Mosse, Y.P.; Del Pozo, V.; Earley, E.J.; Erickson, S.W.; Groff, D.; Kolb, E.A.; Krytska, K.; Smith, M.A.; Tsang, M.; et al. Testing of B7-H3 Targeting Antibody-Drug Conjugate (ADC) MGC018 in Models of Pediatric Solid Tumors by the Pediatric Preclinical Testing Consortium (PPTC). J. Clin. Oncol. 2021, 39, 10037. [Google Scholar] [CrossRef]
- Brignole, C.; Calarco, E.; Bensa, V.; Giusto, E.; Perri, P.; Ciampi, E.; Corrias, M.V.; Astigiano, S.; Cilli, M.; Loo, D.; et al. Antitumor Activity of the Investigational B7-H3 Antibody-Drug Conjugate, Vobramitamab Duocarmazine, in Preclinical Models of Neuroblastoma. J. Immunother. Cancer 2023, 11, e007174. [Google Scholar] [CrossRef] [PubMed]
- Wienke, J.; Visser, L.L.; Kholosy, W.M.; Keller, K.M.; Barisa, M.; Poon, E.; Munnings-Tomes, S.; Himsworth, C.; Calton, E.; Rodriguez, A.; et al. Integrative Analysis of Neuroblastoma by Single-Cell RNA Sequencing Identifies the NECTIN2-TIGIT Axis as a Target for Immunotherapy. Cancer Cell 2024, 42, 283–300.E8. [Google Scholar] [CrossRef]
- Spel, L.; Schiepers, A.; Boes, M. NFκB and MHC-1 Interplay in Neuroblastoma and Immunotherapy. Trends Cancer 2018, 4, 715–717. [Google Scholar] [CrossRef]
- Forloni, M.; Albini, S.; Limongi, M.Z.; Cifaldi, L.; Boldrini, R.; Nicotra, M.R.; Giannini, G.; Natali, P.G.; Giacomini, P.; Fruci, D. NF-ΚB, and Not MYCN, Regulates MHC Class I and Endoplasmic Reticulum Aminopeptidases in Human Neuroblastoma Cells. Cancer Res. 2010, 70, 916–924. [Google Scholar] [CrossRef]
- Wölfl, M.; Jungbluth, A.A.; Garrido, F.; Cabrera, T.; Meyen-Southard, S.; Spitz, R.; Ernestus, K.; Berthold, F. Expression of MHC Class I, MHC Class II, and Cancer Germline Antigens in Neuroblastoma. Cancer Immunol. Immunother. 2005, 54, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Squire, R.; Fowler, C.L.; Brooks, S.P.; Rich, G.A.; Cooney, D.R. The Relationship of Class I MHC Antigen Expression to Stage IV-S Disease and Survival in Neuroblastoma. J. Pediatr. Surg. 1990, 25, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Li, S.H.; Xu, S.Y.; Chen, K.; Qin, L.J.; Liu, X.Y.; Wang, F.; Fu, S.; Deng, L.; Wang, F.H.; et al. Clinical Significance of a CD3/CD8-Based Immunoscore in Neuroblastoma Patients Using Digital Pathology. Front. Immunol. 2022, 13, 878457. [Google Scholar] [CrossRef]
- Cornel, A.M.; Dunnebach, E.; Hofman, D.A.; Das, S.; Sengupta, S.; van den Ham, F.; Wienke, J.; Strijker, J.G.M.; van den Beemt, D.A.M.H.; Essing, A.H.W.; et al. Epigenetic Modulation of Neuroblastoma Enhances T Cell and NK Cell Immunogenicity by Inducing a Tumor-Cell Lineage Switch. J. Immunother. Cancer 2022, 10, e005002. [Google Scholar] [CrossRef] [PubMed]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.-K.; Gileadi, T.; et al. Antitumor Activity without On-Target off-Tumor Toxicity of GD2–Chimeric Antigen Receptor T Cells in Patients with Neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef]
