
Modelling Cancer Pathophysiology: Mechanisms and Changes in the Extracellular Matrix During Cancer Initiation and Early Tumour Growth

 , ,
, ,  and
and
Simple Summary
Abstract
1. Introduction
2. ECM Remodelling in Cancerogenic Environments
3. Epithelial Cellular Polarity and Early Tumorigenesis
3.1. Dysregulation in Cellular Polarity During Cancer Initiation and Early Tumorigenesis
3.2. In Vitro Models to Recapitulate Cell Polarity Dysregulations in Cancer
3.2.1. Advances in Hydrogel in Vitro Modelling to Study Cell Polarity
3.2.2. Alternative In Vitro Modelling Strategies of Cellular Polarity and Tumorigenesis
4. Cancer miRNAs and Their Impact on Tumour Initiation, Progression and ECM Remodelling
4.1. MicroRNA in Cancer and Cancer Models
Three-Dimensional In Vitro Models to Study MicroRNAs Associated with Cancer Initiation and Growth
5. EMT and EET in Tumour Initiation and Growth
5.1. The Role of EMT in Cancer Initiation and Early Tumour Growth
5.2. Development of EET to Support Tumorigenesis Under Hypoxic Environments
5.3. In Vitro Modelling Methods to Mimic Tumorigenic EMT and EET Processes
5.3.1. Three-Dimensional Modelling Platforms to Study EMT Mediation in Cancer
5.3.2. Three-Dimensional Modelling Platforms to Study EET and VM Processes
6. Vascularisation and Early Tumour Growth
6.1. Vasculogenesis in Cancer and Tumour Development
6.2. Angiogenesis in Cancer and Tumour Development
6.3. Vascularised In Vitro Models to Recapitulate Cancer Agiogenesis and Vasculogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Description | Advantages | Limitations | References |
|---|---|---|---|---|
| Matrigel™ Assays | Basement membrane extracts from Engelbreth-Holm-Swarm used to study vascular-like networks | - Easy to use and widely available - Allows rapid assessment of vascular-like network formation | - Lacks in vivo 3D cellular organisation - Its composition is undefined/heterogeneous, which can hinder reproducibility | [186,187,192] |
| Hydrogel Models | Uses natural or synthetic polymers to form 3D hydrophilic ECM-like matrices | - Better mimics 3D architecture of ECM than Matrigel™ - Can incorporate well-defined chemical and mechanical factors to study vascularised cancer models | - Still lacks full complexity of in vivo settings - Difficulty in controlling the exact architecture of natural hydrogels - Some synthetic hydrogels may not fully support cellular functions | [189,193,194,195] |
| Microfluidic Models | Use of organ-on-chip platforms to recreate vascularised tumour microenvironments | - Enables dynamic control of fluids, nutrients, and signalling molecules via perfusion mechanics - Precise control of microenvironments and perfusion systems | - Often simpler vascular architectures than those observed in vivo - Achieving physiologically relevant conditions can be challenging | [24,196,197] |
| 3D Bioprinting | Uses bioinks to spatially organised cells and fabricate complex vascularised constructs | - Precise spatial control of cells and scaffold/matrix components - Can precisely deposit multiple cell types and ECM components during fabrication | - Current models are still relatively simple compared to in vivo - Fabrication of bioprinted constructs can be technically challenging and remains relatively costly | [169,198,199] |
6.3.1. Hydrogels Models for Tumorigenic Vascular Development
6.3.2. Microfluidics to Model Vascularised Tumour Environments
6.3.3. 3D Bioprinting to Model Vascularised Tumour Environments
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ECM | Extracellular matrix |
| 3D | Three-dimensional |
| miRNA | Micro-RNA |
| mRNA | Messenger-RNA |
| EMT | Epithelial-to-mesenchymal transition |
| CSC | Cancer stem cells |
| GAG | Glycosaminoglycan |
| MMP | Matrix metalloproteinase |
| MAPL | Mitogen-activated protein kinase |
| PI3K | Phosphoinositide 3-kinase |
| Crumbs3 | Crumbs cell polarity complex component 3 |
| Patj | Pals1-associated tight junction protein |
| Par3 | Partitioning defective 3 |
| Par6 | Partitioning defective 6 |
| aPKC | Atypical protein kinase |
| E-cadherin | Epithelial cadherin |
| Cdc42 | Cell division cycle 42 |
| Lgl | Lethal 2 giant larvae |
| Dlg | Discs large |
| JAM | Junction adhesion molecules |
| PTEN | Phosphatase and tensin homolog |
| MDCK | Madin–Darby canine kidney |
| 2D | Two-dimensional |
| RGD | Arg-Gly-Asp |
| SAPH | Self-assembling peptide hydrogel |
| YGSIR | Tyr–Ile–Gly–Ser–Arg |
| VEGFA | Vascular endothelial growth factor A |
| oncomiRs | Cancer-related miRNAs |
| PDCD4 | Programmed cell death protein 4 |
| TIMP3 | Tissue inhibitor of metalloproteinase 3 |
| RECK | Reversion-inducing cysteine-rich protein with Kazal motifs |
| PEG | Polyethylene glycol |
| MMP-2 | Metalloproteinase-2 |
| MMP-9 | Metalloproteinase-9 |
| Col1A1 | Collagen type I alpha 1 |
| SqCa | Squamous cell carcinoma |
| AdCa | Adenocarcinoma |
| HPV | Human papillomavirus |
| EV | Extracellular vesicle |
| GelMA | Gelatin methacryloyl |
| TGF-β | Transforming growth factor-beta |
| CAF | Cancer-associated fibroblast |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| EC | Endothelial cell |
| EPC | Endothelial progenitor cell |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| SDF-1 | Stromal cell-derived factor 1 |
| FGF | Fibroblast growth factor |
| VSMC | Vascular smooth muscle cell |
| HUVEC | Human umbilical vein endothelial cell |
| Ang-1 | Angiopoietin-1 |
| CLIC3 | Chloride intracellular channel protein 3 |
| ECFC-EC | Endothelial colony forming cell-derived EC |
| GSC | Glioma stem cell |
| ADSC | Adipose-derived stromal cells |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Joyce, J.A. The Evolving Tumor Microenvironment: From Cancer Initiation to Metastatic Outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Catalano, V.; Turdo, A.; Di, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and Its Microenvironment: A Synergistic Interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef]
- Marangio, A.; Biccari, A.; Angelo, E.D.; Sensi, F.; Spolverato, G.; Pucciarelli, S.; Agostini, M. The Study of the Extracellular Matrix in Chronic Inflammation: A Way to Prevent Cancer Initiation? Cancers 2022, 14, 5903. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Duca, L.; Durbeej, M. A Guide to the Composition and Functions of the Extracellular Matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef]
