
The Basis for Targeting the Tumor Macrophage Compartment in Glioblastoma Immunotherapy

 , ,
, ,  and
and
Simple Summary
Abstract
1. Introduction
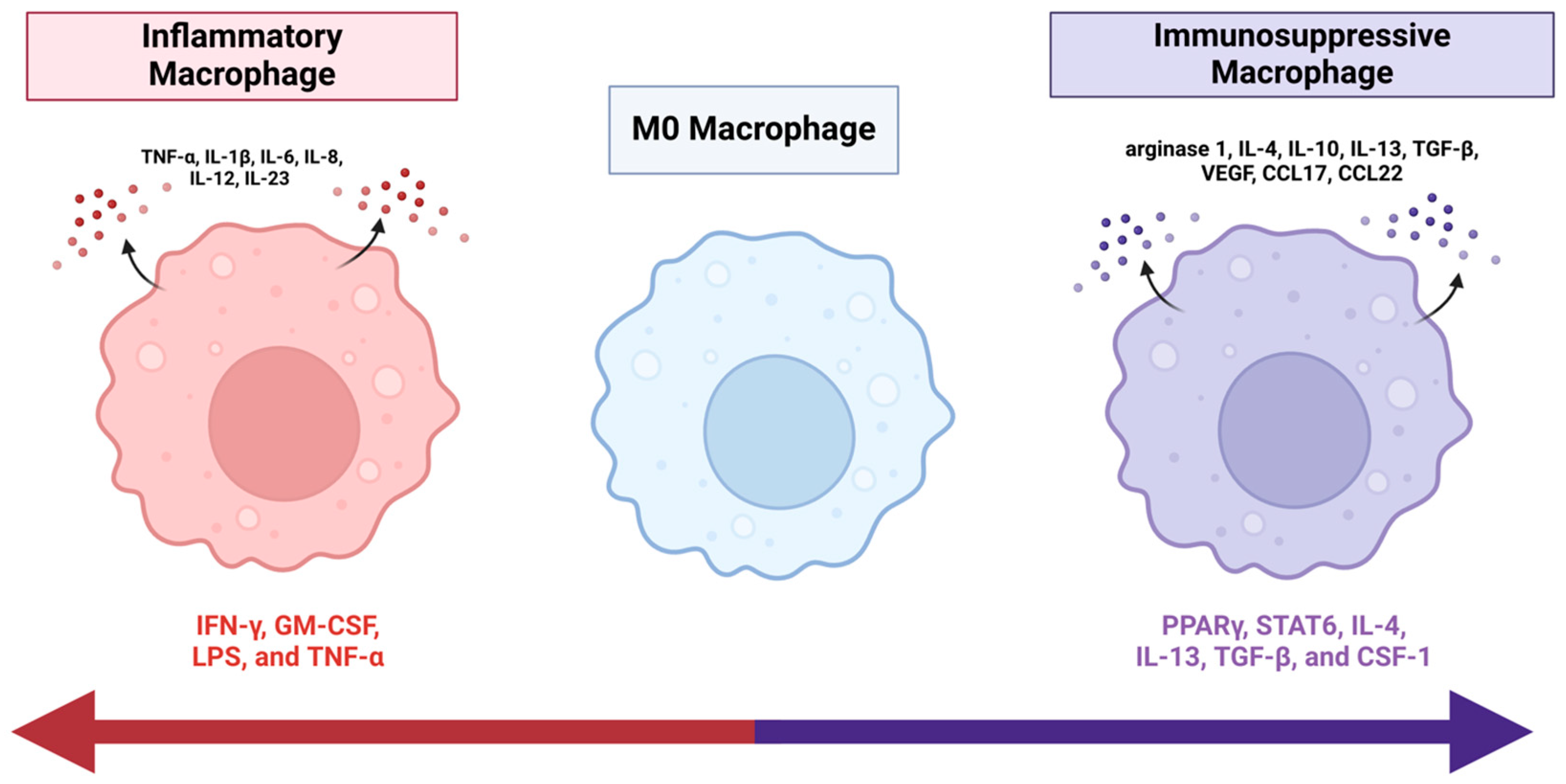
2. Nomenclature
3. Barriers to Immunotherapy
4. GBM–TAM Interactions
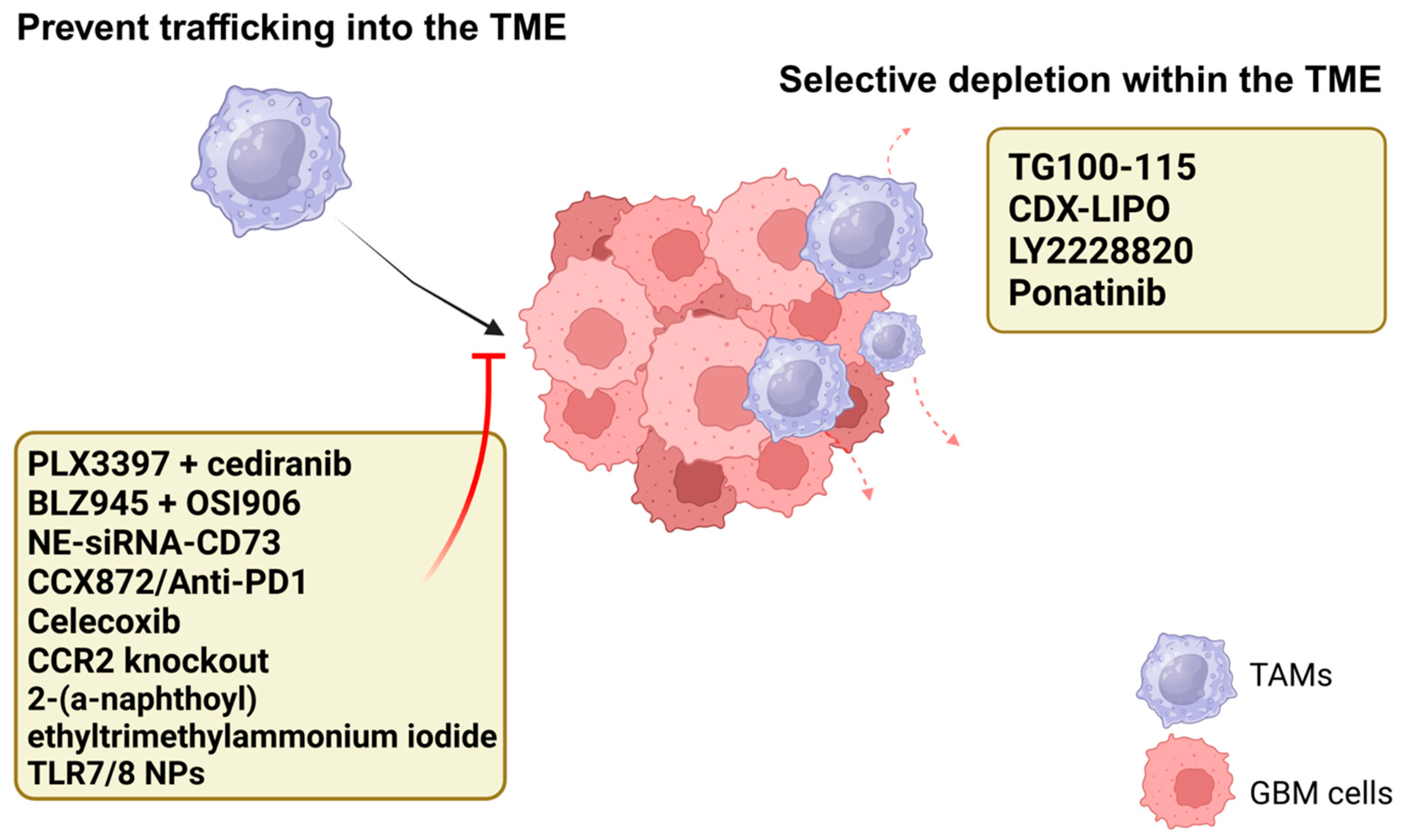
5. Depleting the TAM Population
5.1. Prevent Trafficking into the TME
5.2. Selective Depletion Within the TME
6. Reprogramming Macrophages
6.1. Immune Checkpoints
6.2. Intracellular Signaling Pathways
6.3. Nanotechnology and Delivery Systems
6.4. Emerging Targets
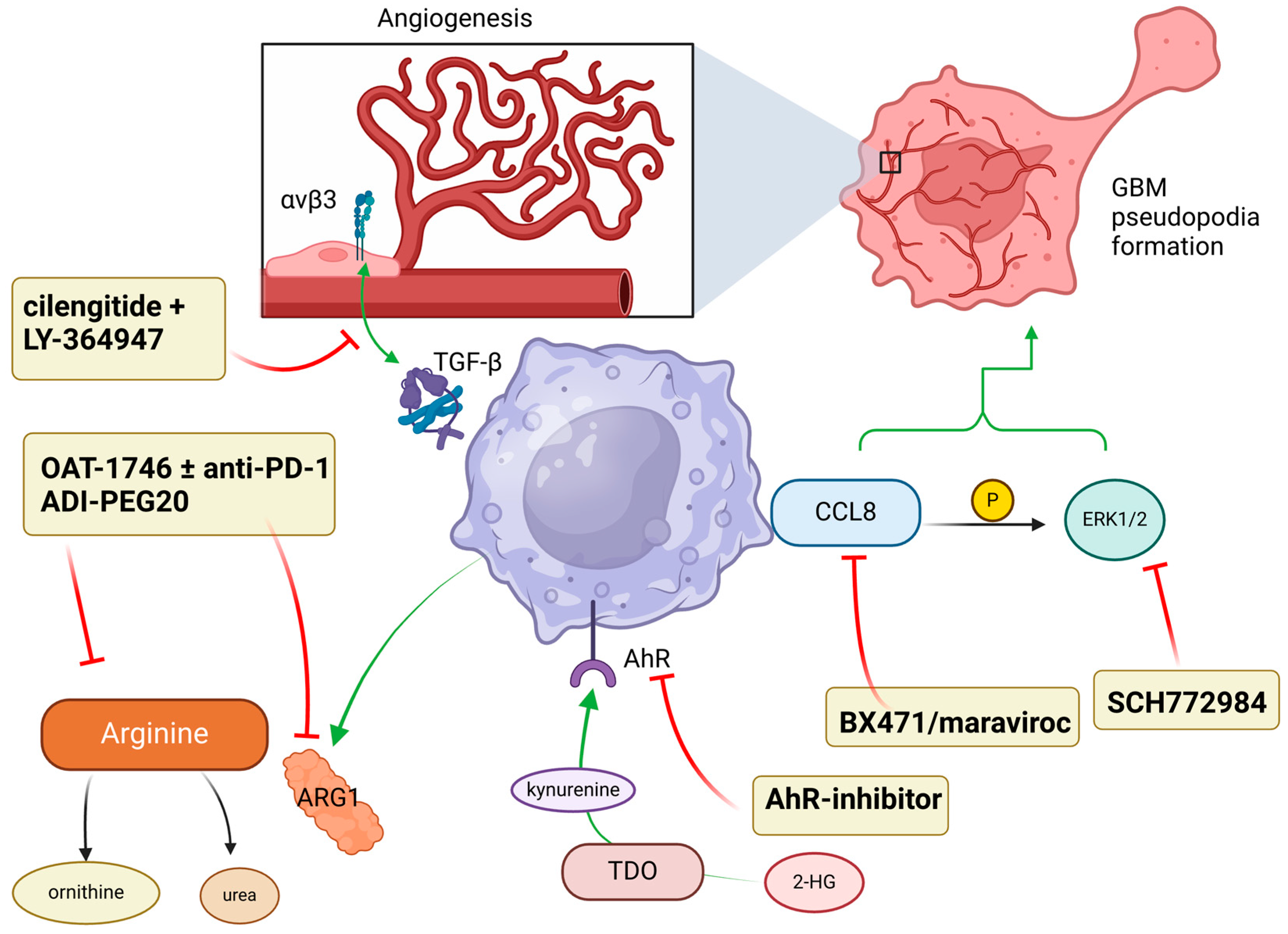
7. Targeting Downstream Pro-Tumor Signals
8. In Human Trials
9. Discussion
10. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Liu, Y.; Zhou, F.; Ali, H.; Lathia, J.D.; Chen, P. Immunotherapy for glioblastoma: Current state, challenges, and future perspectives. Cell. Mol. Immunol. 2024, 21, 1354–1375. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Yalamarty, S.S.K.; Filipczak, N.; Li, X.; Subhan, M.A.; Parveen, F.; Ataide, J.A.; Rajmalani, B.A.; Torchilin, V.P. Mechanisms of Resistance and Current Treatment Options for Glioblastoma Multiforme (GBM). Cancers 2023, 15, 2116. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Previously Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Qu, J.; Kalyani, F.S.; Shen, Q.; Yang, G.; Cheng, T.; Liu, L.; Zhou, J.; Zhou, J. Efficacy and Safety of PD-L1 Inhibitors plus Chemotherapy versus Chemotherapy Alone in First-Line Treatment of Extensive-Stage Small-Cell Lung Cancer: A Retrospective Real-World Study. J. Oncol. 2022, 2022, 3645489. [Google Scholar] [CrossRef] [PubMed]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van de Bent, M.J.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro-Oncol. 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot trial of adoptive transfer of chimeric antigen receptor transduced T cells targeting EGFRvIII in patients with glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Woroniecka, K.; Fecci, P.E. T-cell exhaustion in glioblastoma. Oncotarget 2018, 9, 35287–35288. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef]
