

Growth Hormone-Releasing Hormone (GHRH) Antagonist Peptides Combined with PI3K Isoform Inhibitors Enhance Cell Death in Prostate Cancer


 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. MIA-602 and MIA-690 GHRH Peptide Antagonists
2.2. Reagents
2.3. Cell Culture
2.4. Drug Treatments
2.5. Trypan Blue Exclusion Assay to Measure Total Cell Death
2.6. Cell Proliferation Assay and Determination of Synergy Combination Index (CI)
2.7. Western Blot Analysis
2.8. Statistics
3. Results
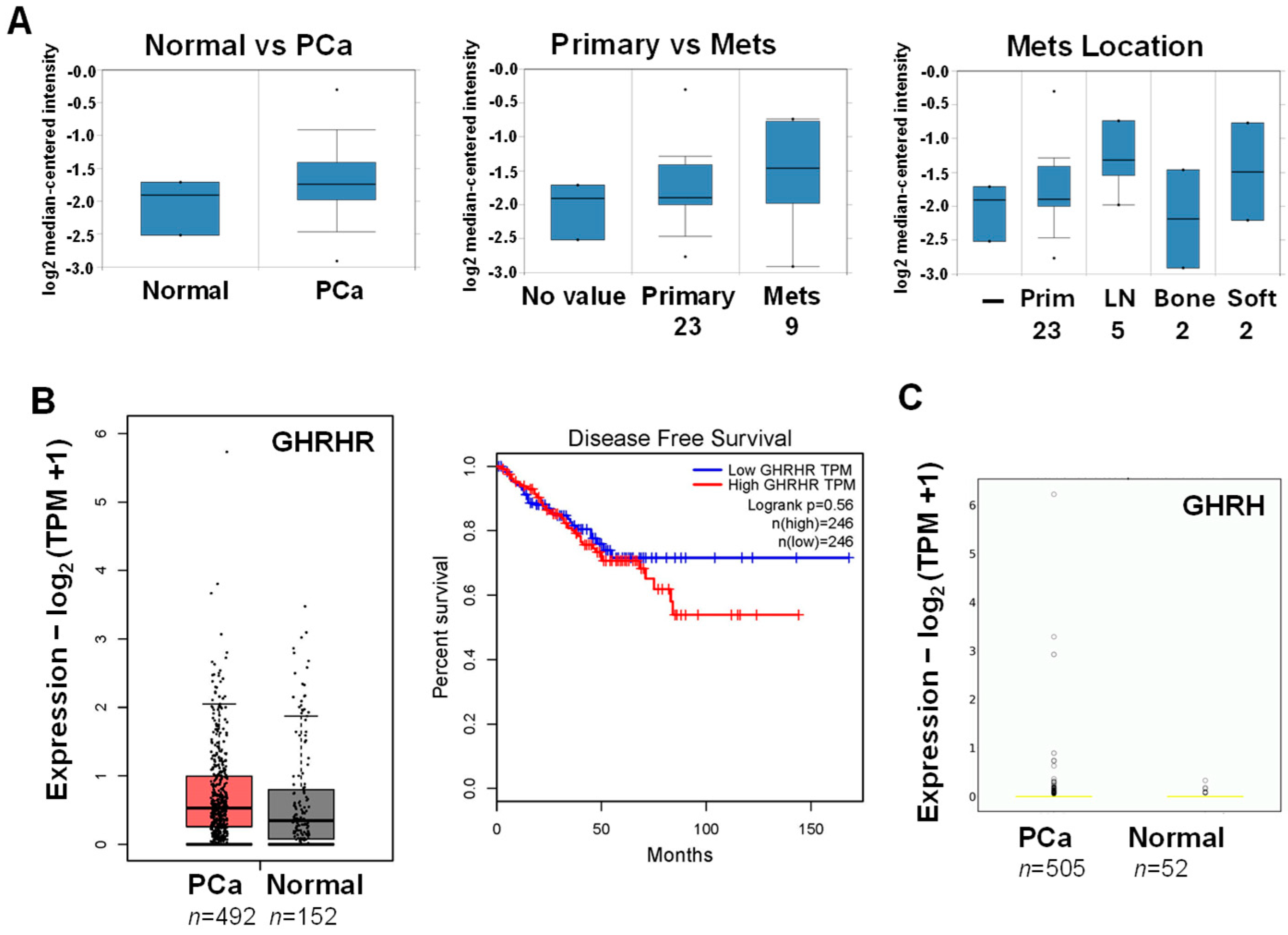
3.1. Database Analysis of GHRHR and GHRH mRNA Expression in PCa
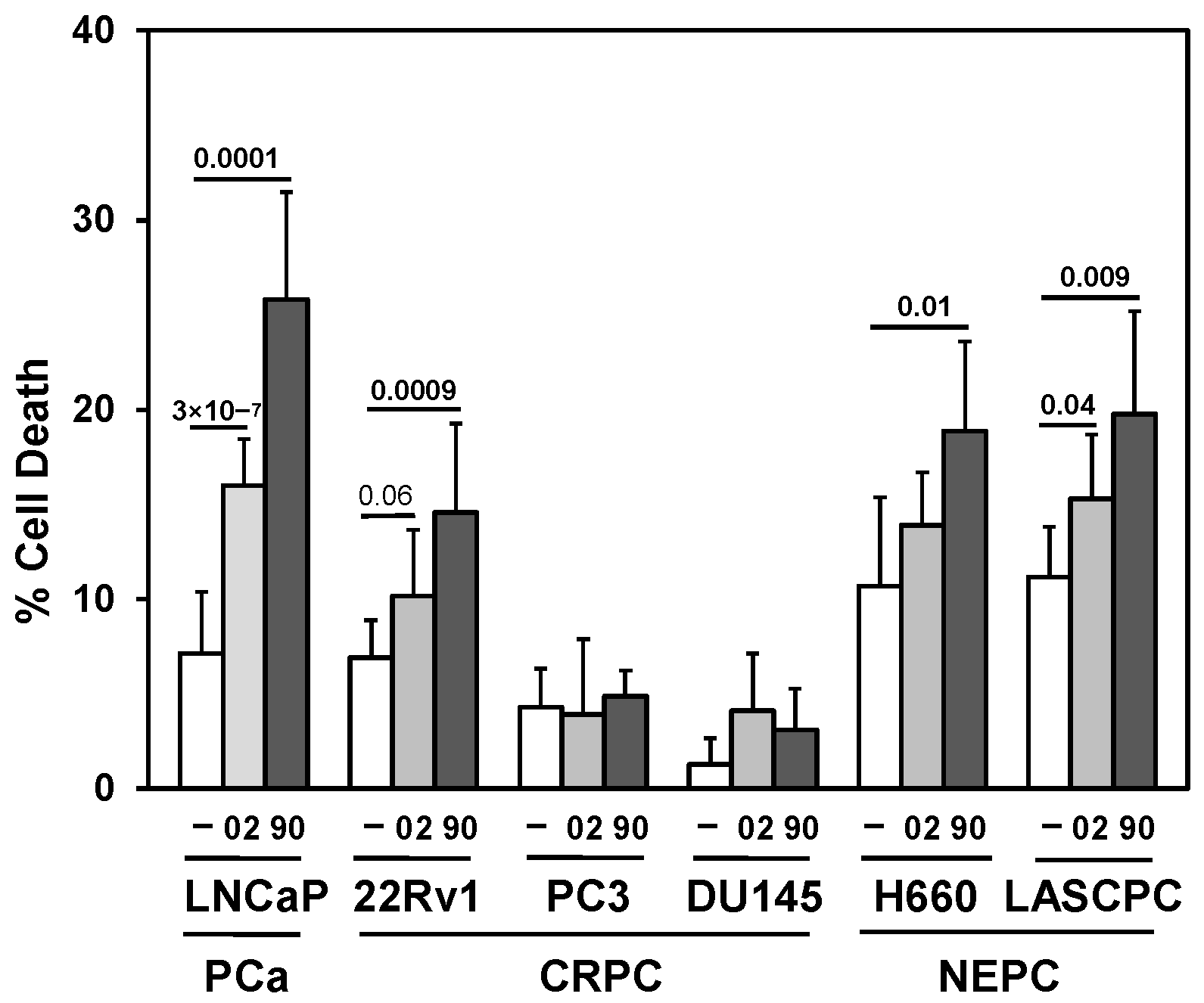
3.2. Searching for a Drug Combination with MIA-602 and MIA-690 GHRH Antagonist Peptides to Increase Cell Death in PCa/CRPC/NEPC
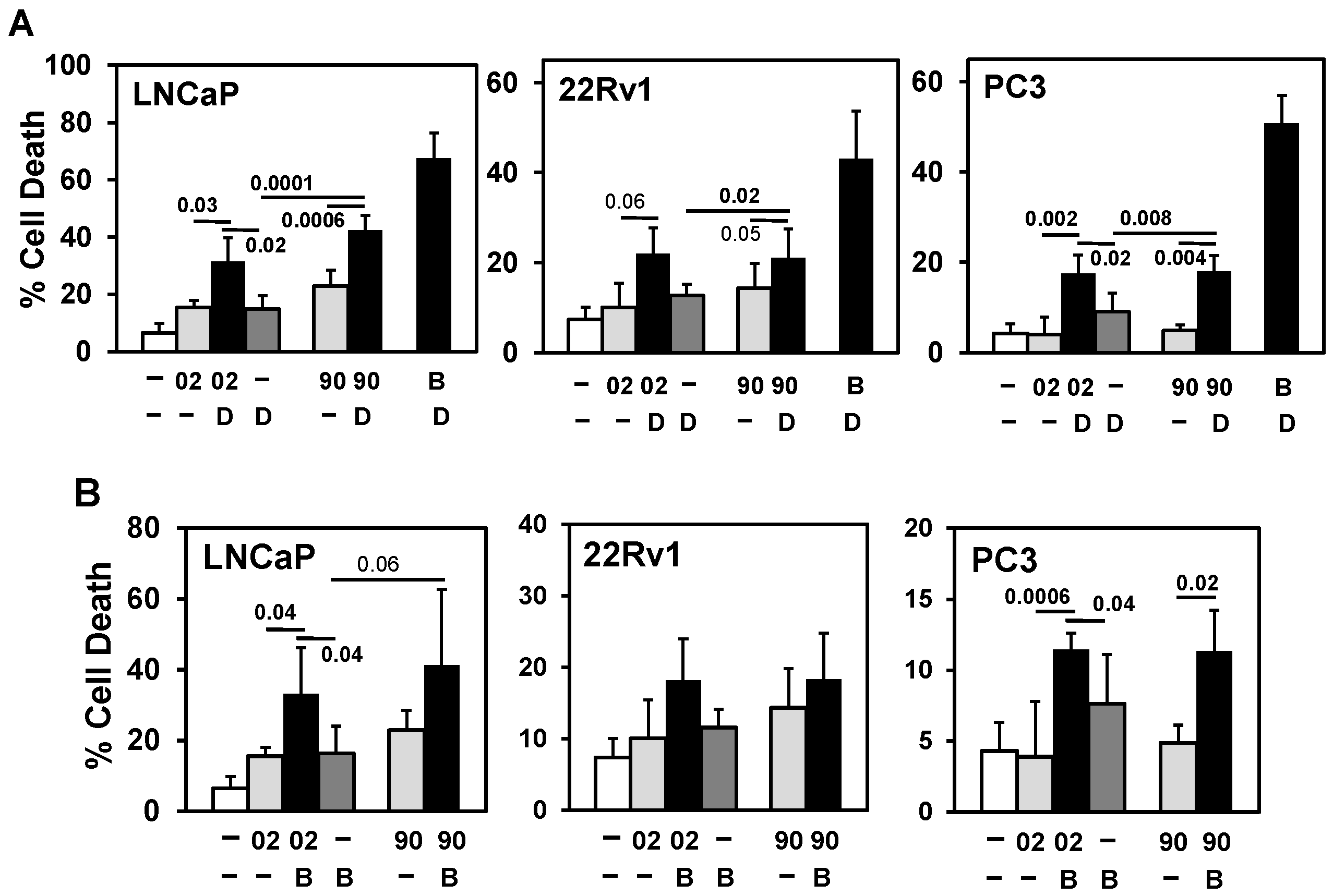
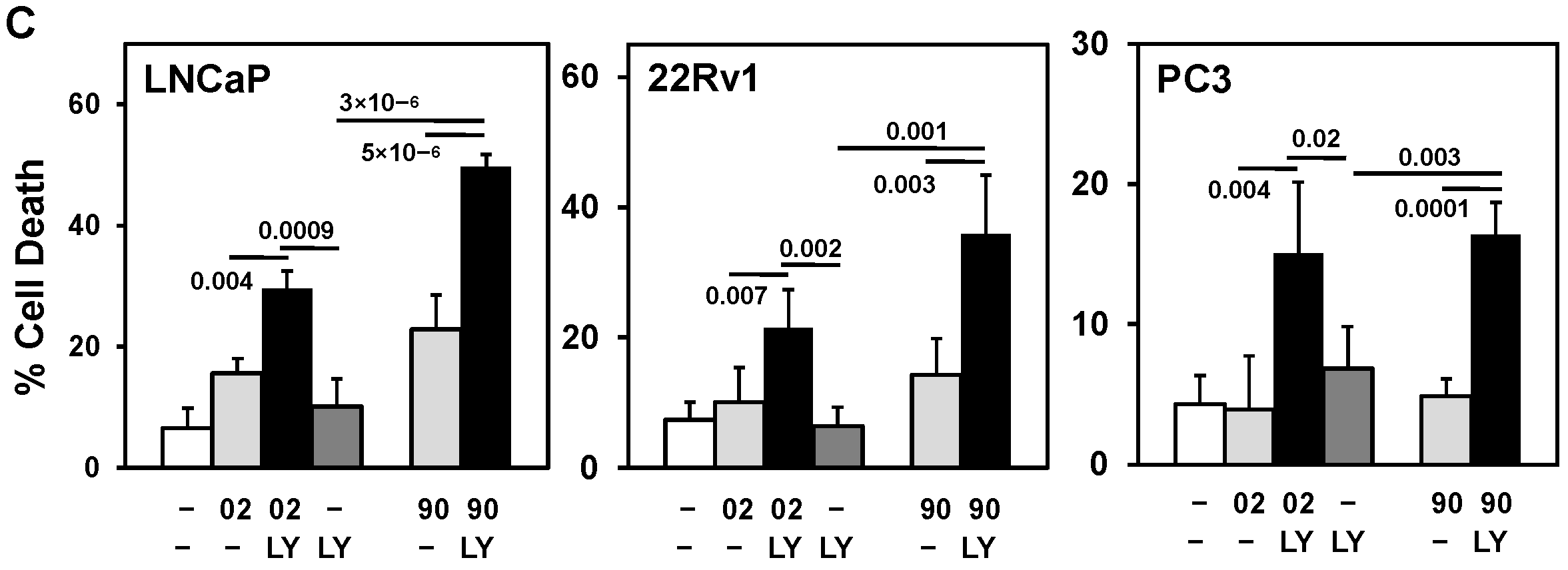
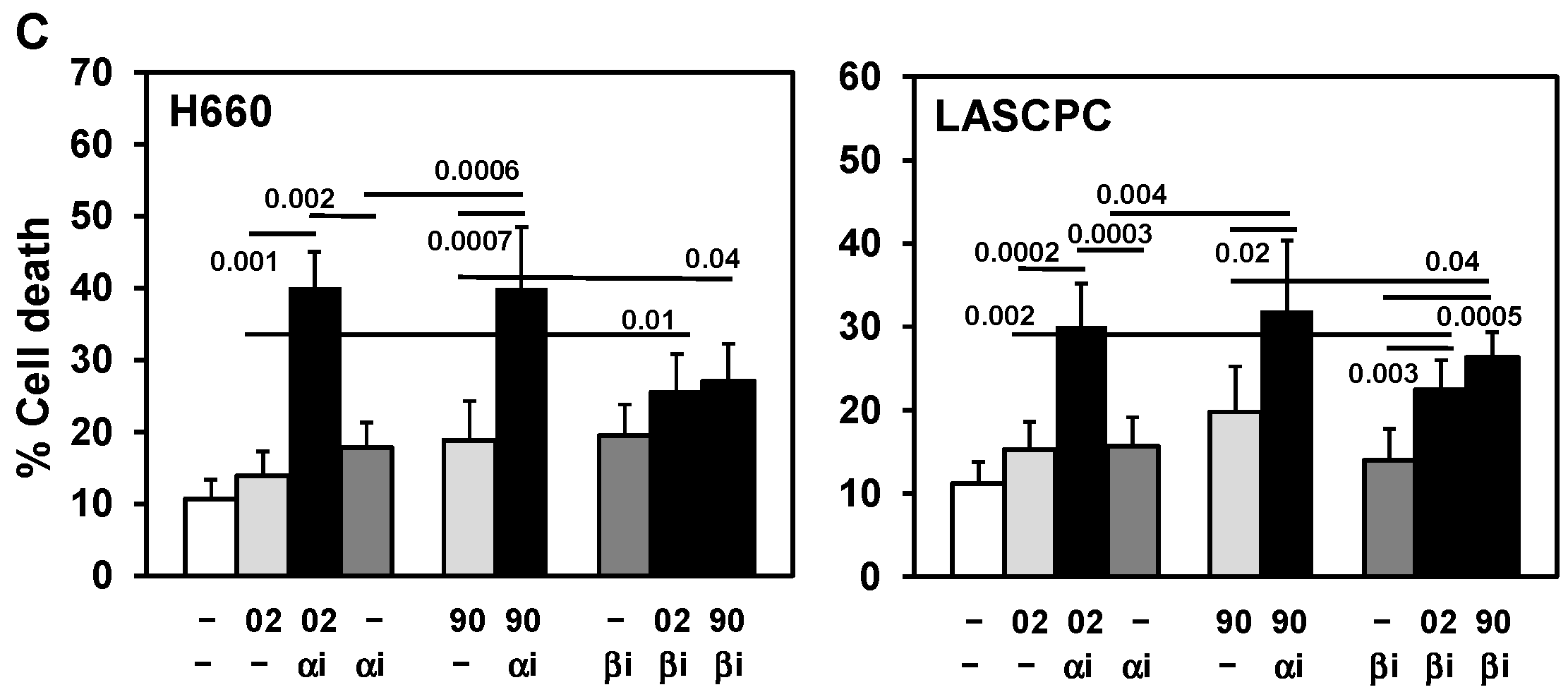
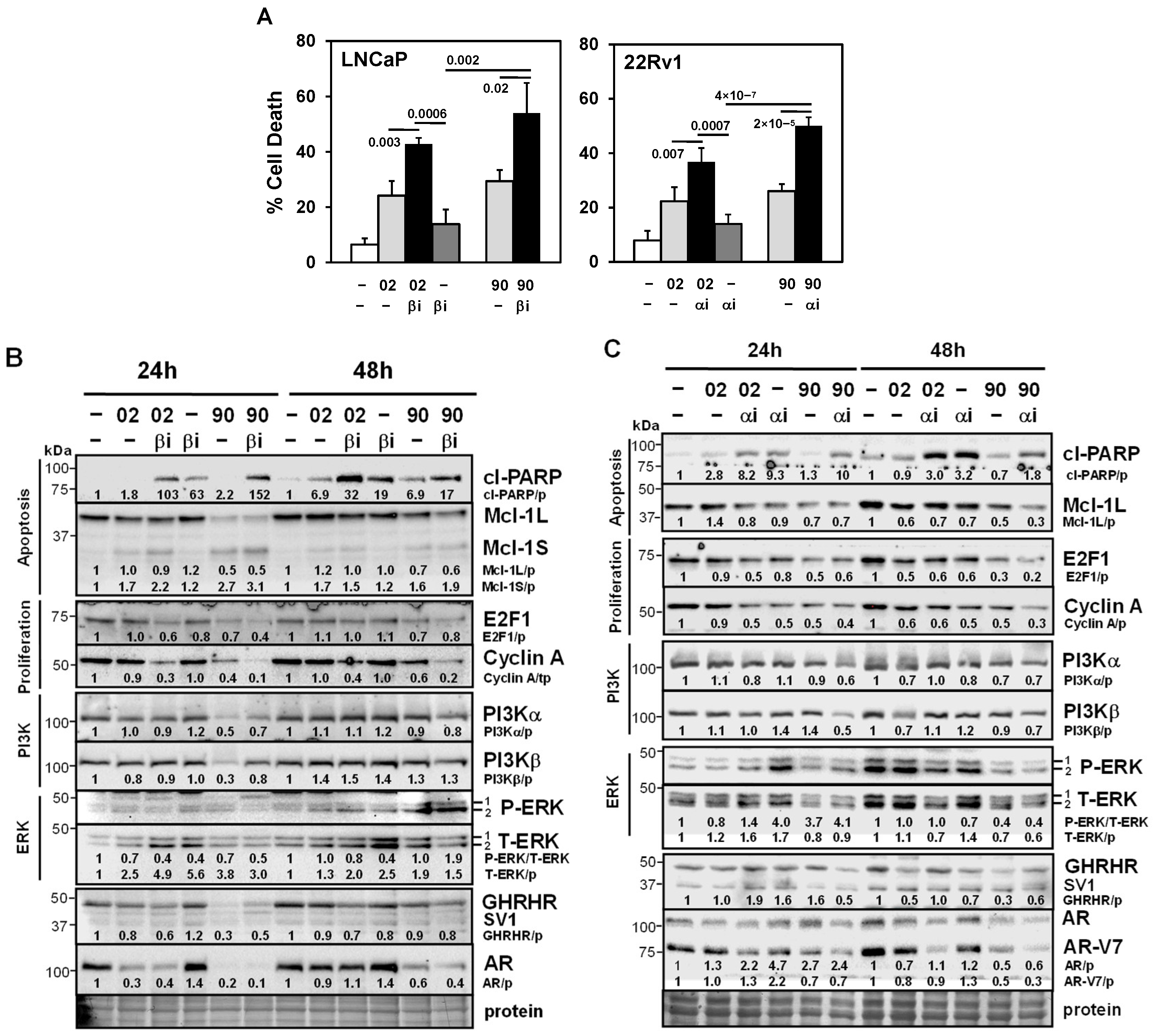
3.3. PI3Kα or β Isoform Inhibitors + MIA-602 or -690 Increases Cell Death in PCa/CRPC/NEPC
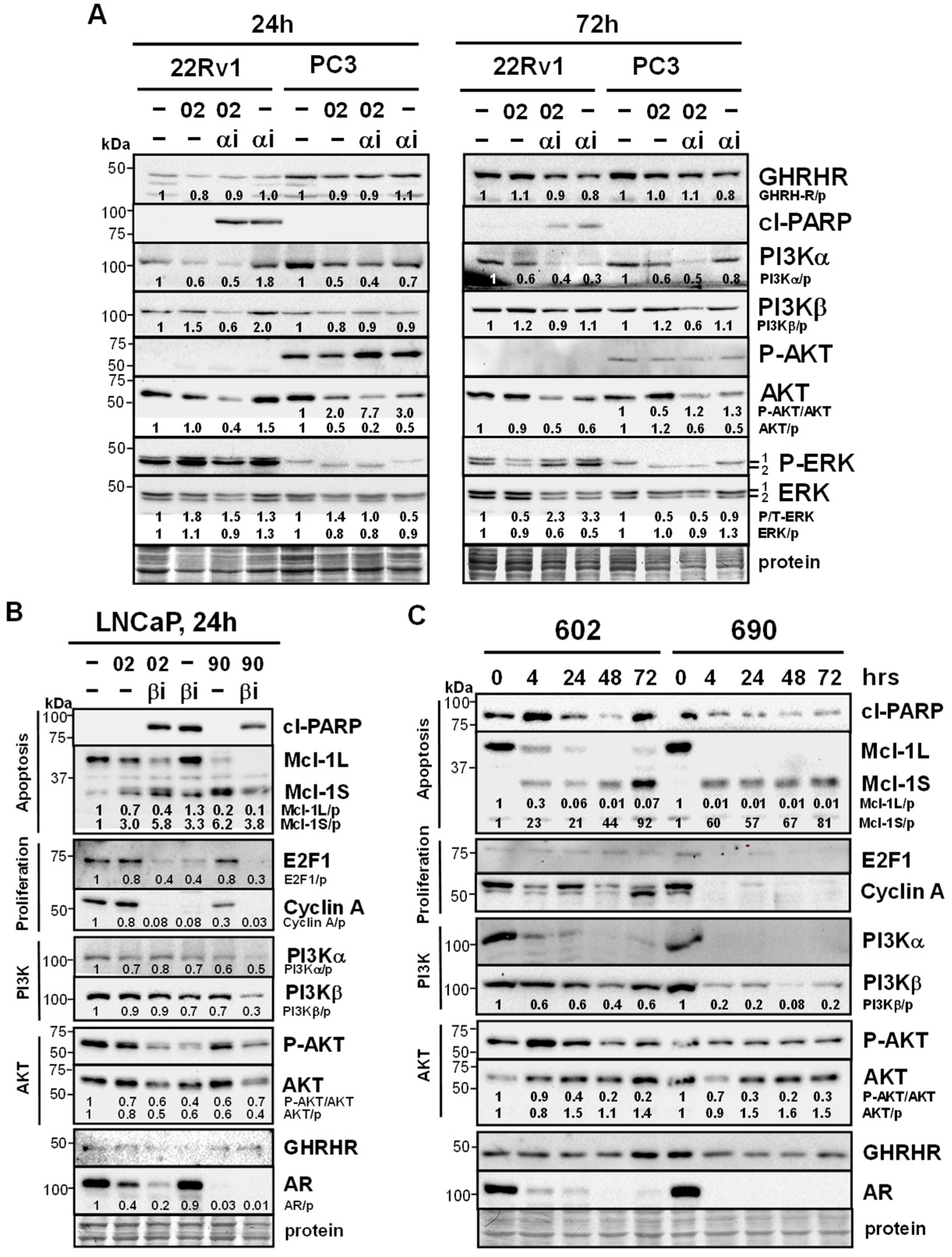
3.4. MIA-602/690 Alone and + PI3K Inhibitors Alters Multiple Signaling Pathways and AR Expression
3.5. Testing MIA-602 and -690 Acetate Salt (Ac) Forms for Future Clinical Applications
3.6. AR Antagonist Enzalutamide + MIA-602 or -690 Has No Effect
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ac | Acetate |
| AKT | Protein Kinase B |
| AR | Androgen Receptor |
| ATCC | American Type Culture Collection |