- Winkler, J.; Werb, Z.; Metcalf, K.J. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Plachot, C.; Chaboub, L.S.; Adissu, H.A.; Wang, L.; Urazaev, A.; Sturgis, J.; Asem, E.K.; Lelièvre, S.A. Factors Necessary to Produce Basoapical Polarity in Human Glandular Epithelium Formed in Conventional and High-Throughput Three-Dimensional Culture: Example of the Breast Epithelium. BMC Biol. 2009, 7, 77. [Google Scholar] [CrossRef]
- Wang, B.; Hsu, S.H.; Majumder, S.; Kutay, H.; Huang, W.; Jacob, S.T.; Ghoshal, K. TGFΒ-Mediated Upregulation of Hepatic MiR-181b Promotes Hepatocarcinogenesis by Targeting TIMP3. Oncogene 2010, 29, 1787–1797. [Google Scholar] [CrossRef]
- Mescher, M.; Jeong, P.; Knapp, S.K.; Rübsam, M.; Saynisch, M.; Kranen, M.; Landsberg, J.; Schlaak, M.; Mauch, C.; Tüting, T.; et al. The Epidermal Polarity Protein Par3 Is a Non-Cell Autonomous Suppressor of Malignant Melanoma. J. Exp. Med. 2017, 214, 339–358. [Google Scholar] [CrossRef]
- Peglion, F.; Etienne-Manneville, S. Cell Polarity Changes in Cancer Initiation and Progression. J. Cell Biol. 2023, 223, e202308069. [Google Scholar] [CrossRef] [PubMed]
- Feigin, M.E.; Akshinthala, S.D.; Araki, K.; Rosenberg, A.Z.; Muthuswamy, L.B.; Martin, B.; Lehmann, B.D.; Berman, H.K.; Pietenpol, J.A.; Cardiff, R.D.; et al. Mislocalization of the Cell Polarity Protein Scribble Promotes Mammary Tumorigenesis and Is Associated with Basal Breast Cancer. Cancer Res. 2014, 74, 3180–3194. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.M.; Midwood, K.S. Illustrating the Interplay between the Extracellular Matrix and MicroRNAs. Int. J. Exp. Pathol. 2014, 95, 158–180. [Google Scholar] [CrossRef] [PubMed]
- Humphries, B.; Yang, C. The MicroRNA-200 Family: Small Molecules with Novel Roles in Cancer Development, Progression and Therapy. Oncotarget 2015, 6, 6472–6498. [Google Scholar] [CrossRef]
- Azimzadeh, M.; Rahaie, M.; Nasirizadeh, N.; Ashtari, K.; Naderi-Manesh, H. An Electrochemical Nanobiosensor for Plasma MiRNA-155, Based on Graphene Oxide and Gold Nanorod, for Early Detection of Breast Cancer. Biosens. Bioelectron. 2016, 77, 99–106. [Google Scholar] [CrossRef]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma Tumor Stem-like Cells Promote Tumor Angiogenesis and Vasculogenesis via Vascular Endothelial Growth Factor and Stromal-Derived Factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar] [CrossRef]
- Paprocka, M.; Kieda, C.; Kantor, A.; Bielawska-Pohl, A.; Dus, D.; Czekanski, A.; Heimrath, J. Increased Endothelial Progenitor Cell Number in Early Stage of Endometrial Cancer. Int. J. Gynecol. Cancer 2017, 27, 947–952. [Google Scholar] [CrossRef]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of Angiogenesis during the Transition from Hyperplasia to Neoplasia. Nature 1989, 339, 58–61. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The Role of Microenvironment in Tumor Angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Kaur, G.; Roy, B. Decoding Tumor Angiogenesis for Therapeutic Advancements: Mechanistic Insights. Biomedicines 2024, 12, 827. [Google Scholar] [CrossRef]
- Magdeldin, T.; López-Dávila, V.; Pape, J.; Cameron, G.W.W.; Emberton, M.; Loizidou, M.; Cheema, U. Engineering a Vascularised 3D In Vitro Model of Cancer Progression. Sci. Rep. 2017, 7, 44045. [Google Scholar] [CrossRef]
- Micalet, A.; Moeendarbary, E.; Cheema, U. 3D In Vitro Models for Investigating the Role of Stiffness in Cancer Invasion. ACS Biomater. Sci. Eng. 2021, 9, 3729–3741. [Google Scholar] [CrossRef] [PubMed]
- Madsen, N.H.; Nielsen, B.S.; Larsen, J.; Gad, M. In Vitro 2D and 3D Cancer Models to Evaluate Compounds That Modulate Macrophage Polarization. Cell. Immunol. 2022, 378, 104574. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, S.R.; Cristante, J.; Guyon, L.; Denis, J.; Chabre, O.; Cherradi, N. Microrna Therapeutics in Cancer: Current Advances and Challenges. Cancers 2021, 13, 2680. [Google Scholar] [CrossRef]
- Brassard-Jollive, N.; Monnot, C.; Muller, L.; Germain, S. In Vitro 3D Systems to Model Tumor Angiogenesis and Interactions with Stromal Cells. Front. Cell Dev. Biol. 2020, 8, 594903. [Google Scholar] [CrossRef]
- Pompili, S.; Latella, G.; Gaudio, E.; Sferra, R.; Vetuschi, A. The Charming World of the Extracellular Matrix: A Dynamic and Protective Network of the Intestinal Wall. Front. Med. 2021, 8, 610189. [Google Scholar] [CrossRef]
- Dzobo, K.; Dandara, C. The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics 2023, 8, 146. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular Matrix and Its Therapeutic Potential for Cancer Treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef]
- Taufalele, P.V.; VanderBurgh, J.A.; Muñoz, A.; Zanotelli, M.R.; Reinhart-King, C.A. Fiber Alignment Drives Changes in Architectural and Mechanical Features in Collagen Matrices. PLoS ONE 2019, 14, e0216537. [Google Scholar] [CrossRef]
- Itoh, Y.; Takehara, Y.; Kawase, T.; Terashima, K.; Ohkawa, Y.; Hirose, Y.; Koda, A.; Hyodo, N.; Ushio, T.; Hirai, Y.; et al. Feasibility of Magnetic Resonance Elastography for the Pancreas at 3T. J. Magn. Reson. Imaging 2016, 43, 384–390. [Google Scholar] [CrossRef]
- Boyd, N.F.; Li, Q.; Melnichouk, O.; Huszti, E.; Martin, L.J.; Gunasekara, A.; Mawdsley, G.; Yaffe, M.J.; Minkin, S. Evidence That Breast Tissue Stiffness Is Associated with Risk of Breast Cancer. PLoS ONE 2014, 9, e100937. [Google Scholar] [CrossRef]
- Mieulet, V.; Garnier, C.; Kieffer, Y.; Guilbert, T.; Nemati, F.; Marangoni, E.; Renault, G.; Chamming’s, F.; Vincent-Salomon, A.; Mechta-Grigoriou, F. Stiffness Increases with Myofibroblast Content and Collagen Density in Mesenchymal High Grade Serous Ovarian Cancer. Sci. Rep. 2021, 11, 4219. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, O.; Koshy, S.T.; Branco Da Cunha, C.; Shin, J.W.; Verbeke, C.S.; Allison, K.H.; Mooney, D.J. Extracellular Matrix Stiffness and Composition Jointly Regulate the Induction of Malignant Phenotypes in Mammary Epithelium. Nat. Mater. 2014, 13, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.; Sun, H.; Jones, M.; Percival, H.; Broadberry, E.; Zindy, E.; Lawless, C.; Streuli, C.; Swift, J.; Brennan, K.; et al. Increased Microenvironment Stiffness Leads to Altered Aldehyde Metabolism and DNA Damage in Mammary Epithelial Cells through a RhoA-Dependent Mechanism. bioRxiv 2020. [Google Scholar] [CrossRef]
- You, Y.; Zheng, Q.; Dong, Y.; Xie, X.; Wang, Y.; Wu, S.; Zhang, L.; Wang, Y.; Xue, T.; Wang, Z.; et al. Matrix Stiffness-Mediated Effects on Stemness Characteristics Occurring in HCC Cells. Oncotarget 2016, 7, 32221. [Google Scholar] [CrossRef] [PubMed]
