
Short Tandem Repeat (STR) Somatic Mutation in Non-Melanoma Skin Cancer (NMSC): Association with Transcriptomic Profile and Potential Implications for Therapy

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Next Generation Sequencing (NGS)
2.2. Somatic Mutation Detection
2.3. Transcriptome Wide Gene Expression and Statistical Analysis
2.4. Artificial Intelligence (AI) Modeling for Prediction
3. Results
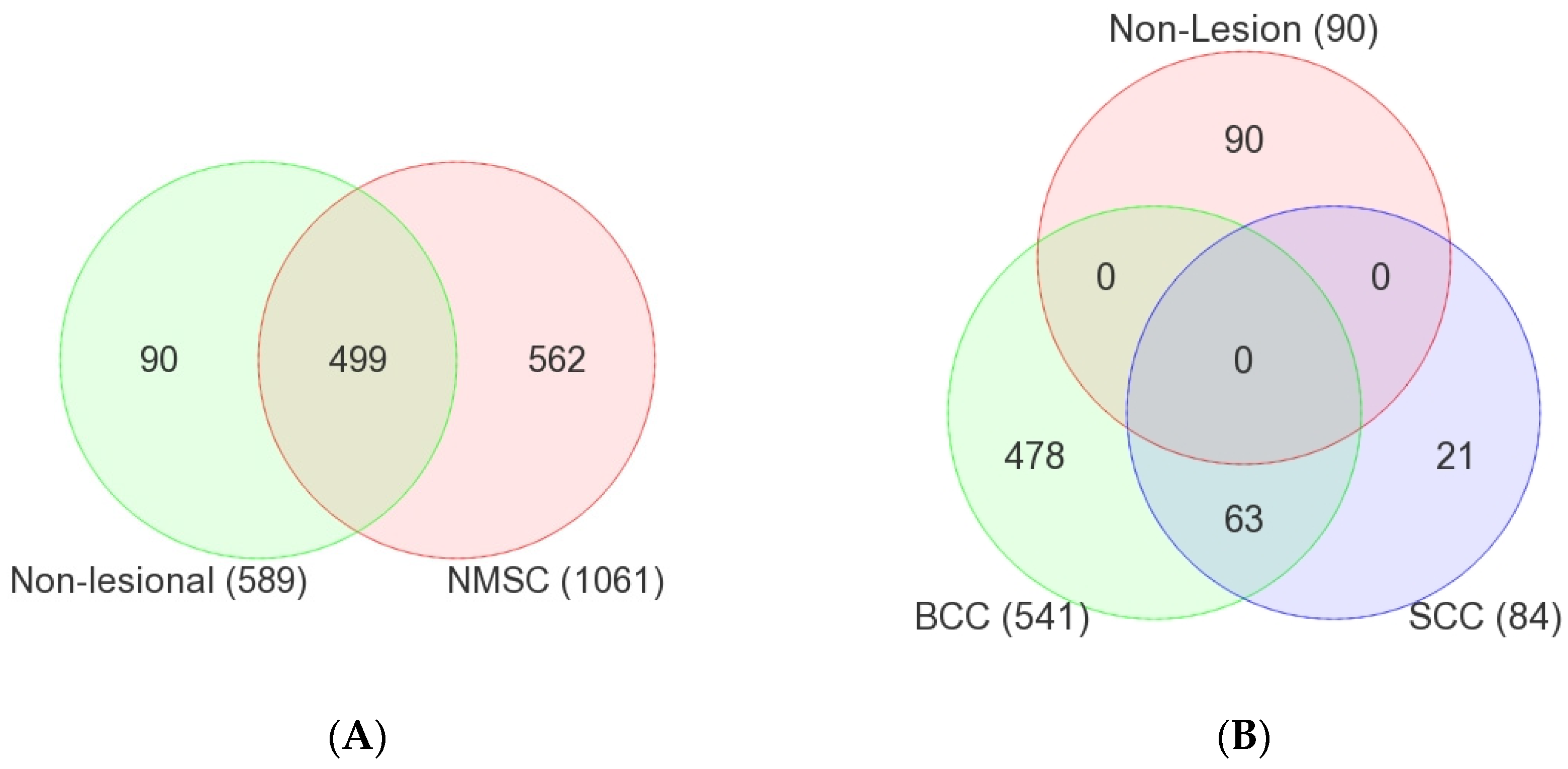
3.1. Common Non-Cancer-Specific STR Somatic Mutations
3.2. NMSC-Associated STR Somatic Mutations
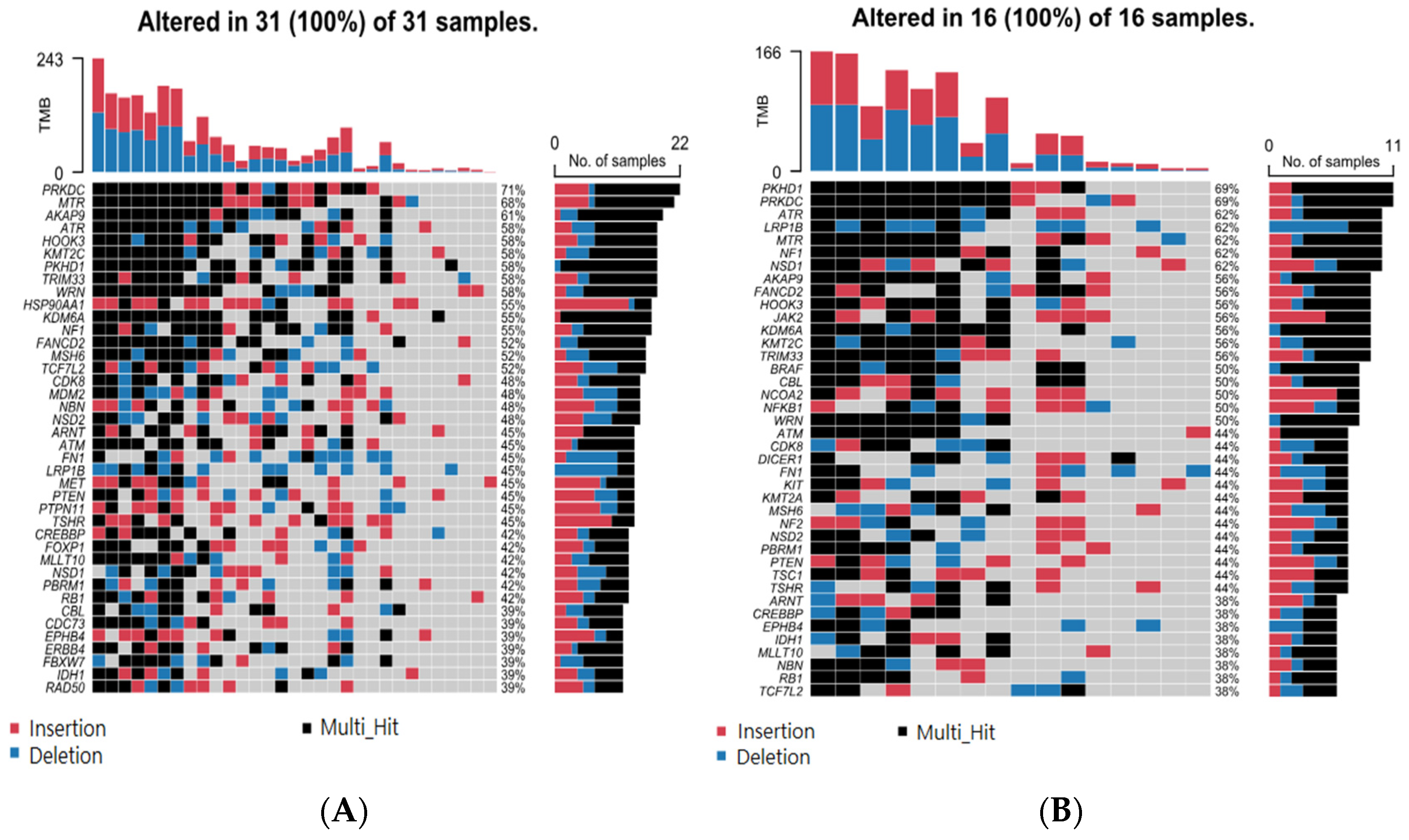
3.3. Association of BCC-Associated STR Somatic Mutations with MSI-Related Gene Mutation
3.4. Association of BCC-Associated STR Somatic Mutations and Differential Gene Expression in BCC Tissue
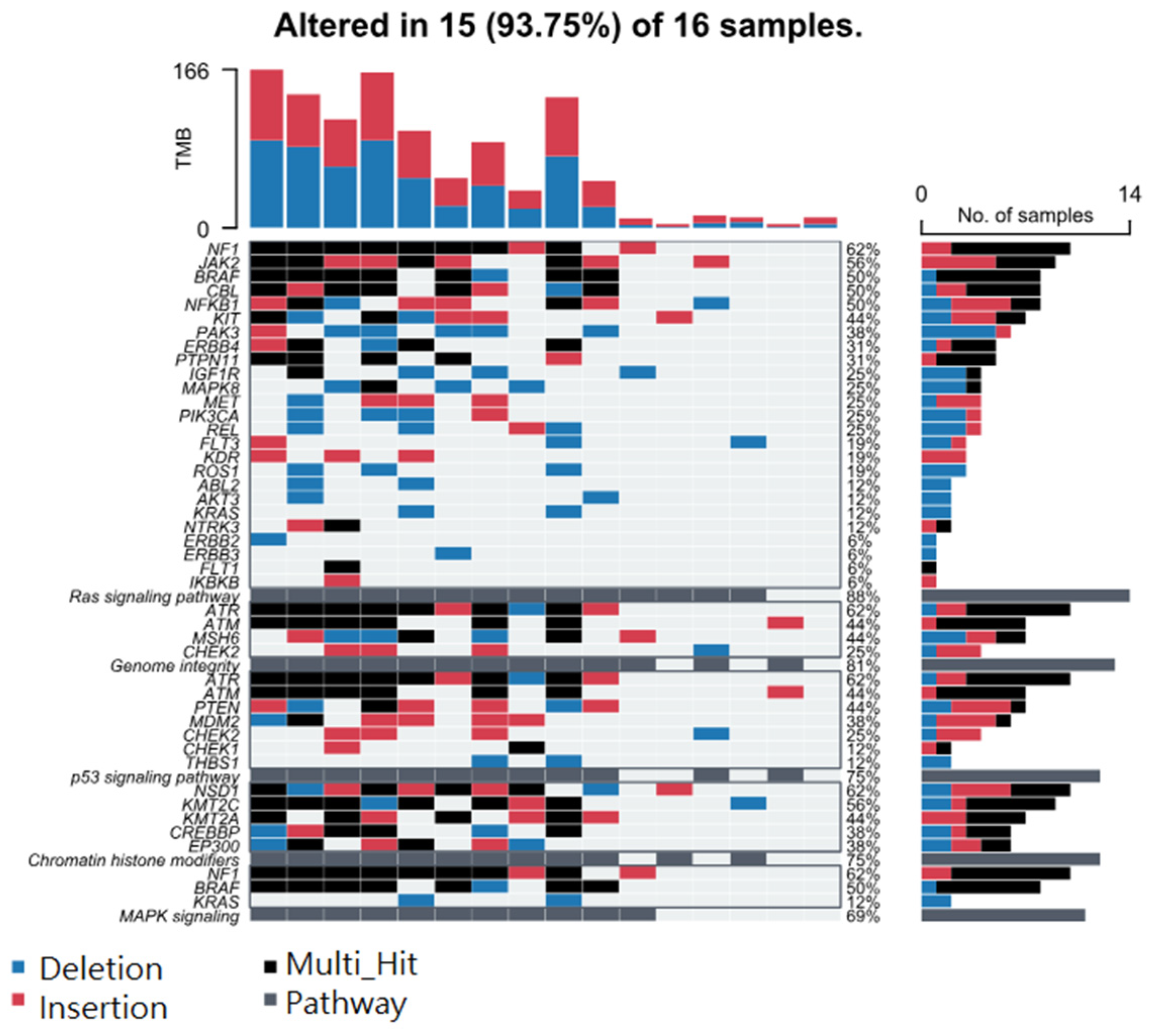
3.5. Association of BCC-Associated STR Somatic Mutations and Known Cancer-Related Pathways
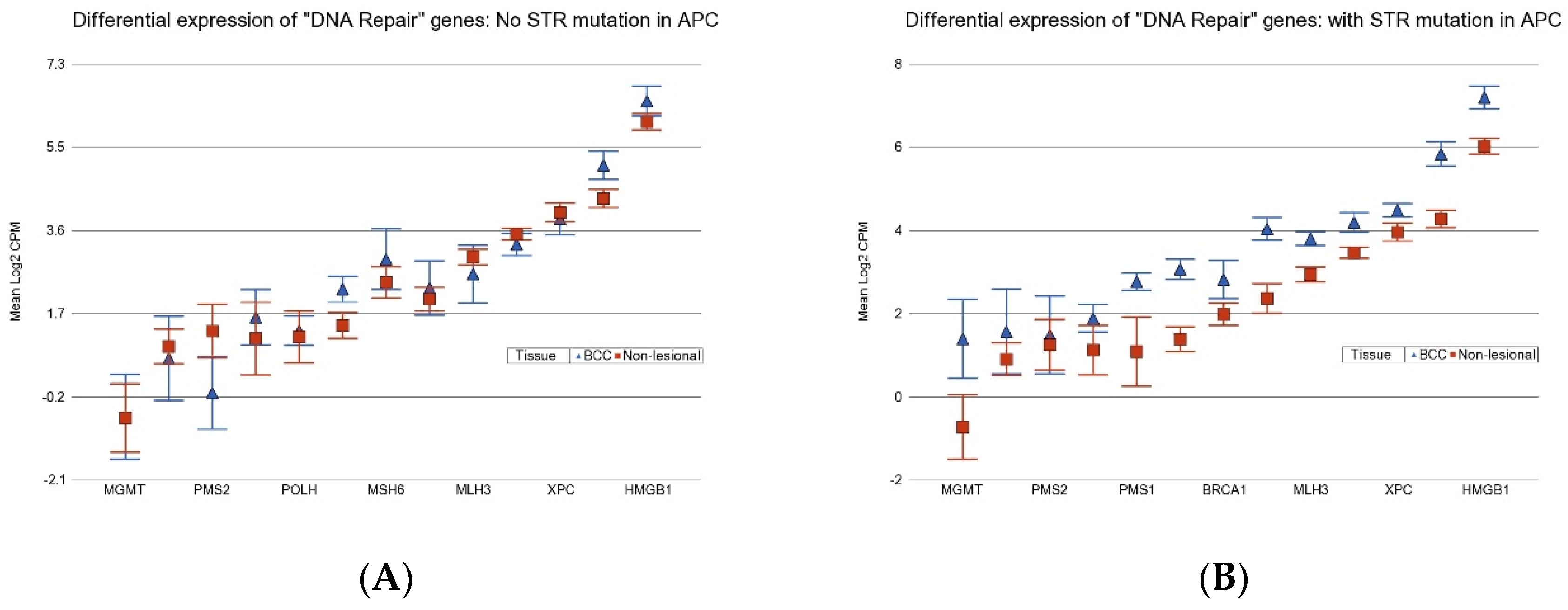
3.6. Association of BCC-Associated STR Somatic Mutations and Gene Pathways Known to Be Dysregulated in BCC
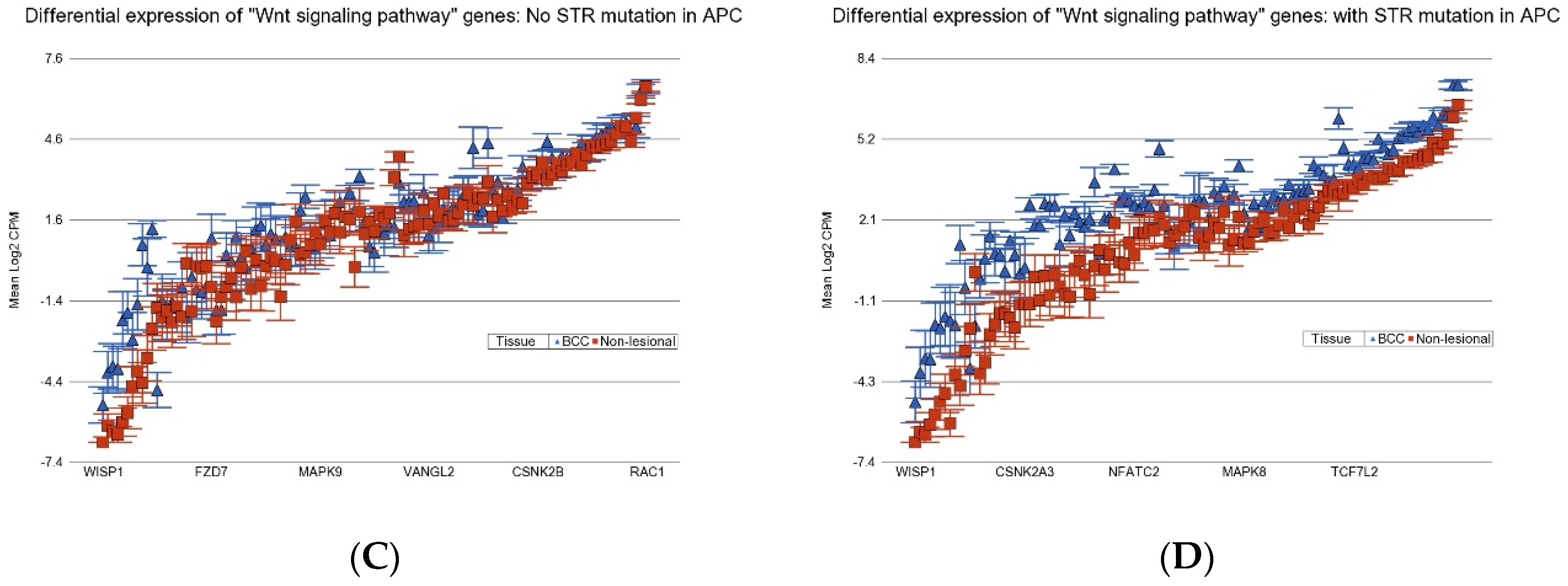
3.7. Potential Utility of These STR Somatic Mutations in BRAF and APC in Therapeutic Consideration
3.7.1. Hedgehog Inhibitors
3.7.2. Gamma-Secretase Inhibitors
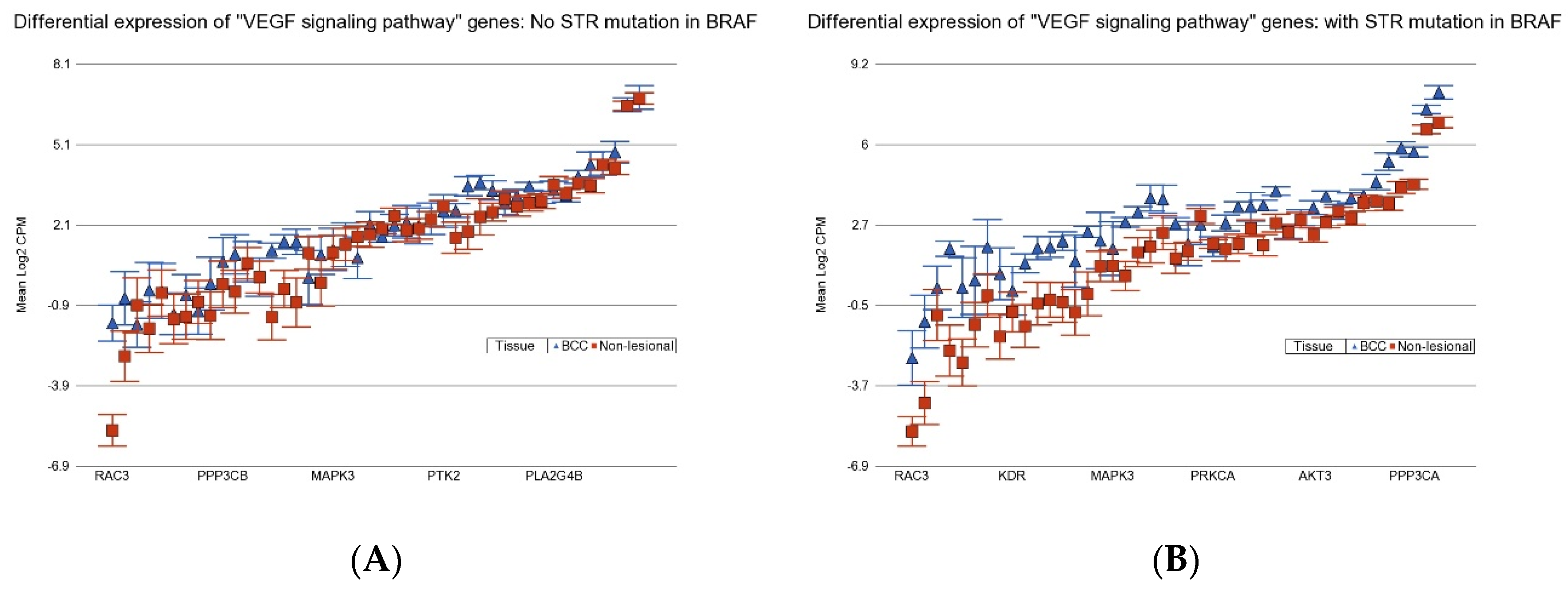
3.7.3. Anti-Vascular Endothelial Growth Factor (VEGF) Therapy
3.7.4. Proteasome Inhibitors
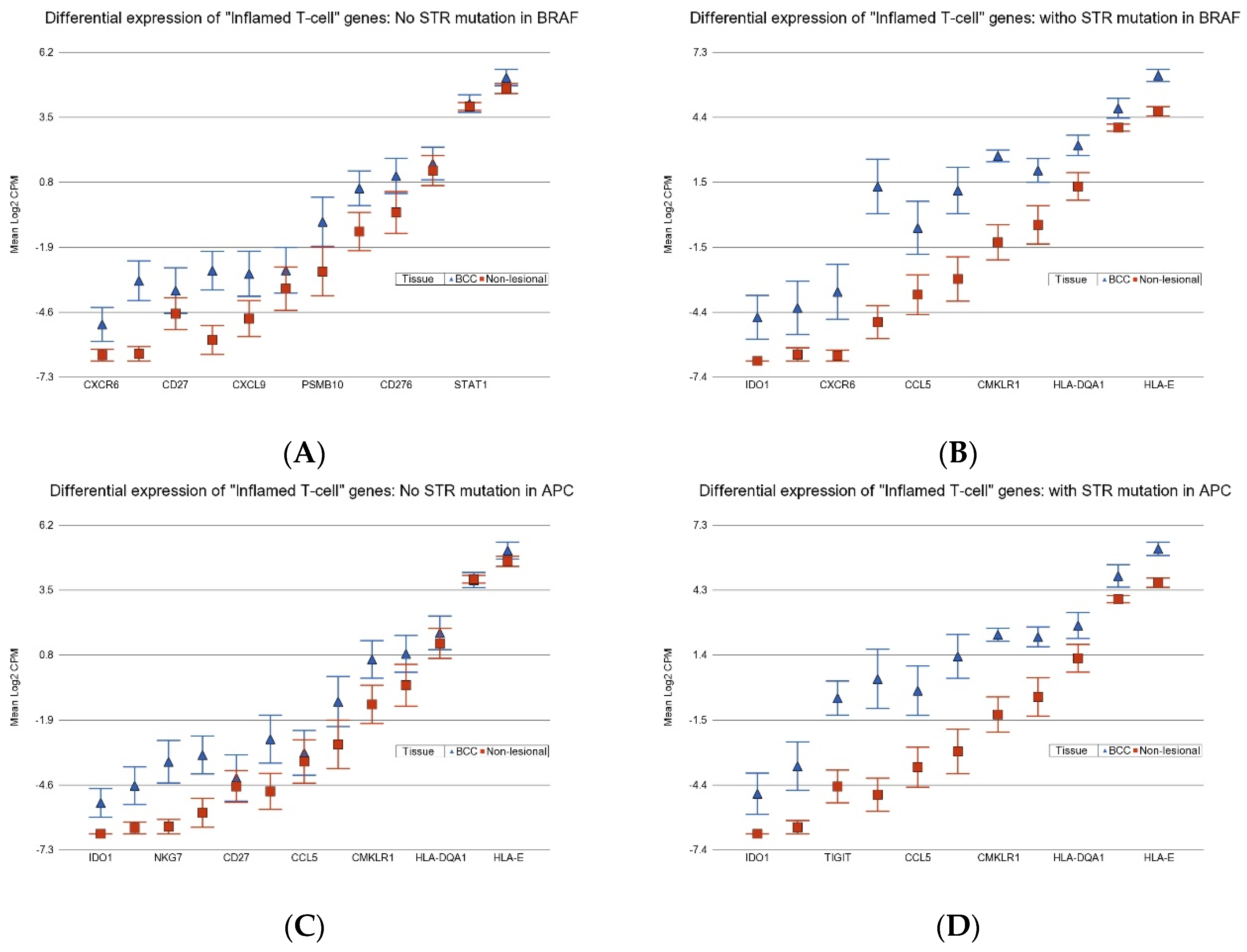
3.7.5. Immune Checkpoint Inhibitors (ICIs)
3.8. Predictive Modeling Using AI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NMSC | Non-melanoma skin cancer |
| As | Arsenic |
| SNV | Single-nucleotide variation |
| DEL | Small deletion |
| SSR | Simple sequence repeat |
| STR | Short tandem repeat |
| INDEL | Insertion or deletion |
| CNV | Copy number variation |
| DNA | Deoxyribonucleic acid |
| MSI | Microsatellite instability |
| MMR | Mismatch repair |
| CRC | Colorectal cancer |
| LOH | Loss of heterozygosity |
| SCC | Squamous cell carcinoma |
| RNA | Ribonucleic acid |
| NGS | Next-generation sequencing |
| EDTA | Ethylenediaminetetraacetic acid |
| Mb | Megabase |
| TAS | Targeted amplicon sequencing |
| AI | Artificial intelligence |
| DL | Deep learning |