| ca | Constitutively Active |
| CDK | Cyclin-Dependent Kinase |
| CI | Combination Index |
| CRPC | Castration-Resistant Prostate Cancer |
| EGFR | Epidermal Growth Factor Receptor |
| ERK | Extracellular Signal-Regulated Kinase |
| FDA | Food and Drug Administration |
| GH | Growth Hormone |
| GHRH | Growth Hormone-Releasing Hormone |
| GHRHR | Growth Hormone-Releasing Hormone Receptor |
| GnRH | Gonadotropin-Releasing Hormone |
| GPCR | G Protein-Coupled Receptor |
| IGF1 | Insulin Growth Factor 1 |
| kDa | Kilodalton |
| mTOR | Mammalian Target of Rapamycin |
| NFkB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| NEPC | Neuroendocrine Prostate Cancer |
| P | Phospho |
| PCa | Prostate Cancer |
| PDK1 | Phosphoinositide-Dependent Kinase-1 |
| PI3K | Phosphoinositide 3-Kinase |
| PIP3 | Phosphatidylinositol (3,4,5)-Trisphosphate |
| PTEN | Phosphatase and Tensin Homolog |
| RTK | Receptor Tyrosine Kinase |
| SV1 | Splice Variant 1 of GHRHR |
| TFA | Trifluoroacetic Acid |
| T | Total |
References
- Sartor, O.; de Bono, J.S. Metastatic prostate cancer. N. Engl. J. Med. 2018, 378, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Prekovic, S.; van den Broeck, T.; Linder, S.; van Royen, M.E.; Houtsmuller, A.B.; Handle, F.; Joniau, S.; Zwart, W.; Claessens, F. Molecular underpinnings of enzalutamide resistance. Endocr. Relat. Cancer 2018, 25, R545–R557. [Google Scholar] [CrossRef]