- Shieh, A.C. Biomechanical Forces Shape the Tumor Microenvironment. Ann. Biomed. Eng. 2011, 39, 1379–1389. [Google Scholar] [CrossRef]
- Mitchell, M.J.; King, M.R. Computational and Experimental Models of Cancer Cell Response to Fluid Shear Stress. Front. Oncol. 2013, 3, 44. [Google Scholar] [CrossRef]
- Mpekris, F.; Angeli, S.; Pirentis, A.P.; Stylianopoulos, T. Stress-Mediated Progression of Solid Tumors: Effect of Mechanical Stress on Tissue Oxygenation, Cancer Cell Proliferation, and Drug Delivery. Biomech. Model. Mechanobiol. 2015, 14, 1391–1402. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 Mediates Metabolic Responses to Intratumoral Hypoxia and Oncogenic Mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.L.; Sivridis, E. Comparison of Metabolic Pathways between Cancer Cells and Stromal Cells in Colorectal Carcinomas: A Metabolic Survival Role for Tumor-Associated Stroma. Cancer Res. 2006, 66, 632–637. [Google Scholar] [CrossRef]
- Levayer, R. Solid Stress, Competition for Space and Cancer: The Opposing Roles of Mechanical Cell Competition in Tumour Initiation and Growth. Semin. Cancer Biol. 2020, 63, 69–80. [Google Scholar] [CrossRef]
- Lee, H.J.; Diaz, M.F.; Price, K.M.; Ozuna, J.A.; Zhang, S.; Sevick-Muraca, E.M.; Hagan, J.P.; Wenzel, P.L. Fluid Shear Stress Activates YAP1 to Promote Cancer Cell Motility. Nat. Commun. 2017, 8, 14122. [Google Scholar] [CrossRef] [PubMed]
- Hyler, A.R.; Baudoin, N.C.; Brown, M.S.; Stremler, M.A.; Cimini, D.; Davalos, R.V.; Schmelz, E.M. Fluid Shear Stress Impacts Ovarian Cancer Cell Viability, Subcellular Organization, and Promotes Genomic Instability. PLoS ONE 2018, 13, e0194170. [Google Scholar] [CrossRef] [PubMed]
- Godde, N.J.; Galea, R.C.; Elsum, I.A.; Humbert, P.O. Cell Polarity in Motion: Redefining Mammary Tissue Organization through EMT and Cell Polarity Transitions. J. Mammary Gland Biol. Neoplasia 2010, 15, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Halaoui, R.; McCaffrey, L. Rewiring Cell Polarity Signaling in Cancer. Oncogene 2015, 34, 939–950. [Google Scholar] [CrossRef]
- Campanale, J.P.; Sun, T.Y.; Montell, D.J. Development and Dynamics of Cell Polarity at a Glance. J. Cell Sci. 2017, 130, 1201–1207. [Google Scholar] [CrossRef]
- Thottacherry, J.J.; Chen, J.; Johnston, D.S. Apical-Basal Polarity in the Gut. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2023; Volume 150–151, pp. 15–22. [Google Scholar]
- Buckley, C.E.; St Johnston, D. Apical–Basal Polarity and the Control of Epithelial Form and Function. Nat. Rev. Mol. Cell Biol. 2022, 23, 559–577. [Google Scholar] [CrossRef]
- Whitford, M.K.M.; McCaffrey, L. Chapter Nine—Polarity in Breast Development and Cancer; Elsevier: Amsterdam, The Netherlands, 2023; Volume 154, ISBN 9780128201589. [Google Scholar]
- Russ, A.; Louderbough, J.M.V.; Zarnescu, D.; Schroeder, J.A. Hugl1 and Hugl2 in Mammary Epithelial Cells: Polarity, Proliferation, and Differentiation. PLoS ONE 2012, 7, e47734. [Google Scholar] [CrossRef]
- Stelwagen, K.; Singh, K. The Role of Tight Junctions in Mammary Gland Function. J. Mammary Gland Biol. Neoplasia 2014, 19, 131–138. [Google Scholar] [CrossRef]
- Otani, T.; Furuse, M. Tight Junction Structure and Function Revisited. Trends Cell Biol. 2020, 30, 805–817. [Google Scholar] [CrossRef]
- Chatterjee, S.J.; Mccaffrey, L.; Mccaffrey, L. Emerging Role of Cell Polarity Proteins in Breast Cancer Progression and Metastasis. Breast Cancer Targets Ther. 2014, 6, 15–27. [Google Scholar]
- Pearson, H.; Perez-Mancera, P.; Dow, L.; Ryan, A.; Tennstedt, P.; Bogani, D.; Elsum, I.; Greenfield, A.; Tuveson, D.; Simon, R.; et al. SCRIB Expression Is Deregulated in Human Prostate Cancer, and Its Deficiency in Mice Promotes Prostate Neoplasia. J. Clin. Investig. 2011, 121, 4257–4267. [Google Scholar] [CrossRef] [PubMed]
- Shelton, P.M.; Duran, A.; Nakanishi, Y.; Caceres, J.F.; Diaz-meco, M.T.; Moscat, J.; Shelton, P.M.; Duran, A.; Nakanishi, Y.; Reina-campos, M.; et al. The Secretion of MiR-200s by a PKC z / ADAR2 Signaling Axis Promotes Liver Metastasis in Colorectal Cancer Article The Secretion of MiR-200s by a PKC z / ADAR2 Signaling Axis Promotes Liver Metastasis in Colorectal Cancer. Cell Rep. 2018, 23, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Fattet, L.; Tsai, J.H.; Kajimoto, T.; Chang, Q.; Newton, A.C.; Yang, J. Apical–Basal Polarity Inhibits Epithelial–Mesenchymal Transition and Tumour Metastasis by PAR-Complex-Mediated SNAI1 Degradation. Nat. Cell Biol. 2019, 21, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Sonoshita, M.; Cagan, R.L. Modeling Human Cancers in Drosophila, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 121. [Google Scholar]
- Zhou, P.J.; Wang, X.; An, N.; Wei, L.; Zhang, L.; Huang, X.; Zhu, H.H.; Fang, Y.X.; Gao, W.Q. Loss of Par3 Promotes Prostatic Tumorigenesis by Enhancing Cell Growth and Changing Cell Division Modes. Oncogene 2019, 38, 2192–2205. [Google Scholar] [CrossRef]
- Portela, M.; Mukherjee, S.; Paul, S.; La Marca, J.E.; Parsons, L.M. The Drosophila Tumour Suppressor Lgl and Vap33 Activate the Hippo Pathway through a Dual Mechanism. J. Cell Sci. 2024, 137, jcs261917. [Google Scholar] [CrossRef]
- Saito, Y.; Li, L.; Coyaud, E.; Luna, A.; Sander, C.; Raught, B.; Asara, J.M.; Brown, M.; Muthuswamy, S.K. LLGL2 Rescues Nutrient Stress by Promoting Leucine Uptake in ER+ Breast Cancer. Nature 2019, 569, 275–279. [Google Scholar] [CrossRef]
- Zhan, L.; Rosenberg, A.; Bergami, K.C.; Yu, M.; Xuan, Z.; Jaffe, A.B.; Allred, C.; Muthuswamy, S.K. Deregulation of Scribble Promotes Mammary Tumorigenesis and Reveals a Role for Cell Polarity in Carcinoma. Cell 2008, 135, 865–878. [Google Scholar] [CrossRef]
- Roignot, J.; Peng, X.; Mostov, K. Polarity in Mammalian Epithelial Morphogenesis. Cold Spring Harb. Perspect. Biol. 2013, 5, a013789. [Google Scholar] [CrossRef]
- Horikoshi, Y.; Suzuki, A.; Yamanaka, T.; Sasaki, K.; Mizuno, K.; Sawada, H.; Yonemura, S.; Ohno, S. Interaction between PAR-3 and the APKC-PAR-6 Complex Is Indispensable for Apical Domain Development of Epithelial Cells. J. Cell Sci. 2009, 122, 1595–1606. [Google Scholar] [CrossRef]
- Yu, W.; Datta, A.; Leroy, P.; O’Brien, L.E.; Mak, G.; Jou, T.S.; Matlin, K.S.; Mostov, K.E.; Zegers, M.M.P. Β1-Integrin Orients Epithelial Polarity via Rac1 and Laminin. Mol. Biol. Cell 2005, 16, 433–445. [Google Scholar] [CrossRef]