- Butler, M.; Prasad, S.; Srivastava, S.K. Targeting Glioblastoma Tumor Microenvironment. In Tumor Microenvironments in Organs; Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2021; pp. 1–9. [Google Scholar] [CrossRef]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Larkin, C.J.; Arrieta, V.A.; Najem, H.; Li, G.; Zhang, P.; Miska, J.; Chen, P.; James, C.D.; Sonabend, A.M.; Heimberger, A.B. Myeloid Cell Classification and Therapeutic Opportunities Within the Glioblastoma Tumor Microenvironment in the Single Cell-Omics Era. Front. Immunol. 2022, 13, 907605. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xu, B.; Ren, J.; Liu, Z.; Cai, L.; Zhang, X.; Wang, W.; Li, S.; Jin, L.; Ding, L. The Importance of M1-and M2-Polarized Macrophages in Glioma and as Potential Treatment Targets. Brain Sci. 2023, 13, 1269. [Google Scholar] [CrossRef]
- Khan, F.; Pang, L.; Dunterman, M.; Lesniak, M.S.; Heimberger, A.B.; Chen, P. Macrophages and microglia in glioblastoma: Heterogeneity, plasticity, and therapy. J. Clin. Investig. 2023, 133, e163446. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, 263. [Google Scholar] [CrossRef]
- Luo, H.; Shusta, E.V. Blood–brain barrier modulation to improve glioma drug delivery. Pharmaceutics 2020, 12, 1085. [Google Scholar] [CrossRef] [PubMed]
- Bausart, M.; Préat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Waibl Polania, J.; Hoyt-Miggelbrink, A.; Tomaszewski, W.H.; Wachsmuth, L.P.; Lorrey, S.J.; Wilkinson, D.S.; Lerner, E.; Woroniecka, K.; Finlay, J.B.; Ayasoufi, K.; et al. Antigen presentation by tumor-associated macrophages drives T cells from a progenitor exhaustion state to terminal exhaustion. Immunity 2025, 58, 232–246.e6. [Google Scholar] [CrossRef]