- Beltran, H.; Demichelis, F. Therapy considerations in neuroendocrine prostate cancer: What next? Endocr. Relat. Cancer 2021, 28, T67–T78. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Ci, X.; Choi, S.Y.C.; Crea, F.; Lin, D.; Wang, Y. Molecular events in neuroendocrine prostate cancer development. Nat. Rev. Urol. 2021, 18, 581–596. [Google Scholar] [CrossRef]
- Liu, S.; Alabi, B.R.; Yin, Q.; Stoyanova, T. Molecular mechanisms underlying the development of neuroendocrine prostate cancer. Semin. Cancer Biol. 2022, 86, 57–68. [Google Scholar] [CrossRef]
- Conn, P.M.; Crowley, W.F., Jr. Gonadotropin-releasing hormone and its analogues. N. Engl. J. Med. 1991, 324, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Cai, R.; Zhang, X.; Sha, W.; Wangpaichitr, M. The development of growth hormone-releasing hormone analogs: Therapeutic advances in cancer, regenerative medicine, and metabolic disorders. Rev. Endocr. Metab. Disord. 2024. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef]
- Fahrenholtz, C.D.; Rick, F.G.; Garcia, M.I.; Zarandi, M.; Cai, R.Z.; Block, N.L.; Schally, A.V.; Burnstein, K.L. Preclinical efficacy of growth hormone-releasing hormone antagonists for androgen-dependent and castration-resistant human prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1084–1089. [Google Scholar] [CrossRef]