| GBM | Gradient boosting machine |
| DRF | Distributed random forest |
| GLM | Generalized linear model |
| XGBoost | eXtreme gradient boosting |
| SE | Stacked ensemble |
| autoML | Automated machine learning |
| VAF | Variant allele frequency |
| MNV | Multi-nucleotide variant |
| BCC | Basal cell carcinoma |
| UACR | Urine albumin–creatinine ratio |
| CPM | Count per million |
| BER | Base excision repair |
| TLS | Translesion synthesis |
| NHEJ | Non-homologous end joining |
| Hh | Hedgehog |
| AUC | Area under curve |
| ROC | Receiver operating characteristic |
| FC | Fold change |
| VEGF | Vascular endothelial growth factor |
| ANOVA | Analysis of variance |
| GCI | Gamma-secretase inhibitor |
| ICI | Immune checkpoint inhibitor |
References
- Jasmine, F.; Argos, M.; Khamkevych, Y.; Islam, T.; Rakibuz-Zaman, M.; Shahriar, M.; Shea, C.R.; Ahsan, H.; Kibriya, M.G. Molecular Profiling and the Interaction of Somatic Mutations with Transcriptomic Profiles in Non-Melanoma Skin Cancer (NMSC) in a Population Exposed to Arsenic. Cells 2024, 13, 1056. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jasmine, F.; Almazan, A.; Khamkevych, Y.; Argos, M.; Shahriar, M.; Islam, T.; Shea, C.R.; Ahsan, H.; Kibriya, M.G. Gene-Environment Interaction: Small Deletions (DELs) and Transcriptomic Profiles in Non-Melanoma Skin Cancer (NMSC) and Potential Implications for Therapy. Cells 2025, 14, 95. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Gemayel, R.; Vinces, M.D.; Legendre, M.; Verstrepen, K.J. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 2010, 44, 445–477. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.X.; Helgason, A.; Masson, G.; Ebenesersdóttir, S.S.; Li, H.; Mallick, S.; Gnerre, S.; Patterson, N.; Kong, A.; Reich, D.; et al. A direct characterization of human mutation based on microsatellites. Nat. Genet. 2012, 44, 1161–1165. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fujimoto, A.; Fujita, M.; Hasegawa, T.; Wong, J.H.; Maejima, K.; Oku-Sasaki, A.; Nakano, K.; Shiraishi, Y.; Miyano, S.; Yamamoto, G.; et al. Comprehensive analysis of indels in whole-genome microsatellite regions and microsatellite instability across 21 cancer types. Genome Res. 2020, 30, 334–346. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Casares, S.; Ionov, Y.; Ge, H.Y.; Stanbridge, E.; Perucho, M. The microsatellite mutator phenotype of colon cancer cells is often recessive. Oncogene 1995, 11, 2303–2310. [Google Scholar] [PubMed]