- Chatterjee, S.; Seifried, L.; Feigin, M.E.; Gibbons, D.L.; Scuoppo, C.; Lin, W.; Rizvi, Z.H.; Lind, E.; Dissanayake, D.; Kurie, J.; et al. Dysregulation of Cell Polarity Proteins Synergize with Oncogenes or the Microenvironment to Induce Invasive Behavior in Epithelial Cells. PLoS ONE 2012, 7, e34343. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Cukierman, E. Modeling Tissue Morphogenesis and Cancer in 3D. Cell 2007, 130, 601–610. [Google Scholar] [CrossRef]
- Barros, P.; Oliveira, R.J.A.; Araújo, M.; Caires, H.R.; Bidarra, S.J.; Barrias, C. An Integrative Alginate-Based 3D in Vitro Model to Explore Epithelial-Stromal Cell Dynamics in the Breast Tumor Microenvironment. Carbohydr. Polym. 2024, 342, 122363. [Google Scholar] [CrossRef] [PubMed]
- Bidarra, S.J.; Oliveira, P.; Rocha, S.; Saraiva, D.P.; Oliveira, C.; Barrias, C.C. A 3D in Vitro Model to Explore the Inter-Conversion between Epithelial and Mesenchymal States during EMT and Its Reversion. Sci. Rep. 2016, 6, 27072. [Google Scholar] [CrossRef]
- Nowak, M.; Freudenberg, U.; Tsurkan, M.V.; Werner, C.; Levental, K.R. Biomaterials Modular GAG-Matrices to Promote Mammary Epithelial Morphogenesis in Vitro. Biomaterials 2017, 112, 20–30. [Google Scholar] [CrossRef]
- Weiss, M.S.; Peñalver, B.; Shikanov, A.; Bluver, D.A.; Mui, M.D.; Shin, S.; Broadbelt, L.J.; Shea, L.D. Biomaterials The Impact of Adhesion Peptides within Hydrogels on the Phenotype and Signaling of Normal and Cancerous Mammary Epithelial Cells. Biomaterials 2012, 33, 3548–3559. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, C.; Span, P.N.; Rowan, A.E.; Aalders, T.W.; Schalken, J.A.; Adema, G.J.; Kouwer, P.H.J.; Zegers, M.M.P.; Ansems, M. Polyisocyanide Hydrogels as a Tunable Platform for Mammary Gland Organoid Formation. Biomaterials 2020, 112, 20–30. [Google Scholar] [CrossRef]
- Clough, H.C.; Brien, M.O.; Zhu, X.; Miller, A.F.; Saiani, A.; Tsigkou, O. Materials Science & Engineering C Neutrally Charged Self-Assembling Peptide Hydrogel Recapitulates in Vitro Mechanisms of Breast Cancer Progression. Mater. Sci. Eng. C 2021, 127, 112200. [Google Scholar]
- Lingard, E.; Dong, S.; Hoyle, A.; Appleton, E.; Hales, A.; Skaria, E.; Lawless, C.; Taylor-hearn, I.; Saadati, S.; Chu, Q.; et al. Biomaterials Advances Optimising a Self-Assembling Peptide Hydrogel as a Matrigel Alternative for 3-Dimensional Mammary Epithelial Cell Culture. Biomater. Adv. 2024, 160, 213847. [Google Scholar] [CrossRef]
- Mirshafiei, M.; Rashedi, H.; Yazdian, F.; Rahdar, A.; Baino, F. Advancements in Tissue and Organ 3D Bioprinting: Current Techniques, Applications, and Future Perspectives. Mater. Des. 2024, 240, 112853. [Google Scholar] [CrossRef]
- Swaminathan, S.; Hamid, Q.; Sun, W.; Clyne, A. Bioprinting of 3D Breast Epithelial Spheroids for Human Cancer Models. Biofabrication 2019, 11, 025003. [Google Scholar] [CrossRef] [PubMed]
- Creff, J.; Courson, R.; Mangeat, T.; Foncy, J.; Souleille, S.; Thibault, C.; Besson, A.; Malaquin, L. Fabrication of 3D Scaffolds Reproducing Intestinal Epithelium Topography by High-Resolution 3D Stereolithography. Biomaterials 2019, 221, 119404. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.S.; Sun, X.; Baird, M.A.; Hourwitz, M.J.; Ri, B.; Pasapera, A.M.; Mehta, S.B.; Losert, W.; Fischbach, C.; Fourkas, J.T.; et al. Contractility, Focal Adhesion Orientation, and Stress Fiber Orientation Drive Cancer Cell Polarity and Migration along Wavy ECM Substrates. Proc. Natl. Acad. Sci. USA 2021, 118, 2021135118. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Wanunu, M.; Wang, M.X.; McReynolds, L.; Wang, Y. Detection of MiRNAs with a Nanopore Single-Molecule Counter. Expert Rev. Mol. Diagn. 2012, 12, 573–584. [Google Scholar] [CrossRef]
- Ambros, V.; Horvitz, H.R. Heterochronic Mutants of the Nematode Caenorhabditis Elegans. Science 1984, 226, 409–416. [Google Scholar] [CrossRef]
- Ambros, V.; Horvitz, H.R. The Lin-14 Locus of Caenorhabditis Elegans Controls the Time of Expression of Specific Postembryonic Developmental Events. Genes Dev. 1987, 1, 398–414. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Ono, S.; Lam, S.; Nagahara, M.; Hoon, D.S.B. Circulating MicroRNA Biomarkers as Liquid Biopsy for Cancer Patients: Pros and Cons of Current Assays. J. Clin. Med. 2015, 4, 1890–1907. [Google Scholar] [CrossRef]
- Wu, W. MicroRNA and Cancer. Methods Mol. Biol. 2011, 676, 59–70. [Google Scholar]
- Cui, M.; Wang, H.; Yao, X.; Zhang, D.; Xie, Y.; Cui, R.; Zhang, X. Circulating MicroRNAs in Cancer: Potential and Challenge. Front. Genet. 2019, 10, 626. [Google Scholar] [CrossRef]
- Gao, Y.; Feng, B.; Han, S.; Zhang, K.; Chen, J.; Li, C.; Wang, R.; Chen, L. The Roles of MicroRNA-141 in Human Cancers: From Diagnosis to Treatment. Cell. Physiol. Biochem. 2016, 38, 427–448. [Google Scholar] [CrossRef] [PubMed]
- Zaravinos, A. The Regulatory Role of MicroRNAs in EMT and Cancer. J. Oncol. 2015, 1, 865816. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.C.; Yoon, S.; Jeong, Y.; Yoon, J.; Baek, K. Regulation of Vascular Endothelial Growth Factor Signaling by MiR-200b. Mol. Cells 2011, 32, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Kuehbacher, A.; Urbich, C.; Zeiher, A.M.; Dimmeler, S. Role of Dicer and Drosha for Endothelial MicroRNA Expression and Angiogenesis. Circ. Res. 2007, 101, 59–68. [Google Scholar] [CrossRef]
- Song, Y.; Zeng, S.; Zheng, G.; Chen, D.; Li, P.; Yang, M.; Luo, K. FOXO3a-Driven MiRNA Signatures Suppresses VEGF-A/NRP1 Signaling and Breast Cancer Metastasis. Oncogene 2021, 40, 777–790. [Google Scholar] [CrossRef]
- Gao, F.; Li, X.; Xu, K.; Wang, R.; Guan, X. C-MYC Mediates the Crosstalk between Breast Cancer Cells and Tumor Microenvironment. Cell Commun. Signal. 2023, 21, 28. [Google Scholar] [CrossRef]
- Humphries, B.; Wang, Z.; Yang, C. MicroRNA Regulation of Epigenetic Modifiers in Breast Cancer. Cancers 2019, 11, 897. [Google Scholar] [CrossRef]
- Lin, S.; Gregory, R.I. MicroRNA Biogenesis Pathways in Cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Grimaldi, A.M.; Nuzzo, S.; Condorelli, G.; Salvatore, M.; Incoronato, M. Prognostic and Clinicopathological Significance of MiR-155 in Breast Cancer: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 5834. [Google Scholar] [CrossRef]
- Chang, S.; Wang, R.H.; Akagi, K.; Kim, K.A.; Martin, B.K.; Cavallone, L.; Haines, D.C.; Basik, M.; Mai, P.; Poggi, E.; et al. Tumor Suppressor BRCA1 Epigenetically Controls Oncogenic MicroRNA-155. Nat. Med. 2011, 17, 1275–1282. [Google Scholar] [CrossRef]