- Schally, A.V.; Perez, R.; Block, N.L.; Rick, F.G. Potentiating effects of GHRH analogs on the response to chemotherapy. Cell Cycle 2015, 14, 699–704. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Yu, M.; Chen, J.; Xu, Z.; Yang, B.; He, Q.; Luo, P.; Yan, H.; Yang, X. Development and safety of PI3K inhibitors in cancer. Arch. Toxicol. 2023, 97, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Shan, K.S.; Bonano-Rios, A.; Theik, N.W.Y.; Hussein, A.; Blaya, M. Molecular targeting of the phosphoinositide-3-protein kinase (PI3K) pathway across various cancers. Int. J. Mol. Sci. 2024, 25, 1973. [Google Scholar] [CrossRef]
- Li, H.; Wen, X.; Ren, Y.; Fan, Z.; Zhang, J.; He, G.; Fu, L. Targeting PI3K family with small-molecule inhibitors in cancer therapy: Current clinical status and future directions. Mol. Cancer 2024, 23, 164. [Google Scholar] [CrossRef]
- Dekant, W.; Dekant, R. Mammalian toxicity of trifluoroacetate and assessment of human health risks due to environmental exposures. Arch. Toxicol. 2023, 97, 1069–1077. [Google Scholar] [CrossRef]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef] [PubMed]
- VanDeusen, H.R.; Ramroop, J.R.; Morel, K.L.; Bae, S.Y.; Sheahan, A.V.; Sychev, Z.; Lau, N.A.; Cheng, L.C.; Tan, V.M.; Li, Z.; et al. Targeting RET kinase in neuroendocrine prostate cancer. Mol. Cancer Res. 2020, 18, 1176–1188. [Google Scholar] [CrossRef]
- McMenamin, M.E.; Soung, P.; Perera, S.; Kaplan, I.; Loda, M.; Sellers, W.R. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999, 59, 4291–4296. [Google Scholar] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Sramkoski, R.M.; Pretlow, T.G.; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A new human prostate carcinoma cell line, 22Rv1. Vitr. Cell Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef]