- Gambichler, T.; Ganjuur, N.; Tannapfel, A.; Vogt, M.; Scholl, L.; Abu Rached, N.; Bruckmüller, S.; Skrygan, M.; Becker, J.C.; Käfferlein, H.U.; et al. Mismatch Repair Protein Expression and Microsatellite Instability in Cutaneous Squamous Cell Carcinoma. Curr. Oncol. 2021, 28, 3316–3322. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McIver, L.J.; Fonville, N.C.; Karunasena, E.; Garner, H.R. Microsatellite genotyping reveals a signature in breast cancer exomes. Breast Cancer Res. Treat. 2014, 145, 791–798. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sardi, I.; Piazzini, M.; Palleschi, G.; Pinzi, C.; Taddei, I.; Arrigucci, S.; Guazzelli, R.; Fabbri, P.; Moretti, S. Molecular detection of microsatellite instability in basal cell carcinoma. Oncol. Rep. 2000, 7, 1119–1141. [Google Scholar] [CrossRef] [PubMed]
- Shibata, D.; Peinado, M.A.; Lonov, Y.; Malkhosyan, S.; Perucho, M. Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat. Genet. 1994, 6, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Boyer, J.C.; Thomas, D.C.; Nguyen, D.C.; Risinger, J.I.; Boyd, J.; Ionov, Y.; Perucho, M.; Kunkel, T. Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J. Biol. Chem. 1994, 269, 14367–14370. [Google Scholar] [CrossRef] [PubMed]
- Verbiest, M.A.; Lundström, O.; Xia, F.; Baudis, M.; Sonay, T.B.; Anisimova, M. Short tandem repeat mutations regulate gene expression in colorectal cancer. Sci. Rep. 2024, 14, 3331. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wooster, R.; Cleton-Jansen, A.-M.; Collins, N.; Mangion, J.; Cornelis, R.; Cooper, C.; Gusterson, B.; Ponder, B.; von Deimling, A.; Wiestler, O.; et al. Instability of short tandem repeats (microsatellites) in human cancers. Nat. Genet. 1994, 6, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Reuschenbach, M.; Sommerer, C.; Hartschuh, W.; Zeier, M.; von Knebel Doeberitz, M.; Kloor, M. Absence of mismatch repair deficiency-related microsatellite instability in non-melanoma skin cancer. J. Investig. Dermatol. 