- Kersten, K.; Hu, K.H.; Combes, A.J.; Samad, B.; Harwin, T.; Ray, A.; Rao, A.A.; Cai, E.; Marchuk, K.; Artichoker, J.; et al. Spatiotemporal co-dependency between macrophages and exhausted CD8+ T cells in cancer. Cancer Cell 2022, 40, 624–638.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kaneda, M.M.; Ma, J.; Li, M.; Shepard, R.M.; Patel, K.; Koga, T.; Sarver, A.; Furnari, F.; Xu, B.; et al. PI3Kγ inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proc. Natl. Acad. Sci. USA 2021, 118, e2009290118. [Google Scholar] [CrossRef]
- Andersen, R.S.; Anand, A.; Harwood, D.S.L.; Kristensen, B.W. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers 2021, 13, 4255. [Google Scholar] [CrossRef]
- Wei, J.; Chen, P.; Gupta, P.; Ott, M.; Zamler, D.; Kassab, C.; Bhat, K.P.; Curran, M.A.; de Groot, J.F.; Heimberger, A.M. Immune biology of glioma-associated macrophages and microglia: Functional and therapeutic implications. Neuro-Oncol. 2020, 22, 180–194. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Chitu, V.; Gokhan, Ş.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Han, R.; Ogurek, S.; Xue, C.; Wu, L.M.; Zhang, L.; Zhang, L.; Hu, J.; Phoenix, T.N.; Waggoner, S.N.; et al. Glioblastoma genetic drivers dictate the function of tumor-associated macrophages/microglia and responses to CSF1R inhibition. Neuro-Oncol. 2022, 24, 584–597. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Schuh, R.S.; Michels, L.R.; Iser, I.C.; Beckenkamp, L.R.; Roliano, G.G.; Lenz, G.S.; Scholl, J.N.; Sévigny, J.; Wink, M.R.; et al. Blockade of CD73 delays glioblastoma growth by modulating the immune environment. Cancer Immunol. Immunother. 2020, 69, 1801–1812. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef]
- Shono, K.; Yamaguchi, I.; Mizobuchi, Y.; Kagusa, H.; Sumi, A.; Fujihara, T.; Nakajima, K.; Kitazato, K.T.; Matsuzaki, K.; Saya, H.; et al. Downregulation of the CCL2/CCR2 and CXCL10/CXCR3 axes contributes to antitumor effects in a mouse model of malignant glioma. Sci. Rep. 2020, 10, 15286. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shen, S.; Liu, T.; Ren, X.; Zhu, C.; Liang, Q.; Cui, X.; Chen, L.; Cheng, P.; Cheng, W.; et al. Chemerin enhances mesenchymal features of glioblastoma by establishing autocrine and paracrine networks in a CMKLR1-dependent manner. Oncogene 2022, 41, 3024–3036. [Google Scholar] [CrossRef]