- Kim, J.G.; Islam, R.; Cho, J.Y.; Jeong, H.; Cap, K.C.; Park, Y.; Hossain, A.J.; Park, J.B. Regulation of RhoA GTPase and Various Transcription Factors in the RhoA Pathway. J. Cell. Physiol. 2018, 233, 6381–6392. [Google Scholar] [CrossRef] [PubMed]
- Armand-Labit, V.; Pradines, A. Circulating Cell-Free MicroRNAs as Clinical Cancer Biomarkers. Biomol. Concepts 2017, 8, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Zhang, R.; Li, Y.; Pu, J.; Lu, Y.; Jiao, J.; Li, K.; Yu, B.; Li, Z.; Wang, R.; et al. Circulating MicroRNA-1 as a Potential Novel Biomarker for Acute Myocardial Infarction. Biochem. Biophys. Res. Commun. 2010, 391, 73–77. [Google Scholar] [CrossRef]
- Bautista-Sánchez, D.; Arriaga-Canon, C.; Pedroza-Torres, A.; La Rosa-Velázquez, I.A.D.; González-Barrios, R.; Contreras-Espinosa, L.; Montiel-Manríquez, R.; Castro-Hernández, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. The Promising Role of MiR-21 as a Cancer Biomarker and Its Importance in RNA-Based Therapeutics. Mol. Ther. Nucleic Acids 2020, 20, 409–420. [Google Scholar] [CrossRef]
- Li, J.; Fu, R.; Yang, L.; Tu, W. MiR-21 Expression Predicts Prognosis in Diffuse Large B-Cell Lymphoma. Int. J. Clin. Exp. Pathol. 2015, 8, 15019. [Google Scholar] [PubMed]
- Masoudi, M.S. MiR-21: A Key Player in Glioblastoma Pathogenesis. J. Cell. Biochem. 2018, 119, 1285–1290. [Google Scholar] [CrossRef]
- Fang, H.; Xie, J.; Zhang, M.; Zhao, Z.; Wan, Y.; Yao, Y. MiRNA-21 Promotes Proliferation and Invasion of Triple-Negative Breast Cancer Cells through Targeting PTEN. Am. J. Transl. Res. 2017, 9, 953–961. [Google Scholar]
- Selves, J.; Al Saati, T.; Souque, A.; Tsongalis, G.J.; Suriawinata, A.A.; Carre, N.; Buscail, L.; Cordelier, P. MicroRNA-21 Is Induced Early in Pancreatic Ductal Adenocarcinoma Precursor Lesions. Clin. Chem. 2010, 56, 603–612. [Google Scholar]
- Hua, Y.T.; Xu, W.X.; Li, H.; Xia, M. Emerging Roles of MiR-133a in Human Cancers. J. Cancer 2021, 12, 198. [Google Scholar] [CrossRef]
- Dal Bo, M.; Bomben, R.; Hernández, L.; Gattei, V. The MYC/MiR-17-92 Axis in Lymphoproliferative Disorders: A Common Pathway with Therapeutic Potential. Oncotarget 2015, 6, 19381. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. MicroRNAs as Oncogenes and Tumor Suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative Disease and Autoimmunity in Mice with Increased MiR-17-92 Expression in Lymphocytes. Nat. Immunol. 2008, 9, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Nguyen, L.H.; Zhou, K.; De Soysa, T.Y.; Li, L.; Miller, J.B.; Tian, J.; Locker, J.; Zhang, S.; Shinoda, G.; et al. Precise Let-7 Expression Levels Balance Organ Regeneration against Tumor Suppression. Elife 2015, 4, e09431. [Google Scholar] [CrossRef]
- Li, X.; Roslan, S.; Johnstone, C.N.; Wright, J.A.; Bracken, C.P.; Anderson, M.; Bert, A.G.; Selth, L.A.; Anderson, R.L.; Goodall, G.J. MiR-200 Can Repress Breast Cancer Metastasis through ZEB1-Independent but Moesin-Dependent Pathways. Oncogene 2014, 33, 4077–4088. [Google Scholar] [CrossRef]
- Banyard, J.; Chung, I.; Wilson, A.M.; Vetter, G.; Le Béchec, A.; Bielenberg, D.R.; Zetter, B.R. Regulation of Epithelial Plasticity by MiR-424 and MiR-200 in a New Prostate Cancer Metastasis Model. Sci. Rep. 2013, 3, 3151. [Google Scholar] [CrossRef]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and Oncogenesis of MCF-10A Mammary Epithelial Acini Grown in Three-Dimensional Basement Membrane Cultures. Methods 2023, 30, 256–268. [Google Scholar] [CrossRef]
- Salinas-Vera, Y.M.; Valdes, J.; Perez-Navarro, Y.; Mandujano-Lazaro, G.; Marchat, L.A.; Ramos-Payan, R.; Nunez-Olvera, S.I.; Perez-Plascencia, C.; Lopez-Camarillo, C. Three-Dimensional 3D Culture Models in Gynecological and Breast Cancer Research. Front. Oncol. 2022, 12, 826113. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Li, C.U.I.; Lin, Z.; Zhuang, Y.A.N.; Flemington, E.K.; Burow, M.E.; Lin, Y.I.; Shan, B.I.N. The MicroRNA Expression Associated with Morphogenesis of Breast Cancer Cells in Three-Dimensional Organotypic Culture. Oncol. Rep. 2012, 28, 117–126. [Google Scholar]
- Salinas-Vera, Y.M.; Hidalgo-Miranda, A.; Cisneros-Villanueva, M.; Arriaga-Pizano, L.A.; Prieto-Chávez, J.L.; López-Camarillo, C. Three-Dimensional Organotypic Cultures Reshape the MicroRNAs Transcriptional Program in Breast Cancer Cells. Cancers 2022, 14, 2490. [Google Scholar] [CrossRef]
- Lõhmussaar, K.; Oka, R.; Valle-Inclan, J.E.; Smits, M.H.; Wardak, H.; Korving, J.; Begthel, H.; Proost, N.; van de Ven, M.; Kranenburg, O.W.; et al. Patient-Derived Organoids Model Cervical Tissue Dynamics and Viral Oncogenesis in Cervical Cancer. Cell Stem Cell 2021, 28, 1380–1396. [Google Scholar] [CrossRef]
- Thippabhotla, S.; Zhong, C.; He, M. 3D Cell Culture Stimulates the Secretion of in Vivo like Extracellular Vesicles. Sci. Rep. 2019, 9, 13012. [Google Scholar] [CrossRef] [PubMed]
- Godbole, N.; Lai, A.; Carrion, F.; Scholz-romero, K.; Ravichandran, A.; Croft, P.K.; Mccart, A.E.; Joshi, V.; Lakhani, S.R.; Kamal, M.; et al. Extracellular Vesicle MiRNAs from Three-Dimensional Ovarian Cancer in Vitro Models and Their Implication in Overall Cancer Survival. Heliyon 2025, 11, e42188. [Google Scholar] [CrossRef] [PubMed]
- Balachander, G.M.; Rajashekar, B.; Sarashetti, P.M.; Rangarajan, A.; Chatterjee, K. MiRNomics Reveals Breast Cancer Cells Cultured on 3D Scaffolds Better Mimic Tumors in Vivo than Conventional 2D Culture. ACS Biomater. Sci. Eng. 2018, 4, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Lu, W.; Wang, G.; Li, Y.; Li, S.; Song, X.; Wang, X.; Chuai, M.; Ka, K.; Lee, H.; Cao, L.; et al. Autophagy Functions on EMT in Gastrulationof Avian Embryo. Cell Cycle 2014, 13, 2752–2764. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Abd, R.; Raouf, E.; Ouf, S.A.; Gabr, M.M.; Zakaria, M.M.; El Yasergy, K.F.; Ali, B.; Dein, E. Escherichia Coli Foster Bladder Cancer Cell Line Progression via Epithelial Mesenchymal Transition, Stemness and Metabolic Reprogramming. Sci. Rep. 2020, 10, 18024. [Google Scholar]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-Endothelial Transition and Cancer Stem Cells: Two Cornerstones of Vasculogenic Mimicry in Malignant Tumors. Oncotarget 2016, 8, 30502. [Google Scholar] [CrossRef]
- Wei, X.; Chen, Y.; Jiang, X.; Peng, M.; Liu, Y.; Mo, Y.; Ren, D.; Hua, Y.; Yu, B.; Zhou, Y.; et al. Mechanisms of Vasculogenic Mimicry in Hypoxic Tumor Microenvironments. Mol. Cancer 2021, 20, 1–18. [Google Scholar] [CrossRef]