2012, 132, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Quinn, A.G.; Campbell, C.; Healy, E.; Rees, J.L. Chromosome 9 allele loss occurs in both basal and squamous cell carcinomas of the skin. J. Investig. Dermatol. 1994, 102, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Laus, A.C.; Gomes, I.N.F.; da Silva, A.L.V.; da Silva, L.S.; Milan, M.B.; Aparecida Teixeira, S.; Martin, A.C.B.M.; Pereira, L.D.N.B.; de Carvalho, C.E.B.; Crovador, C.S.; et al. Establishment and molecular characterization of HCB-541, a novel and aggressive human cutaneous squamous cell carcinoma cell line. Hum. Cell 2024, 37, 1170–1183. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peris, K.; Keller, G.; Chimenti, S.; Amantea, A.; Kerl, H.; Höfler, H. Microsatellite instability and loss of heterozygosity in melanoma. J. Investig. Dermatol. 1995, 105, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Saridaki, Z.; Liloglou, T.; Zafiropoulos, A.; Koumantaki, E.; Zoras, O.; Spandidos, D.A. Mutational analysis of CDKN2A genes in patients with squamous cell carcinoma of the skin. Br. J. Dermatol. 2003, 148, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Halman, A.; Lonsdale, A.; Oshlack, A. Analysis of Tandem Repeats in Short-Read Sequencing Data: From Genotyping Known Pathogenic Repeats to Discovering Novel Expansions. Curr. Protoc. 2024, 4, e70010. [Google Scholar] [CrossRef] [PubMed]
- Tanudisastro, H.A.; Deveson, I.W.; Dashnow, H.; MacArthur, D.G. Sequencing and characterizing short tandem repeats in the human genome. Nat. Rev. Genet. 2024, 25, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Argos, M.; Rahman, M.; Parvez, F.; Dignam, J.; Islam, T.; Quasem, I.; Hore, S.K.; Haider, A.T.; Hossain, Z.; Patwary, T.I.; et al. Baseline comorbidities in a skin cancer prevention trial in Bangladesh. Eur. J. Clin. Investig. 2013, 43, 579–588. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kibriya, M.G.; Jasmine, F.; Munoz, A.; Islam, T.; Ahmed, A.; Tong, L.; Rakibuz-Zaman, M.; Shahriar, M.; Kamal, M.; Shea, C.R.; et al. Interaction of Arsenic Exposure and Transcriptomic Profile in Basal Cell Carcinoma. Cancers 2022, 14, 5598. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kibriya, M.G.; Jasmine, F.; Parvez, F.; Argos, M.; Roy, S.; Paul-Brutus, R.; Islam, T.; Ahmed, A.; Rakibuz-Zaman, M.; Shinkle, J.; et al. Association between genome-wide copy number variation and arsenic-induced skin lesions: A prospective study. Environ. Health 2017, 16, 75. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Ramelyte, E.; Amann, V.C.; Dummer, R. Sonidegib for the treatment of advanced basal cell carcinoma. Expert Opin. Pharmacother. 2016, 17, 1963–1968. [Google Scholar] [CrossRef] [PubMed]
- Ramelyte, E.; Nägeli, M.C.; Hunger, R.; Merat, R.; Gaide, O.; Navarini, A.A.; Cozzio, A.; Wagner, N.B.; Maul, L.V.; Dummer, R. Swiss Recommendations for Cutaneous Basal Cell Carcinoma. Dermatology 2023, 239, 122–131. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ramelyte, E.; Restivo, G.; Imhof, L.; Nägeli, M.C.; Dummer, R. How to break resistance to hedgehog inhibitors in advanced basal cell carcinoma? Br. J. Dermatol. 2021, 184, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shih, I.-M.; Wang, T.-L. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res. 2007, 67, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Çorbacıoğlu, Ş.K.; Aksel, G. Receiver operating characteristic curve analysis in diagnostic accuracy studies: A guide to interpreting the area under the curve value. Turk. J. Emerg. Med. 2023, 23, 195–198. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Bibee, K.P.; Kulkarni, A.; Lee, S.; Ho, J.; Osmanbeyoglu, H.U.; Ferris, R.L.; Zandberg, D.P. Genomic and transcriptomic analysis of cutaneous squamous cell carcinoma arising in immunocompetent and immunosuppressed patients. Oral Oncol. 2024, 148, 106582. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kushida, Y.; Miki, H.; Ohmori, M. Loss of heterozygosity in actinic keratosis, squamous cell carcinoma and sun-exposed normal-appearing skin in Japanese: Difference between Japanese and Caucasians. Cancer Lett. 1999, 140, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Saridaki, Z.; Sourvinos, G.; Spandidos, D.A.; Koumantaki, E.; Liloglou, T.; Papadopoulos, O.; Zoras, O. High frequency of loss of heterozygosity on chromosome region 9p21-p22 but lack of p16INK4a/p19ARF mutations in greek patients with basal cell carcinoma of the skin. J. Investig. Dermatol. 2000, 115, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.J.; Palmer, J.M.; Walters, M.K.; Nancarrow, D.J.; Parsons, P.G.; Hayward, N.K. Simple tandem repeat allelic deletions confirm the preferential loss of distal chromosome 6q in melanoma. Int. J. Cancer 1994, 58, 203–206. [Google Scholar] [CrossRef]