- Muñoz-Moreno, L.; Román, I.D.; Bajo, A.M. GHRH and the prostate. Rev. Endocr. Metab. Disord. 2024. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- de Las Pozas, A.; Reiner, T.; De Cesare, V.; Trost, M.; Perez-Stable, C. Inhibiting multiple deubiquitinases to reduce androgen receptor expression in prostate cancer cells. Sci. Rep. 2018, 8, 13146. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, E.; Giantulli, S.; Sciarra, A.; Silvestri, I. AR and PI3K/AKT in prostate cancer: A tale of two interconnected pathways. Int. J. Mol. Sci. 2023, 24, 2046. [Google Scholar] [CrossRef]
- Raith, F.; O’Donovan, D.H.; Lemos, C.; Politz, O.; Haendler, B. Addressing the reciprocal crosstalk between the AR and the PI3K/AKT/mTOR signaling pathways for prostate cancer treatment. Int. J. Mol. Sci. 2023, 24, 2289. [Google Scholar] [CrossRef]
- Muñoz-Moreno, L.; Carmena, M.J.; Schally, A.V.; Prieto, J.C.; Bajo, A.M. Stimulation of neuroendocrine differentiation in prostate cancer cells by GHRH and its blockade by GHRH antagonists. Investig. N. Drugs 2020, 38, 746–754. [Google Scholar] [CrossRef]
- Gan, J.; Ke, X.; Jiang, J.; Dong, H.; Yao, Z.; Lin, Y.; Lin, W.; Wu, X.; Yan, S.; Zhuang, Y.; et al. Growth hormone-releasing hormone receptor antagonists inhibit human gastric cancer through downregulation of PAK1-STAT3/NF-κB signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 14745–14750. [Google Scholar] [CrossRef]