- Xiao, T.; Zhang, Q.; Zong, S.; Zhong, W.; Qin, Y.; Bi, Z.; Chen, S.; Liu, H.; Wei, J.; Zhou, B.; et al. Protease-Activated Receptor-1 (PAR1) Promotes Epithelial-Endothelial Transition through Twist1 in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 185. [Google Scholar] [CrossRef]
- Chen, F.; Long, Q.; Fu, D.; Zhu, D.; Ji, Y.; Han, L.; Zhang, B.; Xu, Q.; Liu, B.; Li, Y.; et al. Targeting SPINK1 in the Damaged Tumour Microenvironment Alleviates Therapeutic Resistance. Nat. Commun. 2018, 9, 4315. [Google Scholar] [CrossRef] [PubMed]
- Vićovac, L.; Aplin, J.D. Epithelial-Mesenchymal Transition during Trophoblast Differentiation. Cells Tissues Organs 1996, 156, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Debnath, P.; Singh, R.; Bhowmick, A.; Ghosh, A.; Ghosh, D.; Dutta, P.; Maity, D.; Palchaudhuri, S. Biochimie Epithelial Mesenchymal Transition Induced Nuclear Localization of the Extracellular Matrix Protein Fibronectin. Biochimie 2024, 219, 142–145. [Google Scholar] [CrossRef]
- Horejs, C.; Serio, A.; Purvis, A.; Gormley, A.J.; Bertazzo, S.; Poliniewicz, A. Biologically-Active Laminin-111 Fragment That Modulates the Epithelial-to-Mesenchymal Transition in Embryonic Stem Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 5908–5913. [Google Scholar] [CrossRef]
- de Almeida, P.G.; Pinheiro, G.G.; Nunes, A.M.; Gonçalves, A.B.; Thorsteinsdóttir, S. Fibronectin Assembly during Early Embryo Development: A Versatile Communication System between Cells and Tissues. Dev. Dyn. 2016, 245, 520–535. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Hayward, M.K.; Muncie, J.M.; Weaver, V.M. Tissue Mechanics in Stem Cell Fate, Development, and Cancer. Dev. Cell 2021, 56, 1833–1847. [Google Scholar] [CrossRef]
- Spicer, A.P.; Tien, J.Y.L. Hyaluronan and Morphogenesis. Birth Defects Res. 2004, 72, 89–108. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling Mechanisms of the Epithelial-Mesenchymal Transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.S.; Ram Padam, K.S.; Sharma, M.; Kudva, A.; Patel, P.; Radhakrishnan, R. Novel Transcripts of EMT Driving the Malignant Transformation of Oral Submucous Fibrosis. Sci. Rep. 2025, 15, 3294. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Barrio, M.G.; Portillo, F.; Nieto, M.A. The Transcription Factor Snail Controls Epithelial—Mesenchymal Transitions by Repressing E-Cadherin Expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Leight, J.L.; Wozniak, M.A.; Chen, S.; Lynch, M.L.; Chen, C.S.; Wang, Y. Matrix Rigidity Regulates a Switch between TGF-Β1–Induced Apoptosis and Epithelial–Mesenchymal Transition. Mol. Biol. Cell 2012, 23, 781–791. [Google Scholar] [CrossRef]
- Krstic, J.; Santibanez, J.F. Transforming Growth Factor-Beta and Matrix Metalloproteinases: Functional Interactions in Tumor Stroma-Infiltrating Myeloid Cells. Sci. World J. 2014, 1, 521754. [Google Scholar] [CrossRef]
- Espinosa Neira, R.; Salazar, E.P. Native Type IV Collagen Induces an Epithelial to Mesenchymal Transition-like Process in Mammary Epithelial Cells MCF10A. Int. J. Biochem. Cell Biol. 2012, 44, 2194–2203. [Google Scholar] [CrossRef]
- Walter, C.; Davis, J.T.; Mathur, J.; Pathak, A. Physical Defects in Basement Membrane-Mimicking Collagen-IV Matrices Trigger Cellular EMT and Invasion. Integr. Biol. 2018, 10, 342–355. [Google Scholar] [CrossRef]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-schaller, N.; Cornille, K.; Hopfer, U.; Bentires-alj, M.; et al. VEGF-Mediated Angiogenesis Links EMT-Induced Cancer Stemness to Tumor Initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef]
- Jokela, T.A.; LaBarge, M.A. Integration of Mechanical and ECM Microenvironment Signals in the Determination of Cancer Stem Cell States. Curr. Stem Cell Rep. 2021, 7, 39–47. [Google Scholar] [CrossRef]
- Landen, C.N.; Goodman, B.; Katre, A.A.; Steg, A.D.; Nick, A.M.; Stone, R.L.; Miller, L.D.; Mejia, P.V.; Jennings, N.B.; Gershenson, D.M.; et al. Targeting Aldehyde Dehydrogenase Cancer Stem Cells in Ovarian Cancer. Mol. Cancer Ther. 2010, 9, 3186–3199. [Google Scholar] [CrossRef]
- Sultan, M.; Coyle, K.M.; Vidovic, D.; Thomas, M.L.; Gujar, S.; Marcato, P. Hide-and-Seek: The Interplay between Cancer Stem Cells and the Immune System. Carcinogenesis 2017, 2, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Larrea Murillo, L.; Sugden, C.J.; Ozsvari, B.; Moftakhar, Z.; Hassan, G.S.; Sotgia, F.; Lisanti, M.P. ALDH High Breast Cancer Stem Cells Exhibit a Mesenchymal—Senescent Hybrid Phenotype, with Elevated Metabolic and Migratory Activities. Cells 2024, 13, 2059. [Google Scholar] [CrossRef]
- Luo, X.; Zou, W.; Wei, Z.; Yu, S.; Zhao, Y.; Wu, Y.; Wang, A.; Lu, Y. Inducing Vascular Normalization: A Promising Strategy for Immunotherapy. Int. Immunopharmacol. 2022, 112, 109167. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zhong, W.; Zhao, J.; Qian, B.; Liu, H.; Chen, S.; Qiao, K.; Lei, Y.; Zong, S.; Wang, H.; et al. Polyphyllin I Suppresses the Formation of Vasculogenic Mimicry via Twist1/VE-Cadherin Pathway. Cell Death Dis. 2018, 9, 906. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, A.; Peri, S.; Versienti, G.; Fiorillo, C.; Becatti, M.; Magnelli, L.; Papucci, L. Gastric Cancer Vascularization and the Contribution of Reactive Oxygen Species. Biomolecules 2023, 13, 886. [Google Scholar] [CrossRef]
- Sun, D.; Sun, B.; Liu, T.; Zhao, X.; Che, N.; Gu, Q.; Dong, X.; Yao, Z.; Li, R.; Li, J.; et al. Slug Promoted Vasculogenic Mimicry in Hepatocellular Carcinoma. J. Cell. Mol. Med. 2013, 17, 1038–1047. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Tang, H.; Chen, L.; Liu, X.; Zeng, S.; Tan, H.; Chen, G. Pan-Cancer Dissection of Vasculogenic Mimicry Characteristic to Provide Potential Therapeutic Targets. Front. Pharmacol. 2024, 15, 1346719. [Google Scholar] [CrossRef]
- Yang, J.; Lu, Y.; Lin, Y.Y.; Zheng, Z.Y.; Fang, J.H.; He, S.; Zhuang, S.M. Vascular Mimicry Formation Is Promoted by Paracrine TGF-β and SDF1 of Cancer-Associated Fibroblasts and Inhibited by MiR-101 in Hepatocellular Carcinoma. Cancer Lett. 2016, 383, 18–27. [Google Scholar] [CrossRef]
- Song, H.; Ci, H.; Xu, J.; Xu, Z.; Zhang, Y.; Wang, Y.; Wu, S.; Tao, Y. Vasculogenic Mimicry and Expression of Slug and Vimentin Correlate with Metastasis and Prognosis in Non-Small Cell Lung Cancer. Int. J. Clin. Exp. Pathol. 2018, 11, 2749. [Google Scholar]
- Cannell, I.G.; Sawicka, K.; Pearsall, I.; Wild, S.A.; Deighton, L.; Pearsall, S.M.; Lerda, G.; Joud, F.; Khan, S.; Bruna, A.; et al. FOXC2 Promotes Vasculogenic Mimicry and Resistance to Anti-Angiogenic Therapy. Cell Rep. 2023, 42, 112791. [Google Scholar] [CrossRef] [PubMed]