- Quinn, A.G.; Healy, E.; Rehman, I.; Sikkink, S.; Rees, J.L. Microsatellite instability in human non-melanoma and melanoma skin cancer. J. Investig. Dermatol. 1995, 104, 309–312. [Google Scholar] [CrossRef]
- Mitra, I.; Huang, B.; Mousavi, N.; Ma, N.; Lamkin, M.; Yanicky, R.; Shleizer-Burko, S.; Lohmueller, K.E.; Gymrek, M. Patterns of de novo tandem repeat mutations and their role in autism. Nature 2021, 589, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Mishra, R.K.; Singh, L. Genome-wide analysis of microsatellite repeats in humans: Their abundance and density in specific genomic regions. Genome Biol. 2003, 4, R13. [Google Scholar] [CrossRef]
- Kelkar, Y.D.; Eckert, K.A.; Chiaromonte, F.; Makova, K.D. A matter of life or death: How microsatellites emerge in and vanish from the human genome. Genome Res. 2011, 21, 2038–2048. [Google Scholar] [CrossRef] [PubMed]
- Willems, T.; Gymrek, M.; Highnam, G.; The 1000 Genomes Project Consortium; Mittelman, D.; Erlich, Y. The landscape of human STR variation. Genome Res. 2014, 24, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Gymrek, M.; Willems, T.; Guilmatre, A.; Zeng, H.; Markus, B.; Georgiev, S.; Daly, M.J.; Price, A.L.; Pritchard, J.K.; Sharp, A.J.; et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 2016, 48, 22–29. [Google Scholar] [CrossRef]
- Fotsing, S.F.; Margoliash, J.; Wang, C.; Saini, S.; Yanicky, R.; Shleizer-Burko, S.; Goren, A.; Gymrek, M. The impact of short tandem repeat variation on gene expression. Nat. Genet. 2019, 51, 1652–1659. [Google Scholar] [CrossRef]
- Martin-Trujillo, A.; Garg, P.; Patel, N.; Jadhav, B.; Sharp, A.J. Genome-wide evaluation of the effect of short tandem repeat variation on local DNA methylation. Genome Res. 2023, 33, 184–196. [Google Scholar] [CrossRef]