- Turco, V.; Pfleiderer, K.; Hunger, J.; Horvat, N.K.; Karimian-Jazi, K.; Schregel, K.; Fischer, M.; Brugnara, G.; Jähne, K.; Sturm, V.; et al. T cell-independent eradication of experimental glioma by intravenous TLR7/8-agonist-loaded nanoparticles. Nat. Commun. 2023, 14, 771. [Google Scholar] [CrossRef]
- Dang, W.; Xiao, J.; Ma, Q.; Miao, J.; Cao, M.; Chen, L.; Shi, Y.; Yao, X.; Yu, S.; Liu, X.; et al. Combination of p38 MAPK inhibitor with PD-L1 antibody effectively prolongs survivals of temozolomide-resistant glioma-bearing mice via reduction of infiltrating glioma-associated macrophages and PD-L1 expression on resident glioma-associated microglia. Brain Tumor Pathol. 2021, 38, 189–200. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, J.; Jiang, J.; He, Y.; Zhang, W.; Mo, X.; Kang, X.; Xu, Q.; Wang, B.; Huang, Y.; et al. Remodeling tumor immune microenvironment (TIME) for glioma therapy using multi-targeting liposomal codelivery. J. Immunother. Cancer 2020, 8, e000207. [Google Scholar] [CrossRef]
- Barnwal, A.; Tamang, R.; Sanjeev, D.; Bhattacharyya, J. Ponatinib delays the growth of solid tumours by remodelling immunosuppressive tumour microenvironment through the inhibition of induced PD-L1 expression. Br. J. Cancer 2023, 129, 1007–1021. [Google Scholar] [CrossRef]
- Tang, F.; Wang, Y.; Zeng, Y.; Xiao, A.; Tong, A.; Xu, J. Tumor-associated macrophage-related strategies for glioma immunotherapy. npj Precis. Oncol. 2023, 7, 78. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, H.; Chen, B.; Liu, X.; Zhang, S.; Zong, Z.; Gao, M. PD-L1-Mediated Immunosuppression in Glioblastoma Is Associated with the Infiltration and M2-Polarization of Tumor-Associated Macrophages. Front. Immunol. 2020, 11, 588552. [Google Scholar] [CrossRef]
- Dual-sgRNA CRISPR/Cas9 Knockout of PD-L1 in Human U87 Glioblastoma Tumor Cells Inhibits Proliferation, Invasion, and Tumor-Associated Macrophage Polarization. Scientific Reports. Available online: https://www.nature.com/articles/s41598-022-06430-1 (accessed on 25 February 2025).