- Kirschmann, D.A.; Seftor, E.A.; Hardy, K.M.; Seftor, R.E.B.; Hendrix, M.J.C. Molecular Pathways: Vasculogenic Mimicry in Tumor Cells: Diagnostic and Therapeutic Implications. Clin. Cancer Res. 2012, 18, 2726–2732. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; Herreros, A.G. De The Transcription Factor Snail Is a Repressor of E-Cadherin Gene Expression in Epithelial Tumour Cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Abuwatfa, W.H.; Pitt, W.G.; Husseini, G.A. Scaffold-Based 3D Cell Culture Models in Cancer Research. J. Biomed. Sci. 2024, 31, 7. [Google Scholar] [CrossRef]
- Cho, Y.; Yu, S.J.; Kim, J.; Ko, U.H.; Park, E.Y.; Choung, J.S.; Choi, G.; Kim, D.; Lee, E.; Im, S.G.; et al. Remodeling of Adhesion Network within Cancer Spheroids via Cell-Polymer Interaction. ACS Biomater. Sci. Eng. 2020, 6, 5632–5644. [Google Scholar] [CrossRef]
- Wang, Y.; Mirza, S.; Wu, S.; Zeng, J.; Shi, W.; Band, H.; Band, V.; Duan, B. 3D Hydrogel Breast Cancer Models for Studying the Effects of Hypoxia on Epithelial to Mesenchymal Transition. Oncotarget 2018, 9, 32191–32203. [Google Scholar] [CrossRef]
- Pal, M.; Chen, H.; Lee, B.H.; Lee, J.Y.H.; Yip, Y.S.; Tan, N.S.; Tan, L.P. Epithelial-Mesenchymal Transition of Cancer Cells Using Bioengineered Hybrid Scaffold Composed of Hydrogel/3D-Fibrous Framework. Sci. Rep. 2019, 9, 8997. [Google Scholar] [CrossRef]
- Szostakowska-Rodzoś, M.; Chmielarczyk, M.; Zacharska, W.; Fabisiewicz, A.; Kurzyk, A.; Myśliwy, I.; Kozaryna, Z.; Postek, E.; Grzybowska, E.A. Plasticity of Expression of Stem Cell and EMT Markers in Breast Cancer Cells in 2D and 3D Culture Depend on the Spatial Parameters of Cell Growth; Mathematical Modeling of Mechanical Stress in Cell Culture in Relation to ECM Stiffness. Bioengineering 2025, 12, 147. [Google Scholar] [CrossRef]
- Shuai, Q.; Xu, X.; Liang, Y.; Halbiyat, Z.; Lu, X.; Hu, Z.; Peng, Z.; An, J.; Feng, Z.; Huang, T.; et al. Engineered in Vivo and in Vitro Tumor Model Recapitulates Vasculogenic Mimicry Signatures in Melanoma. Bioeng. Transl. Med. 2024, 9, e10648. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J.C. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Trimm, E.; Red-Horse, K. Vascular Endothelial Cell Development and Diversity. Nat. Rev. Cardiol. 2023, 20, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Tirziu, D.; Simons, M. Endothelium as Master Regulator of Organ Development and Growth. Vascul. Pharmacol. 2009, 50, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Patel-Hett, S.; D’Amore, P.A. Signal Transduction in Vasculogenesis and Developmental Angiogenesis. Int. J. Dev. Biol. 2011, 55, 353. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Puranik, A.S.; Gauvin, R.; Edalat, F.; Carrillo-Conde, B.; Peppas, N.A.; Khademhosseini, A. Building Vascular Networks. Sci. Transl. Med. 2012, 4, 160ps23. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Bhat, S.M.; Badiger, V.A.; Vasishta, S.; Chakraborty, J.; Prasad, S.; Ghosh, S.; Joshi, M.B. 3D Tumor Angiogenesis Models: Recent Advances and Challenges. J. Cancer Res. Clin. Oncol. 2021, 147, 3477–3494. [Google Scholar] [CrossRef]
- Rhodes, J.M.; Simons, M. The Extracellular Matrix and Blood Vessel Formation: Not Just a Scaffold: Angiogenesis Review Series. J. Cell. Mol. Med. 2007, 11, 176–205. [Google Scholar] [CrossRef]
- Vailhé, B.; Vittet, D.; Feige, J.J. In Vitro Models of Vasculogenesis and Angiogenesis. Lab. Investig. 2001, 81, 439–452. [Google Scholar] [CrossRef]
- Kopp, H.G.; Ramos, C.A.; Rafii, S. Contribution of Endothelial Progenitors and Proangiogenic Hematopoietic Cells to Vascularization of Tumor and Ischemic Tissue. Curr. Opin. Hematol. 2006, 13, 175–181. [Google Scholar] [CrossRef]
- Chang, E.I.; Chang, E.I.; Thangarajah, H.; Hamou, C.; Gurtner, G.C. Hypoxia, Hormones, and Endothelial Progenitor Cells in Hemangioma. Lymphat. Res. Biol. 2007, 5, 237–244. [Google Scholar] [CrossRef]
- Spring, H.; Schüler, T.; Arnold, B.; Hämmerling, G.J.; Ganss, R. Chemokines Direct Endothelial Progenitors into Tumor Neovessels. Proc. Natl. Acad. Sci. USA 2005, 102, 18111–18116. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, E.A.; Finley, S.D.; Popel, A.S.; Gabhann, F. Mac A Systems Biology View of Blood Vessel Growth and Remodelling. J. Cell. Mol. Med. 2014, 18, 1491–1508. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Rundhaug, J.E. Matrix Metalloproteinases and Angiogenesis Angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The Role of Pericytes in Blood-Vessel Formation and Maintenance. Neuro. Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef]
- Betsholtz, C.; Lindblom, P.; Gerhardt, H. Role of Pericytes in Vascular Morphogenesis. Mech. Angiogenes. 2005, 20, 115–125. [Google Scholar]
- Folkman, J. Angiogenesis. Annu. Rev. Med. 2006, 57, 1–18. [Google Scholar] [CrossRef]
- Zhao, X.I.N.; Liu, H.Q.I.U.; Li, J.I.; Liu, X.L. Endothelial Progenitor Cells Promote Tumor Growth and Progression by Enhancing New Vessel Formation. Oncology 2016, 12, 793–799. [Google Scholar] [CrossRef]
- Prakash, J.; Shaked, Y. The Interplay between Extracellular Matrix Remodeling and Cancer Therapeutics. Cancer Discov. 2024, 14, 1375–1388. [Google Scholar] [CrossRef]
- Yuan, Z.; Li, Y.; Zhang, S.; Wang, X.; Dou, H.; Yu, X.; Zhang, Z.; Yang, S.; Xiao, M. Extracellular Matrix Remodeling in Tumor Progression and Immune Escape: From Mechanisms to Treatments. Mol. Cancer 2023, 22, 48. [Google Scholar] [CrossRef]
- Furler, R.L.; Nixon, D.F.; Brantner, C.A.; Popratiloff, A.; Uittenbogaart, C.H. TGF-β Sustains Tumor Progression through Biochemical and Mechanical Signal Transduction. Cancers 2018, 10, 199. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Chao, Z.; Wang, Z.; Hao, X.; Xi, Z.; Ma, S.; Guo, X.; Zhang, J.; Zhou, Q.; Qu, G.; et al. Biomechanics in the Tumor Microenvironment: From Biological Functions to Potential Clinical Applications. Exp. Hematol. Oncol. 2025, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Dickerson, E.B.; Auerbach, R. The Sponge/Matrigel Angiogenesis Assay. Angiogenesis 2002, 5, 75–80. [Google Scholar] [CrossRef]
- Mullen, P. The Use of Matrigel to Facilitate the Establishment of Human Cancer Cell Lines as Xenografts. Cancer Cell Cult. Methods Protoc. 2004, 88, 287–292. [Google Scholar]
- Simons, M.; Alitalo, K.; Annex, B.H.; Augustin, H.G.; Beam, C.; Berk, B.C.; Byzova, T.; Carmeliet, P.; Chilian, W.; Cooke, J.P.; et al. State-of-the-Art Methods for Evaluation of Angiogenesis and Tissue Vascularization: A Scientific Statement from The American Heart Association. Circ. Res. 2015, 116, e99–e132. [Google Scholar] [CrossRef]