- Verbiest, M.; Maksimov, M.; Jin, Y.; Anisimova, M.; Gymrek, M.; Bilgin Sonay, T. Mutation and selection processes regulating short tandem repeats give rise to genetic and phenotypic diversity across species. J. Evol. Biol. 2023, 3, 321–336. [Google Scholar] [CrossRef]
- Danaee, H.; Nelson, H.H.; Karagas, M.R.; Schned, A.R.; Ashok, T.D.S.; Hirao, T.; Perry, A.E.; Kelsey, K.T. Microsatellite instability at tetranucleotide repeats in skin and bladder cancer. Oncogene 2002, 21, 4894–4899. [Google Scholar] [CrossRef]
- Maruvka, Y.E.; Mouw, K.W.; Karlic, R.; Parasuraman, P.; Kamburov, A.; Polak, P.; Haradhvala, N.J.; Hess, J.M.; Rheinbay, E.; Brody, Y.; et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat. Biotechnol. 2017, 35, 951–959. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Correlations | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NMSC_asso_MLH1_mut | NMSC_asso_MSH2_mut | NMSC_asso_MSH6_mut | NMSC_asso_PMS2_mut | LRP1B_STR_BCC | SYNE1_STR_BCC | CSMD3_STR_BCC | FANCD2_STR_BCC | MTR_STR_BCC | PRKDC_STR_BCC | APC_STR_BCC | ATM_STR_BCC | NF1_STR_BCC | ||
| LRP1B_STR_BCC | Pearson Correlation | 0.081 | 0.200 | 0.234 | 0.426 * | |||||||||
| Sig. (2-tailed) | 0.695 | 0.327 | 0.251 | 0.030 | ||||||||||
| SYNE1_STR_BCC | Pearson Correlation | 0.299 | 0.216 | 0.300 | 0.247 | 0.617 ** | ||||||||
| Sig. (2-tailed) | 0.137 | 0.289 | 0.136 | 0.224 | 0.001 | |||||||||
| CSMD3_STR_BCC | Pearson Correlation | 0.031 | 0.234 | 0.055 | 0.066 | 0.389 * | 0.456 * | |||||||
| Sig. (2-tailed) | 0.879 | 0.251 | 0.791 | 0.747 | 0.049 | 0.019 | ||||||||
| FANCD2_STR_BCC | Pearson Correlation | 0.195 | 0.234 | 0.370 | 0.066 | 0.389 * | 0.613 ** | 0.370 | ||||||
| Sig. (2-tailed) | 0.340 | 0.251 | 0.063 | 0.747 | 0.049 | 0.001 | 0.063 | |||||||
| MTR_STR_BCC | Pearson Correlation | 0.089 | −0.158 | 0.283 | 0.101 | 0.158 | 0.220 | 0.123 | 0.123 | |||||
| Sig. (2-tailed) | 0.664 | 0.440 | 0.161 | 0.623 | 0.440 | 0.281 | 0.549 | 0.549 | ||||||
| PRKDC_STR_BCC | Pearson Correlation | 0.089 | 0.253 | 0.123 | 0.539 ** | 0.474 * | 0.378 | 0.443 * | 0.283 | 0.513 ** | ||||
| Sig. (2-tailed) | 0.664 | 0.212 | 0.549 | 0.004 | 0.014 | 0.057 | 0.023 | 0.161 | 0.007 | |||||
| APC_STR_BCC | Pearson Correlation | 0.422 * | −0.158 | 0.123 | 0.320 | 0.474 * | 0.537 ** | 0.443 * | 0.283 | 0.350 | 0.350 | |||