- Chen, P.; Zhao, D.; Li, J.; Liang, X.; Li, J.; Chang, A.; Henry, V.K.; Lan, Z.; Spring, D.J.; Rao, G.; et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019, 35, 868–884.e6. [Google Scholar] [CrossRef]
- Ni, X.; Wu, W.; Sun, X.; Ma, J.; Yu, Z.; He, X.; Cheng, J.; Xu, P.; Liu, H.; Shang, T.; et al. Interrogating glioma-M2 macrophage interactions identifies Gal-9/Tim-3 as a viable target against PTEN-null glioblastoma. Sci. Adv. 2022, 8, eabl5165. [Google Scholar] [CrossRef]
- Rivera-Ramos, A.; Cruz-Hernández, L.; Talaverón, R.; Sánchez-Montero, M.T.; García-Revilla, J.; Mulero-Acevedo, M.; Deierborg, T.; Venero, J.L.; Soto, M.S. Galectin-3 depletion tames pro-tumoural microglia and restrains cancer cells growth. Cancer Lett. 2024, 591, 216879. [Google Scholar] [CrossRef]
- Wu, J.; Yang, H.; Cheng, J.; Zhang, L.; Ke, Y.; Zhu, Y.; Wang, C.; Zhang, X.; Zhen, X.; Zheng, L.T. Knockdown of milk-fat globule EGF factor-8 suppresses glioma progression in GL261 glioma cells by repressing microglial M2 polarization. J. Cell. Physiol. 2020, 235, 8679–8690. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Yerneni, S.S.; Maurer, L.M.; Crentsil, H.E.; Debom, G.N.; Klei, L.; Smyers, M.; Sneiderman, C.T.; Schwab, K.E.; Acharya, R.; et al. MALT1 protease inhibition restrains glioblastoma progression by reversing tumor-associated macrophage-dependent immunosuppression. bioRxiv 2024. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, D.; Jiang, H.; Ye, J.; Zhang, L.; Bagley, S.J.; Winkler, J.; Gong, Y.; Fan, Y. Small-molecule toosendanin reverses macrophage-mediated immunosuppression to overcome glioblastoma resistance to immunotherapy. Sci. Transl. Med. 2023, 15, eabq3558. [Google Scholar] [CrossRef]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 3974. [Google Scholar] [CrossRef]
- Sylvestre, M.; Crane, C.A.; Pun, S.H. Progress on Modulating Tumor-Associated Macrophages with Biomaterials. Adv. Mater. 2020, 32, e1902007. [Google Scholar] [CrossRef]
- Petrovic, M.; Majchrzak, O.B.; Marecar, R.A.M.H.; Laingoniaina, A.C.; Walker, P.R.; Borchard, G.; Jordan, O.; Tankov, S. Combining antimiR-25 and cGAMP Nanocomplexes Enhances Immune Responses via M2 Macrophage Reprogramming. Int. J. Mol. Sci. 2024, 25, 12787. [Google Scholar] [CrossRef]
- Chouleur, T.; Emanuelli, A.; Souleyreau, W.; Derieppe, M.A.; Leboucq, T.; Hardy, S.; Mathivet, T.; Tremblay, M.L.; Bikfalvi, A. PTP4A2 Promotes Glioblastoma Progression and Macrophage Polarization under Microenvironmental Pressure. Cancer Res. Commun. 2024, 4, 1702–1714. [Google Scholar] [CrossRef]
- Mu, B.; Jing, J.; Li, R.; Li, C. USP9X deubiquitinates TRRAP to promote glioblastoma cell proliferation and migration and M2 macrophage polarization. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2025, 398, 855–865. [Google Scholar] [CrossRef]
- Sun, R.; Han, R.; McCornack, C.; Khan, S.; Tabor, G.T.; Chen, Y.; Hou, J.; Jiang, H.; Schoch, K.M.; Mao, D.D.; et al. TREM2 inhibition triggers antitumor cell activity of myeloid cells in glioblastoma. Sci. Adv. 2023, 9, eade3559. [Google Scholar] [CrossRef]
- Xiang, Y.; Miao, H. Lipid Metabolism in Tumor-Associated Macrophages. In Lipid Metabolism in Tumor Immunity; Springer: Berlin, Germany, 2025; Available online: https://link.springer.com/chapter/10.1007/978-981-33-6785-2_6 (accessed on 24 March 2025).