- Yu, L.; Huang, L.; Lin, D.; Lai, X.; Wu, L.; Liao, X.; Liu, J.; Zeng, Y.; Liang, L.; Zhang, G.; et al. GD2-Specific Chimeric Antigen Receptor-Modified T Cells for the Treatment of Refractory and/or Recurrent Neuroblastoma in Pediatric Patients. J. Cancer Res. Clin. Oncol. 2022, 148, 2643–2652. [Google Scholar] [CrossRef]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-Tumor Genomic Biomarkers for PD-1 Checkpoint Blockade-Based Immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Bocca, P.; Di Carlo, E.; Caruana, I.; Emionite, L.; Cilli, M.; De Angelis, B.; Quintarelli, C.; Pezzolo, A.; Raffaghello, L.; Morandi, F.; et al. Bevacizumab-Mediated Tumor Vasculature Remodelling Improves Tumor Infiltration and Antitumor Efficacy of GD2-CAR T Cells in a Human Neuroblastoma Preclinical Model. Oncoimmunology 2018, 7, e1378843. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Hirabayashi, K.; Ahn, S.; Kren, N.P.; Montgomery, S.A.; Wang, X.; Tiruthani, K.; Mirlekar, B.; Michaud, D.; Greene, K.; et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019, 35, 221–237.e8. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, B.; Muthugounder, S.; Jambon, S.; Tibbetts, R.; Hung, L.; Bassiri, H.; Hogarty, M.D.; Barrett, D.M.; Shimada, H.; Asgharzadeh, S. Preclinical Assessment of the Efficacy and Specificity of GD2-B7H3 SynNotch CAR-T in Metastatic Neuroblastoma. Nat. Commun. 2021, 12, 511. [Google Scholar] [CrossRef]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef]
- Halliwell, E.; Vitali, A.; Muller, H.; Alonso-Ferrero, M.; Barisa, M.; Gavriil, A.; Piapi, A.; Leboreiro-Babe, C.; Gileadi, T.; Yeung, J.; et al. Targeting of Low ALK Antigen Density Neuroblastoma Using AND Logic-Gate Engineered CAR-T Cells. Cytotherapy 2023, 25, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Esmaeilzadeh, A.; Hadiloo, K.; Jabbari, M.; Elahi, R. Current Progress of Chimeric Antigen Receptor (CAR) T versus CAR NK Cell for Immunotherapy of Solid Tumors. Life Sci. 2024, 337, 122381. [Google Scholar] [CrossRef]
- Pelosi, A.; Fiore, P.F.; Di Matteo, S.; Veneziani, I.; Caruana, I.; Ebert, S.; Munari, E.; Moretta, L.; Maggi, E.; Azzarone, B. Pediatric Tumors-Mediated Inhibitory Effect on NK Cells: The Case of Neuroblastoma and Wilms’ Tumors. Cancers 2021, 13, 2374. [Google Scholar] [CrossRef]
- Raffaghello, L.; Prigione, I.; Airoldi, I.; Camoriano, M.; Levreri, I.; Gambini, C.; Pende, D.; Steinle, A.; Ferrone, S.; Pistoia, V. Downregulation and/or Release of NKG2D Ligands as Immune Evasion Strategy of Human Neuroblastoma. Neoplasia 2004, 6, 558–568. [Google Scholar] [CrossRef]
- McNerney, K.O.; Karageorgos, S.A.; Hogarty, M.D.; Bassiri, H. Enhancing Neuroblastoma Immunotherapies by Engaging INKT and NK Cells. Front. Immunol. 2020, 11, 873. [Google Scholar] [CrossRef]
- Zobel, M.J.; Zamora, A.K.; Wu, H.; Sun, J.; Lascano, D.; Malvar, J.; Wang, L.; Sheard, M.A.; Seeger, R.C.; Kim, E.S. Initiation of Immunotherapy with Activated Natural Killer Cells and Anti-GD2 Antibody Dinutuximab Prior to Resection of Primary Neuroblastoma Prolongs Survival in Mice. J. Immunother. Cancer 2020, 8, e001560. [Google Scholar] [CrossRef]