- Nakatsu, M.N.; Sainson, R.C.A.; Aoto, J.N.; Taylor, K.L.; Aitkenhead, M.; Pérez-del-Pulgar, S.; Carpenter, P.M.; Hughes, C.C.W. Angiogenic Sprouting and Capillary Lumen Formation Modeled by Human Umbilical Vein Endothelial Cells (HUVEC) in Fibrin Gels: The Role of Fibroblasts and Angiopoietin-1. Microvasc. Res. 2003, 66, 102–112. [Google Scholar] [CrossRef]
- Sobrino, A.; Phan, D.T.T.; Datta, R.; Wang, X.; Hachey, S.J.; Romero-López, M.; Gratton, E.; Lee, A.P.; George, S.C.; Hughes, C.C.W. 3D Microtumors In Vitro Supported by Perfused Vascular Networks. Sci. Rep. 2016, 6, 31589. [Google Scholar] [CrossRef]
- Chen, H.; Cheng, Y.; Wang, X.; Wang, J.; Shi, X.; Li, X.; Tan, W.; Tan, Z. 3D Printed in Vitro Tumor Tissue Model of Colorectal Cancer. Theranostics 2020, 10, 12127. [Google Scholar] [CrossRef]
- Kozlowski, M.T.; Crook, C.J.; Ku, H.T. Towards Organoid Culture without Matrigel. Commun. Biol. 2021, 4, 1387. [Google Scholar] [CrossRef]
- Hernandez-Fernaud, J.R.; Ruengeler, E.; Casazza, A.; Neilson, L.J.; Pulleine, E.; Santi, A.; Ismail, S.; Lilla, S.; Dhayade, S.; MacPherson, I.R.; et al. Secreted CLIC3 Drives Cancer Progression through Its Glutathione-Dependent Oxidoreductase Activity. Nat. Commun. 2017, 8, 14206. [Google Scholar] [CrossRef]
- Roudsari, L.C.; Jeffs, S.E.; Witt, A.S.; Gill, B.J.; West, J.L. A 3D Poly(Ethylene Glycol)-Based Tumor Angiogenesis Model to Study the Influence of Vascular Cells on Lung Tumor Cell Behavior. Sci. Rep. 2016, 6, 32726. [Google Scholar] [CrossRef] [PubMed]
- Habanjar, O.; Diab-assaf, M.; Caldefie-chezet, F.; Delort, L. 3D Cell Culture Systems: Tumor Application, Advantages, and Disadvantages. Int. J. Mol. Sci. 2021, 22, 12200. [Google Scholar] [CrossRef] [PubMed]
- Mandrycky, C.J.; Howard, C.C.; Rayner, S.G.; Jung, Y. Organ-on-a-Chip Systems for Vascular Biology. J. Mol. Cell. Cardiol. 2021, 159, 1–13. [Google Scholar] [CrossRef]
- Srivastava, K.S.; Foo, W.G.; Aggarwal, N.; Chang, M.W. Organ-on-Chip Technology: Opportunities and Challenges. Biotechnol. Notes 2024, 5, 8–12. [Google Scholar] [CrossRef]
- Chae, S.; Ha, D.; Lee, H. 3D Bioprinting Strategy for Engineering Vascularized Tissue Models. J. Bioprinting 2023, 9, 748. [Google Scholar] [CrossRef]
- Gnatowski, P.; Piłat, E.; Kucińska-Lipka, J.; Saeb, M.R.; Hamblin, M.R.; Mozafari, M. Recent Advances in 3D Bioprinted Tumor Models for Personalized Medicine. Transl. Oncol. 2023, 37, 101750. [Google Scholar] [CrossRef]
- Schnellmann, R.; Ntekoumes, D.; Choudhury, M.I.; Sun, S. Stiffening Matrix Induces Age-Mediated Microvascular Phenotype Through Increased Cell Contractility and Destabilization of Adherens Junctions. Adv. Sci. 2022, 9, 2201483. [Google Scholar] [CrossRef]
- Nguyen, E.H.; Zanotelli, M.R.; Schwartz, M.P.; Murphy, W.L. Differential Effects of Cell Adhesion, Modulus and VEGFR-2 Inhibition on Capillary Network Formation in Synthetic Hydrogel Arrays. Biomaterials 2014, 35, 2149–2161. [Google Scholar] [CrossRef]
- Truong, D.; Fiorelli, R.; Barrientos, E.S.; Melendez, E.L.; Sanai, N.; Mehta, S.; Nikkhah, M. A Three-Dimensional (3D) Organotypic Microfluidic Model for Glioma Stem Cells—Vascular Interactions. Biomaterials 2019, 198, 63–77. [Google Scholar] [CrossRef]
- Shengjie, L.; Xiong, Z.; Wang, X.; Yan, Y.; Liu, H.; Zhang, R. Direct Fabrication of a Hybrid Cell/Hydrogel Construct by a Double-Nozzle Assembling Technology. J. Bioact. Compat. Polym. 2009, 24, 249–265. [Google Scholar] [CrossRef]
- Cheng, S.; Li, Y.; Yu, C.; Deng, Z.; Huang, J.; Zhang, Z. 3D Bioprinted Tumor-Vessel-Bone Co-Culture Scaffold for Breast Cancer Bone Metastasis Modeling and Drug Testing. Chem. Eng. J. 2023, 476, 146685. [Google Scholar] [CrossRef]
- Cui, H.; Esworthy, T.; Zhou, X.; Hann, S.Y.; Glazer, R.I.; Li, R.; Zhang, L.G. Engineering a Novel 3D Printed Vascularized Tissue Model for Investigating Breast Cancer Metastasis to Bone. Adv. Healthc. Mater. 2020, 9, 1900924. [Google Scholar] [CrossRef]

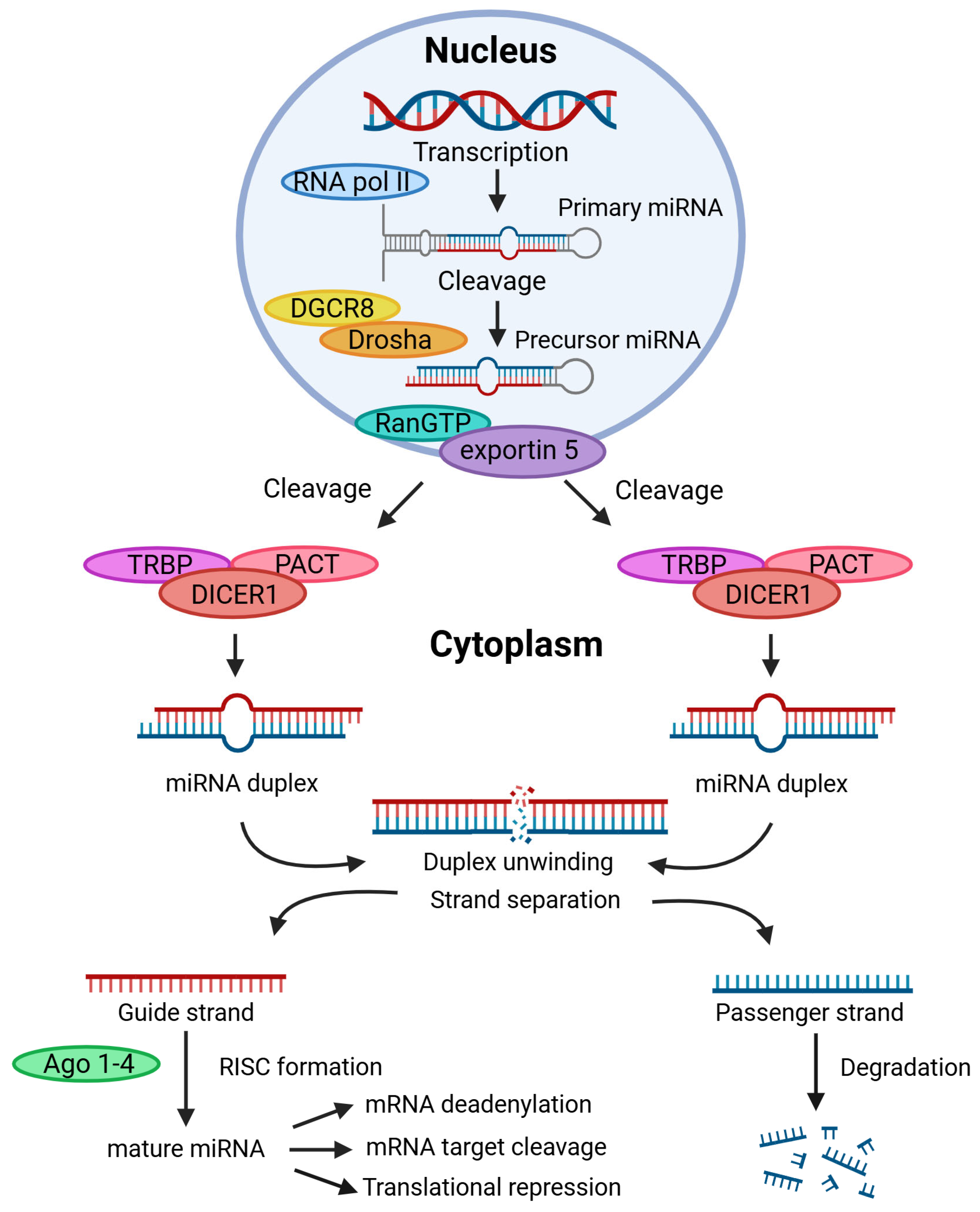
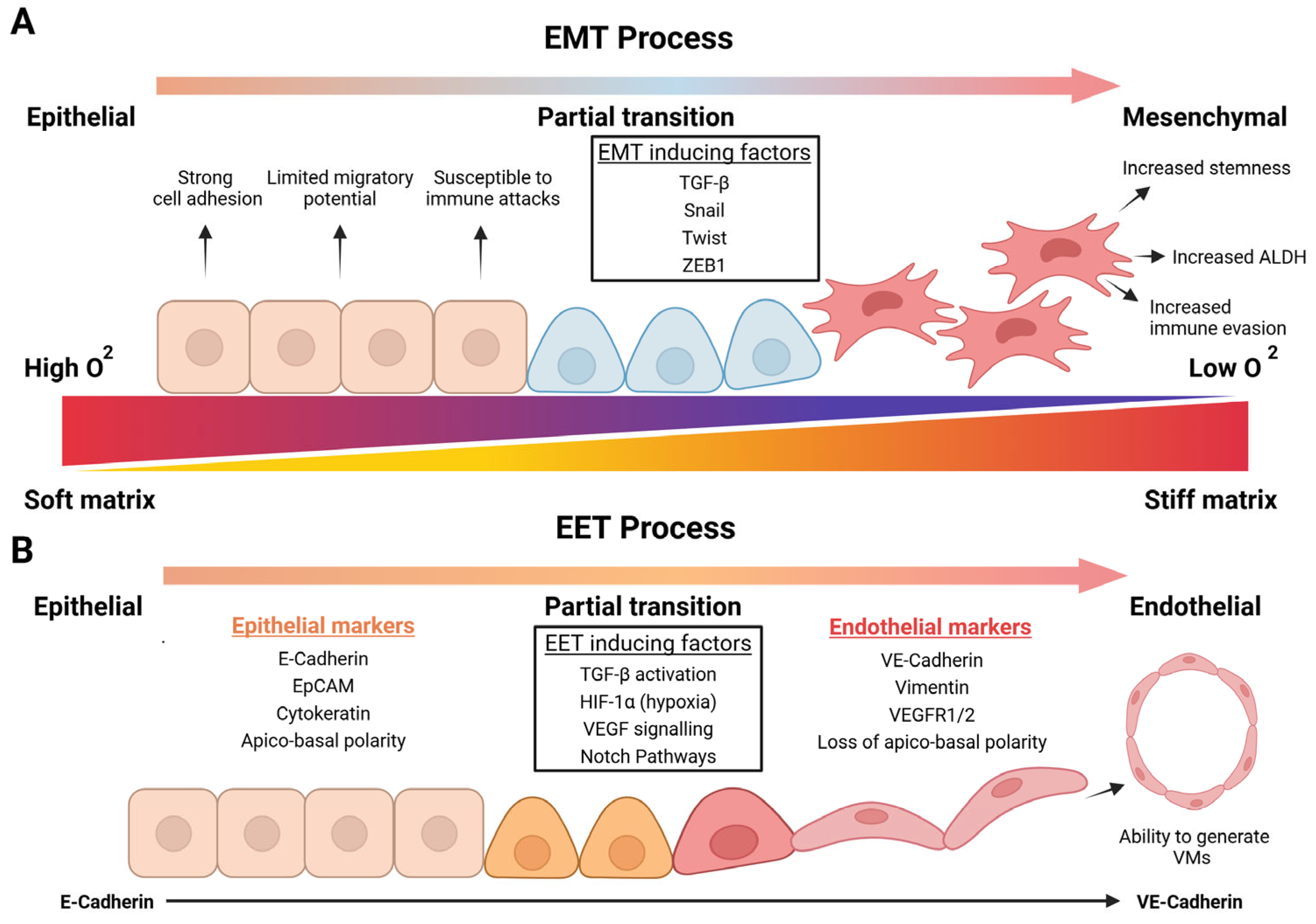
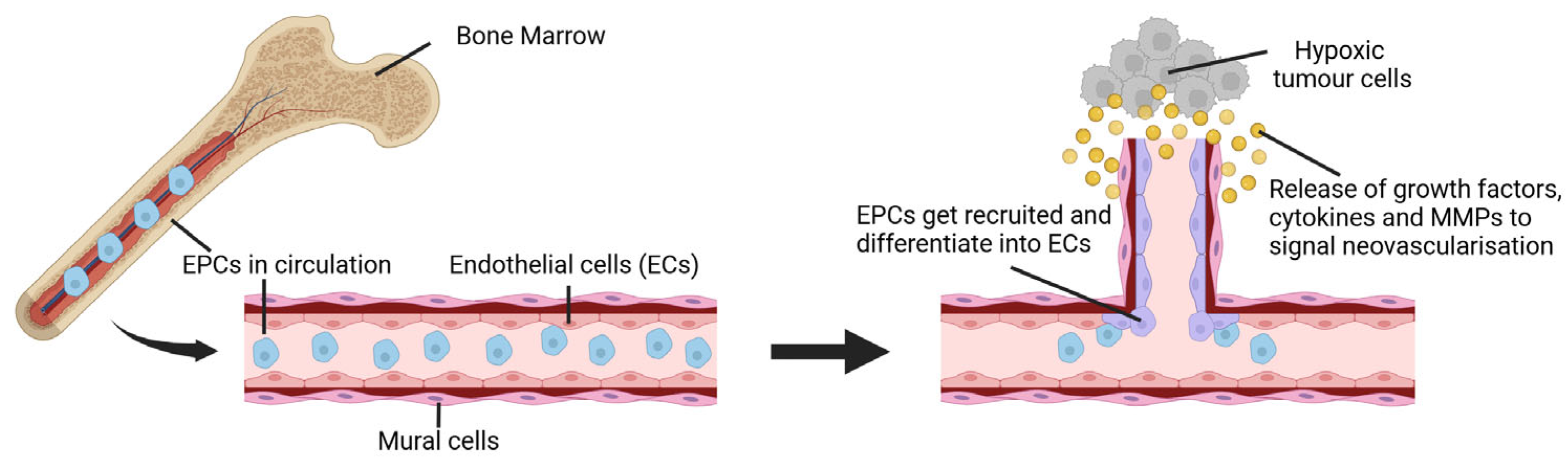

| Cancer Type | Expression Profile | Diagnostic Value | Prognostic Value | Diagnostic and Prognostic Value |
|---|---|---|---|---|
| Breast | Downregulated | let-7b-5p, let-7c-5p | miR-409-3p | |
| Upregulated | miR-195, miR-376c, miR-409-3p, miR-148b, miR-299-5p, miR-145, miR-191, miR-382, miR-215, miR-133a, miR-133b, miR-92a, miR-192, miR-1, miR-411, miR-195, miR-202 | miR-122 miR-141 | miR-21, miR-34a, miR-210, miR-10b, miR-375, miR-125b, miR-801, miR-155 | |
| Pancreatic | Downregulated | miR-100-5p, miR-375 | miR-718 | |
| Upregulated | miR-378 *, miR-409-3p, miR-1290, miR-26a, miR-18a | miR-146b-3p, miR-200a, miR-200c, miR-210, miR-221, miR-21, miR-194 | miR-141, miR-375 | |
| Prostate | Downregulated | miR-16, miR-199a, miR-21 | ||
| Upregulated | miR-378 *, miR-409-3p, miR-1290, miR-26a, miR-18a | miR-146b-3p, miR-210, miR-21, miR-221, miR-19, miR-200a, miR-200c | miR-141, miR-375 | |
| Non-small-cell lung carcinoma | Downregulated | let-7b-5p, let-7c-5p | ||
| Upregulated | miR-20a-5p, miR-141-3p, miR-145-5p, miR-155-5p, miR-223-3p | miR-320b, miR-23b-3p, miR-10b-3p, miR-195-5p | miR-21-5p |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larrea Murillo, L.; Green, M.; Mahon, N.; Saiani, A.; Tsigkou, O. Modelling Cancer Pathophysiology: Mechanisms and Changes in the Extracellular Matrix During Cancer Initiation and Early Tumour Growth. Cancers 2025, 17, 1675. https://doi.org/10.3390/cancers17101675
Larrea Murillo L, Green M, Mahon N, Saiani A, Tsigkou O. Modelling Cancer Pathophysiology: Mechanisms and Changes in the Extracellular Matrix During Cancer Initiation and Early Tumour Growth. Cancers. 2025; 17(10):1675. https://doi.org/10.3390/cancers17101675
Chicago/Turabian StyleLarrea Murillo, Luis, Megan Green, Niall Mahon, Alberto Saiani, and Olga Tsigkou. 2025. "Modelling Cancer Pathophysiology: Mechanisms and Changes in the Extracellular Matrix During Cancer Initiation and Early Tumour Growth" Cancers 17, no. 10: 1675. https://doi.org/10.3390/cancers17101675
APA StyleLarrea Murillo, L., Green, M., Mahon, N., Saiani, A., & Tsigkou, O. (2025). Modelling Cancer Pathophysiology: Mechanisms and Changes in the Extracellular Matrix During Cancer Initiation and Early Tumour Growth. Cancers, 17(10), 1675. https://doi.org/10.3390/cancers17101675