- Qiao, X.; Hu, Z.; Xiong, F.; Yang, Y.; Peng, C.; Wang, D.; Li, X. Lipid metabolism reprogramming in tumor-associated macrophages and implications for therapy. Lipids Health Dis. 2023, 22, 45. [Google Scholar] [CrossRef]
- Wu, H.; Han, Y.; Rodriguez Sillke, Y.; Deng, H.; Siddiqui, S.; Treese, C.; Schmidt, F.; Friedrich, M.; Keye, J.; Wan, J.; et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol. Med. 2019, 11, e10698. [Google Scholar] [CrossRef]
- Huang, J.; Tsang, W.Y.; Fang, X.-N.; Zhang, Y.; Luo, J.; Gong, L.-Q.; Zhang, B.-F.; Wong, C.N.; Li, Z.-H.; Liu, B.-L.; et al. FASN Inhibition Decreases MHC-I Degradation and Synergizes with PD-L1 Checkpoint Blockade in Hepatocellular Carcinoma. Cancer Res. 2024, 84, 855–871. [Google Scholar] [CrossRef]
- Duong, L.K.; Corbali, H.I.; Riad, T.S.; Ganjoo, S.; Nanez, S.; Voss, T.; Barsoumian, H.B.; Welsh, J.; Cortez, M.A. Lipid metabolism in tumor immunology and immunotherapy. Front. Oncol. 2023, 13, 1187279. [Google Scholar] [CrossRef]
- Zhu, H.; Yu, X.; Zhang, S.; Shu, K. Targeting the Complement Pathway in Malignant Glioma Microenvironments. Front. Cell Dev. Biol. 2021, 9, 657472. [Google Scholar] [CrossRef]
- Piao, C.; Zhang, W.M.; Li, T.T.; Zhang, C.C.; Qui, S.; Liu, Y.; Liu, S.; Jin, M.; Jia, L.X.; Song, W.C.; et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp. Cell Res. 2018, 366, 127–138. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Daugan, M.V.; Noé, R.; Petitprez, F.; Vano, Y.A.; Sanchez-Salas, R.; Becht, E.; Meilleroux, J.; Le Clec’h, B.; Giraldo, N.A.; et al. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol. Res. 2019, 7, 1091–1105. [Google Scholar] [CrossRef]
- Deng, Y.; Hu, J.C.; He, S.H.; Lou, B.; Ding, T.B.; Yang, J.T.; Mo, M.G.; Ye, D.Y.; Zhou, L.; Jiang, X.C.; et al. Sphingomyelin synthase 2 facilitates M2-like macrophage polarization and tumor progression in a mouse model of triple-negative breast cancer. Acta Pharmacol. Sin. 2021, 42, 149–159. [Google Scholar] [CrossRef]
- Bi, J.; Khan, A.; Tang, J.; Armando, A.M.; Wu, S.; Zhang, W.; Gimple, R.C.; Reed, A.; Jing, H.; Koga, T.; et al. Targeting glioblastoma signaling and metabolism with a re-purposed brain-penetrant drug. Cell Rep. 2021, 37, 109957. [Google Scholar] [CrossRef]
- Hernández, A.; Domènech, M.; Muñoz-Mármol, A.M.; Carrato, C.; Balana, C. Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment. Cells 2021, 10, 3529. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Ochoa, A.C.; Al-Khami, A.A. Arginine Metabolism in Myeloid Cells Shapes Innate and Adaptive Immunity. Front. Immunol. 2017, 8, 93. [Google Scholar] [CrossRef]
- Pilanc, P.; Wojnicki, K.; Roura, A.J.; Cyranowski, S.; Ellert-Miklaszewska, A.; Ochocka, N.; Gielniewski, B.; Grzybowski, M.M.; Błaszczyk, R.; Stańczak, P.S.; et al. A Novel Oral Arginase 1/2 Inhibitor Enhances the Antitumor Effect of PD-1 Inhibition in Murine Experimental Gliomas by Altering the Immunosuppressive Environment. Front. Oncol. 2021, 11, 703465. [Google Scholar] [CrossRef]
- Hajji, N.; Garcia-Revilla, J.; Soto, M.S.; Perryman, R.; Symington, J.; Quarles, C.C.; Healey, D.R.; Guo, Y.; Orta-Vázquez, M.L.; Mateos-Cordero, S.; et al. Arginine deprivation alters microglial polarity and synergizes with radiation to eradicate non-arginine-auxotrophic glioblastoma tumors. J. Clin. Investig. 2022, 132, e142137. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Dang, W.-Q.; Cao, M.-F.; Xiao, J.-F.; Lv, S.-Q.; Jiang, W.-J.; Yao, Z.-H.; Lu, H.-M.; Miao, J.-Y.; et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Investig. 2020, 100, 619–629. [Google Scholar] [CrossRef]
- Cui, X.; Tan Morales, R.T.; Qian, W.; Wang, H.; Gagner, J.P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking Macrophage-associated Immunosuppression for Regulating Glioblastoma Angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef]
- Halaby, M.J.; McGaha, T.L. 2-HG modulates glioma macrophages via Trp metabolism. Nat. Cancer 2021, 2, 677–679. [Google Scholar] [CrossRef]
- Thomas, R.P.; Nagpal, S.; Iv, M.; Soltys, S.G.; Bertrand, S.; Pelpola, J.S.; Ball, R.; Yang, J.; Sundaram, V.; Lavezo, J.; et al. Macrophage Exclusion after Radiation Therapy (MERT): A First in Human Phase I/II Trial using a CXCR4 Inhibitor in Glioblastoma. Clin. Cancer Res. 2019, 25, 6948–6957. [Google Scholar] [CrossRef]
- Recht, L.D. A Follow-Up Study to Add Whole Brain Radiotherapy (WBRT) to Standard Temozolomide Chemo-Radiotherapy in Newly Diagnosed Glioblastoma (GBM) Treated With 4 Weeks of Continuous Infusion Plerixafor. 2024. Clinicaltrials.gov. Available online: https://clinicaltrials.gov/study/NCT03746080 (accessed on 12 March 2025).