- Chu, Y.; Nayyar, G.; Jiang, S.; Rosenblum, J.M.; Soon-Shiong, P.; Safrit, J.T.; Lee, D.A.; Cairo, M.S. Combinatorial Immunotherapy of N-803 (IL-15 Superagonist) and Dinutuximab with Ex Vivo Expanded Natural Killer Cells Significantly Enhances in Vitro Cytotoxicity against GD2 + Pediatric Solid Tumors and in Vivo Survival of Xenografted Immunodeficient NSG Mice. J. Immunother. Cancer 2021, 9, e002267. [Google Scholar] [CrossRef] [PubMed]
- Modak, S.; Le Luduec, J.-B.; Cheung, I.Y.; Goldman, D.A.; Ostrovnaya, I.; Doubrovina, E.; Basu, E.; Kushner, B.H.; Kramer, K.; Roberts, S.S.; et al. Adoptive Immunotherapy with Haploidentical Natural Killer Cells and Anti-GD2 Monoclonal Antibody M3F8 for Resistant Neuroblastoma: Results of a Phase I Study. Oncoimmunology 2018, 7, e1461305. [Google Scholar] [CrossRef]
- Veneziani, I.; Infante, P.; Ferretti, E.; Melaiu, O.; Battistelli, C.; Lucarini, V.; Compagnone, M.; Nicoletti, C.; Castellano, A.; Petrini, S.; et al. Nutlin-3a Enhances Natural Killer Cell–Mediated Killing of Neuroblastoma by Restoring P53-Dependent Expression of Ligands for NKG2D and DNAM-1 Receptors. Cancer Immunol. Res. 2021, 9, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Focaccetti, C.; Benvenuto, M.; Pighi, C.; Vitelli, A.; Napolitano, F.; Cotugno, N.; Fruci, D.; Palma, P.; Rossi, P.; Bei, R.; et al. DNAM-1-Chimeric Receptor-Engineered NK Cells, Combined with Nutlin-3a, More Effectively Fight Neuroblastoma Cells in Vitro: A Proof-of-Concept Study. Front. Immunol. 2022, 13, 886319. [Google Scholar] [CrossRef] [PubMed]
- Bodden, M.; Häcker, A.; Röder, J.; Kiefer, A.; Zhang, C.; Bhatti, A.; Pfeifer Serrahima, J.; Ullrich, E.; Kühnel, I.; Wels, W.S. Co-Expression of an IL-15 Superagonist Facilitates Self-Enrichment of GD2-Targeted CAR-NK Cells and Mediates Potent Cell Killing in the Absence of IL-2. Cancers 2023, 15, 4310. [Google Scholar] [CrossRef]
- Chu, Y.; Nayyar, G.; Tian, M.; Lee, D.A.; Ozkaynak, M.F.; Ayala-Cuesta, J.; Klose, K.; Foley, K.; Mendelowitz, A.S.; Luo, W.; et al. Efficiently Targeting Neuroblastoma with the Combination of Anti-ROR1 CAR NK Cells and N-803 in Vitro and in Vivo in NB Xenografts. Mol. Ther. Oncol. 2024, 32, 200820. [Google Scholar] [CrossRef]
- Baghery Saghchy Khorasani, A.; Yousefi, A.M.; Bashash, D. CAR NK Cell Therapy in Hematologic Malignancies and Solid Tumors; Obstacles and Strategies to Overcome the Challenges. Int. Immunopharmacol. 2022, 110, 109041. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrade-Perez, V.; Raynal, N.J.-M. Interplay Between the Epigenome, the Microenvironment, and the Immune System in Neuroblastoma. Cancers 2025, 17, 1812. https://doi.org/10.3390/cancers17111812
Andrade-Perez V, Raynal NJ-M. Interplay Between the Epigenome, the Microenvironment, and the Immune System in Neuroblastoma. Cancers. 2025; 17(11):1812. https://doi.org/10.3390/cancers17111812
Chicago/Turabian StyleAndrade-Perez, Valentina, and Noël J.-M. Raynal. 2025. "Interplay Between the Epigenome, the Microenvironment, and the Immune System in Neuroblastoma" Cancers 17, no. 11: 1812. https://doi.org/10.3390/cancers17111812
APA StyleAndrade-Perez, V., & Raynal, N. J.-M. (2025). Interplay Between the Epigenome, the Microenvironment, and the Immune System in Neuroblastoma. Cancers, 17(11), 1812. https://doi.org/10.3390/cancers17111812