| Sig. (2-tailed) | 0.032 | 0.440 | 0.549 | 0.111 | 0.014 | 0.005 | 0.023 | 0.161 | 0.080 | 0.080 | ||||
| ATM_STR_BCC | Pearson Correlation | 0.320 | 0.275 | 0.195 | 0.586 ** | 0.566 ** | 0.624 ** | 0.522 ** | 0.359 | 0.256 | 0.754 ** | 0.588 ** | ||
| Sig. (2-tailed) | 0.111 | 0.174 | 0.340 | 0.002 | 0.003 | 0.001 | 0.006 | 0.072 | 0.207 | 0.000 | 0.002 | |||
| NF1_STR_BCC | Pearson Correlation | 0.150 | 0.275 | 0.359 | 0.138 | 0.243 | 0.299 | 0.359 | 0.195 | −0.077 | 0.256 | 0.256 | 0.490 * | |
| Sig. (2-tailed) | 0.464 | 0.174 | 0.072 | 0.502 | 0.233 | 0.137 | 0.072 | 0.340 | 0.710 | 0.207 | 0.207 | 0.011 | ||
| BRAF_STR_BCC | Pearson Correlation | −0.020 | 0.275 | 0.195 | 0.586 ** | 0.243 | 0.299 | 0.031 | 0.031 | 0.089 | 0.422 * | 0.422 * | 0.490 * | 0.490 * |
| Sig. (2-tailed) | 0.924 | 0.174 | 0.340 | 0.002 | 0.233 | 0.137 | 0.879 | 0.879 | 0.664 | 0.032 | 0.032 | 0.011 | 0.011 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kibriya, M.G.; Almazan, A.; Argos, M.; Islam, T.; Shea, C.R.; Ahsan, H.; Jasmine, F. Short Tandem Repeat (STR) Somatic Mutation in Non-Melanoma Skin Cancer (NMSC): Association with Transcriptomic Profile and Potential Implications for Therapy. Cancers 2025, 17, 1669. https://doi.org/10.3390/cancers17101669
Kibriya MG, Almazan A, Argos M, Islam T, Shea CR, Ahsan H, Jasmine F. Short Tandem Repeat (STR) Somatic Mutation in Non-Melanoma Skin Cancer (NMSC): Association with Transcriptomic Profile and Potential Implications for Therapy. Cancers. 2025; 17(10):1669. https://doi.org/10.3390/cancers17101669
Chicago/Turabian StyleKibriya, Muhammad G., Armando Almazan, Maria Argos, Tariqul Islam, Christopher R. Shea, Habibul Ahsan, and Farzana Jasmine. 2025. "Short Tandem Repeat (STR) Somatic Mutation in Non-Melanoma Skin Cancer (NMSC): Association with Transcriptomic Profile and Potential Implications for Therapy" Cancers 17, no. 10: 1669. https://doi.org/10.3390/cancers17101669
APA StyleKibriya, M. G., Almazan, A., Argos, M., Islam, T., Shea, C. R., Ahsan, H., & Jasmine, F. (2025). Short Tandem Repeat (STR) Somatic Mutation in Non-Melanoma Skin Cancer (NMSC): Association with Transcriptomic Profile and Potential Implications for Therapy. Cancers, 17(10), 1669. https://doi.org/10.3390/cancers17101669