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef]
- Daiichi Sankyo. An Open Label Phase 1b&2 Study of Orally Administered PLX3397 in Combination with Radiation Therapy and Temozolomide in Patients with Newly Diagnosed Glioblastoma. 2020. Clinicaltrials.gov. Available online: https://clinicaltrials.gov/study/NCT01790503 (accessed on 12 March 2025).
- Novartis Pharmaceuticals. A Phase I&II, Open-Label, Multi-Center Study of the Safety and Efficacy of BLZ945 as Single Agent and in Combination with PDR001 in Adults Patients with Advanced Solid Tumors. 2023. Clinicaltrials.gov. Available online: https://clinicaltrials.gov/study/NCT02829723 (accessed on 12 March 2025).
- Jiang, H.; Yu, K.; Cui, Y.; Ren, X.; Li, M.; Yang, C.; Zhao, X.; Zhu, Q.; Lin, S. Combination of Immunotherapy and Radiotherapy for Recurrent Malignant Gliomas: Results from a Prospective Study. Front. Immunol. 2021, 12, 632547. [Google Scholar] [CrossRef]
- Desjardins, A.; Chandramohan, V.; Landi, D.B.; Johnson, M.O.; Khasraw, M.; Peters, K.B.; Low, J.; Herndon, J.E.; Threatt, S.; Bullock, C.A.; et al. A phase 1 trial of D2C7-it in combination with an Fc-engineered anti-CD40 monoclonal antibody (2141-V11) administered intratumorally via convection-enhanced delivery for adult patients with recurrent malignant glioma (MG). J. Clin. Oncol. 2022, 40 (Suppl. S16), e14015. [Google Scholar] [CrossRef]
- Youssef, G.; Lathia, J.; Lee, E.Q.; Chukwueke, U.N.; Lauko, A.; Batchelor, T.; Aquilanti, E.; Nayak, L.; Myers, A.; Russ, A.; et al. Phase 1b/2a study evaluating the combination of MN-166 (ibudilast) and temozolomide in patients with newly diagnosed and recurrent glioblastoma (GBM). J. Clin. Oncol. 2024, 42 (Suppl. S16), 2016. [Google Scholar] [CrossRef]
- Lee, E.Q.; Duda, D.G.; Muzikansky, A.; Gerstner, E.R.; Kuhn, J.G.; Reardon, D.A.; Nayak, L.; Norden, A.D.; Doherty, L.; LaFrankie, D.; et al. Phase I and Biomarker Study of Plerixafor and Bevacizumab in Recurrent High-Grade Glioma. Clin. Cancer Res. 2018, 24, 4643–4649. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Treatment | Mechanism | TAM Effect |
|---|---|---|---|
| Depletion | TG100-115 [27] | PI3K γ inhibition | Suppresses microglia/TAM accumulation and secretion of IL-11 |
| BLZ945 + OSI906 [32] | CSF-1R + IGF-1R inhibition | Prevent immunosuppressive TAM trafficking | |
| PLX3397 + cediranib [33] | CSF-1R + VEGFR2 inhibition | Prevent immunosuppressive TAM trafficking and reduce angiogenesis | |
| NE-siRNA-CD73 [34] | CD73 inhibition | Reduce Tregs, microglia, and macrophages | |
| CCX872/Anti-PD1 [35] | CCL2/CCR2 axis disruption | Reduce tumor-associated MDSCs and T lymphocyte exhaustion | |
| CCR2 knockout [36] | CCL2/CCR2 axis disruption | Decrease TAM infiltration | |
| Celecoxib [37] | CCL2 and CXCL10 inhibition | Reduce microglia and macrophages | |
| 2-(a-naphthoyl) ethyltrimethylammonium iodide [38] | Chemerin/CMKLR1 pathway disruption | Reduce TAM infiltration | |
| R848 via CDNP [49] | TLR7/8 agonism | Increase pro-inflammatory cytokines and reduce immunosuppressive TAM infiltration | |
| LY2228820 [40] | p38 MAPK inhibitor | Inhibit macrophage aggregation | |
| CDX-LIPO [41] | Target PI3K/mTOR | Promote pro-inflammatory phenotype and deplete immunosuppressive TAMs | |
| Ponatinib [42] | Inhibit PD-L1 | Deplete immunosuppressive TAMs | |
| Reprogramming | Nivolumab/Bevacizumab [44] | Inhibit PD-L1 pathway | Regulate TAM polarization |
| PD-L1 knockout [45] | Inhibit PD-L1 pathway via dual-sgRNA CRISPR/Cas9 | Upregulate pro-inflammatory phenotype, downregulate immunosuppressive phenotype | |
| a-lactose [47] | Gal-9/Tim-3 blockade | Inhibit immunosuppressive polarization and VEGF release | |
| Anti-Tim-3 antibody [48] | Gal-9/Tim-3 blockade | Inhibit immunosuppressive phenotype | |
| LGALS9-targeted shRNAs [48] | Gal-9/Tim-3 blockade | Inhibit immunosuppressive phenotype | |
| ITGB3 siRNA/anti-MFG-E8 antibody [50] | MFG-E8 pathway | Increase M1-like microglia, decrease M2-like microglia | |
| MLT-748 [51] | MALT1 inhibition | Reverses immunosuppressive polarization | |
| Toosendanin (TSN) [52] | IL-10 inhibition | Reduce immunosuppressive TAM markers and promote pro-inflammatory cytokines | |