- Cong, Z.; Zhou, F.; Zhang, C.; Zou, X.; Zhang, H.; Wang, Y.; Zhou, Q.; Cai, X.; Liu, Q.; Li, J.; et al. Constitutive signal bias mediated by the human GHRHR splice variant 1. Proc. Natl. Acad. Sci. USA 2021, 118, e2106606118. [Google Scholar] [CrossRef]
- Hartman, M.L. Tipping the balance of cell death: Alternative splicing as a source of MCL-1S in cancer. Cell Death Dis. 2024, 15, 917. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.B.; Li, J.; Meniel, V.S.; Fennell, C.M.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; Macpherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca mutation as a genetic driver of prostate cancer that cooperates with Pten loss to accelerate progression and castration-resistant growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; de Stanchina, E.; Baselga, J.; et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell 2015, 27, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Mao, N.; Zhang, Z.; Lee, Y.S.; Choi, D.; Rivera, A.A.; Li, D.; Lee, C.; Haywood, S.; Chen, X.; Chang, Q.; et al. Defining the therapeutic selective dependencies for distinct subtypes of PI3K pathway-altered prostate cancers. Nat. Commun. 2021, 12, 5053. [Google Scholar] [CrossRef]
- Zheng, N.; Wei, J.; Wu, D.; Xu, Y.; Guo, J. Master kinase PDK1 in tumorigenesis. Biochim. Biophys. Acta. Rev. Cancer 2023, 1878, 188971. [Google Scholar] [CrossRef]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Zhang, X.; Dai, X.; Han, S.; Wu, X.; Wang, L.; Wei, W.; Zhang, N.; Xie, W.; Guo, J. S6K1-mediated phosphorylation of PDK1 impairs AKT kinase activity and oncogenic functions. Nat. Commun. 2022, 13, 1548. [Google Scholar] [CrossRef]
- Nalairndran, G.; Hassan Abdul Razack, A.; Mai, C.W.; Fei-Lei Chung, F.; Chan, K.K.; Hii, L.W.; Lim, W.M.; Chung, I.; Leong, C.O. Phosphoinositide-dependent Kinase-1 (PDPK1) regulates serum/glucocorticoid-regulated Kinase 3 (SGK3) for prostate cancer cell survival. J. Cell Mol. Med. 2020, 24, 12188–12198. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First global approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef]
- Choudhury, A.D.; Higano, C.S.; de Bono, J.S.; Cook, N.; Rathkopf, D.E.; Wisinski, K.B.; Martin-Liberal, J.; Linch, M.; Heath, E.I.; Baird, R.D.; et al. A phase I study investigating AZD8186, a potent and selective inhibitor of PI3Kβ/δ, in patients with advanced solid tumors. Clin. Cancer Res. 2022, 28, 2257–2269. [Google Scholar] [CrossRef]
- Wright, S.C.E.; Vasilevski, N.; Serra, V.; Rodon, J.; Eichhorn, P.J.A. Mechanisms of resistance to PI3K inhibitors in cancer: Adaptive responses, drug tolerance and cellular plasticity. Cancers 2021, 13, 1538. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Type | AR | PTEN | AKT | p53 | Additional |
| LNCaP | AS PCa | + | mut/− | ca | +/+ | ARmut T877A |
| 22Rv1 | CRPC | + | +/+ | wt | +/+ | (1) AR-V7 (splice variant—LBD) (2) PIK3CA mut |