| mRNA-NPs [53] | IRF5/IKKb expression | Upregulate pro-inflammatory phenotype, downregulate immunosuppressive phenotype | |
| CpG-Au-NPs [54] | Enhance immunosuppressive macrophage repolarization | Upregulate pro-inflammatory phenotype, downregulate immunosuppressive phenotype | |
| microRNA-25 antibody (antimiR-25) [55] | STING pathway suppression | Promotes pro-inflammatory phenotype polarization | |
| JMS-053 [56] | PRL inhibition | Induces apoptosis and promotes pro-inflammatory polarization | |
| USP9X knockout [57] | De-stabilizes TRRAP protein | Lowers TRRAP expression and shifts TAMs towards pro-inflammatory phenotype | |
| TREM2 knockout [58] | Decreased TREM2 activation | Promotes pro-inflammatory polarization | |
| SMS2 knockout [67] | Inhibition of sphingomyelin production | Suppress immunosuppressive TAM polarization | |
| Targeting downstream signals | OAT-1746 [71] | Inhibiting ARG1/2 | Polarize TAM to pro-pro-inflammatory phenotype |
| ADI-PEG20 [72] | Arginine depletion | Promote pro-inflammatory polarization and increase radiosensitivity of GBM cells | |
| BX471 and maraviroc [73] | CCL8-antibody and ERK1/2 inhibition | Decrease macrophage-derived CCL8 | |
| SCH772984 [73] | ERK1/2 inhibition | Reduce macrophage-derived CCL8 and decrease pseudopodia formation | |
| Cilengitide + LY-364947 [74] | Inhibition of integrin αvβ3 and TGF-β, respectively | Inhibit endothelial cells-macrophage interaction and reduce angiogenesis | |
| AhR-inhibitor [76] | Inhibit AhR-kynurenine interaction | Reverse immunosuppression |
| ID | Author | Phase | Mechanism | TAM Effect | Patient Population | Outcomes | Recruitment Status |
|---|---|---|---|---|---|---|---|
| NCT01977677 [77] | Thomas et al. | I/II | Inhibition of SDF-1/CXC4 axis | Prevent TAM recruitment | ndGBM | mOS 21.3 months, PFS 14.5 months | Completed |
| NCT03746080 [78] | Recht et al. | II | Inhibition of SDF-1/CXC4 axis + WBRT + SOC | Prevent TAM recruitment | ndGBM | 6-month PFS rate 91.7%, mOS 15.1 months | Active, not recruiting |
| NCT01349036 [79] | Butowski et al. | II | PLX3397 (CSF1-R inhibition) | Deplete immunosuppressive TAMs | rGBM | 6-month PFS rate 8.8% | Terminated |
| NCT01790503 [80] | Sankyo et al. | I/II | PLX3397 + SOC | Deplete immunosuppressive TAMs | ndGBM | mPFS 6.7 months | Completed |
| NCT02829723 [81] | Novartis | I/II | BLZ945 (CSF1-R inhibition) | Deplete immunosuppressive TAMs | rGBM | 6-month PFS rate 15.2% | Terminated |
| NCT03392545 [82] | Jiang et al. | I | GM-CSF + poly I/C + radiation | Reprogramming TAM polarization | rGBM | No results posted | Unknown |
| NCT04547777 [83] | Desjardins et al. | I | Anti-CD40 + D2C7-IT | Reprogramming TAM polarization | rGBM | No results posted | Recruiting |
| NCT03782415 [84] | Youssef et al. | I/II | Inhibition of MIF | Prevent MDSC differentiation into immunosuppressive TAM | ndGBM and rGBM | mOS 21.0 (ndGBM) and 8.6 (rGBM) | Active, not recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eckert, T.; Walton, C.; Bell, M.; Small, C.; Rowland, N.C.; Rivers, C.; Zukas, A.; Lindhorst, S.; Fecci, P.; Strickland, B.A. The Basis for Targeting the Tumor Macrophage Compartment in Glioblastoma Immunotherapy. Cancers 2025, 17, 1631. https://doi.org/10.3390/cancers17101631
Eckert T, Walton C, Bell M, Small C, Rowland NC, Rivers C, Zukas A, Lindhorst S, Fecci P, Strickland BA. The Basis for Targeting the Tumor Macrophage Compartment in Glioblastoma Immunotherapy. Cancers. 2025; 17(10):1631. https://doi.org/10.3390/cancers17101631
Chicago/Turabian StyleEckert, Thomas, Chase Walton, Marcus Bell, Coulter Small, Nathan C. Rowland, Charlotte Rivers, Alicia Zukas, Scott Lindhorst, Peter Fecci, and Ben A. Strickland. 2025. "The Basis for Targeting the Tumor Macrophage Compartment in Glioblastoma Immunotherapy" Cancers 17, no. 10: 1631. https://doi.org/10.3390/cancers17101631
APA StyleEckert, T., Walton, C., Bell, M., Small, C., Rowland, N. C., Rivers, C., Zukas, A., Lindhorst, S., Fecci, P., & Strickland, B. A. (2025). The Basis for Targeting the Tumor Macrophage Compartment in Glioblastoma Immunotherapy. Cancers, 17(10), 1631. https://doi.org/10.3390/cancers17101631