| PC3 | CRPC | − | −/− | ca | −/− | |
| DU145 | CRPC | − | +/− | wt | dn/oe | Bax null |
| H660 | NEPC | − | −/− | ca | mut | |
| LASCPC | NEPC | − | ca | N-myc/AKTmyr oe |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Stable, C.; de las Pozas, A.; Wangpaichitr, M.; Sha, W.; Wang, H.; Cai, R.; Schally, A.V. Growth Hormone-Releasing Hormone (GHRH) Antagonist Peptides Combined with PI3K Isoform Inhibitors Enhance Cell Death in Prostate Cancer. Cancers 2025, 17, 1643. https://doi.org/10.3390/cancers17101643
Perez-Stable C, de las Pozas A, Wangpaichitr M, Sha W, Wang H, Cai R, Schally AV. Growth Hormone-Releasing Hormone (GHRH) Antagonist Peptides Combined with PI3K Isoform Inhibitors Enhance Cell Death in Prostate Cancer. Cancers. 2025; 17(10):1643. https://doi.org/10.3390/cancers17101643
Chicago/Turabian StylePerez-Stable, Carlos, Alicia de las Pozas, Medhi Wangpaichitr, Wei Sha, Haibo Wang, Renzhi Cai, and Andrew V. Schally. 2025. "Growth Hormone-Releasing Hormone (GHRH) Antagonist Peptides Combined with PI3K Isoform Inhibitors Enhance Cell Death in Prostate Cancer" Cancers 17, no. 10: 1643. https://doi.org/10.3390/cancers17101643
APA StylePerez-Stable, C., de las Pozas, A., Wangpaichitr, M., Sha, W., Wang, H., Cai, R., & Schally, A. V. (2025). Growth Hormone-Releasing Hormone (GHRH) Antagonist Peptides Combined with PI3K Isoform Inhibitors Enhance Cell Death in Prostate Cancer. Cancers, 17(10), 1643. https://doi.org/10.3390/cancers17101643