The Metabolic Orchestration of Immune Evasion in Glioblastoma: From Molecular Perspectives to Therapeutic Vulnerabilities


Simple Summary
Abstract
1. Introduction
2. Isocitrate Dehydrogenase Mutational Status Affects Glioma Classification
3. The Role of Glycolysis in GBM Immunosuppression
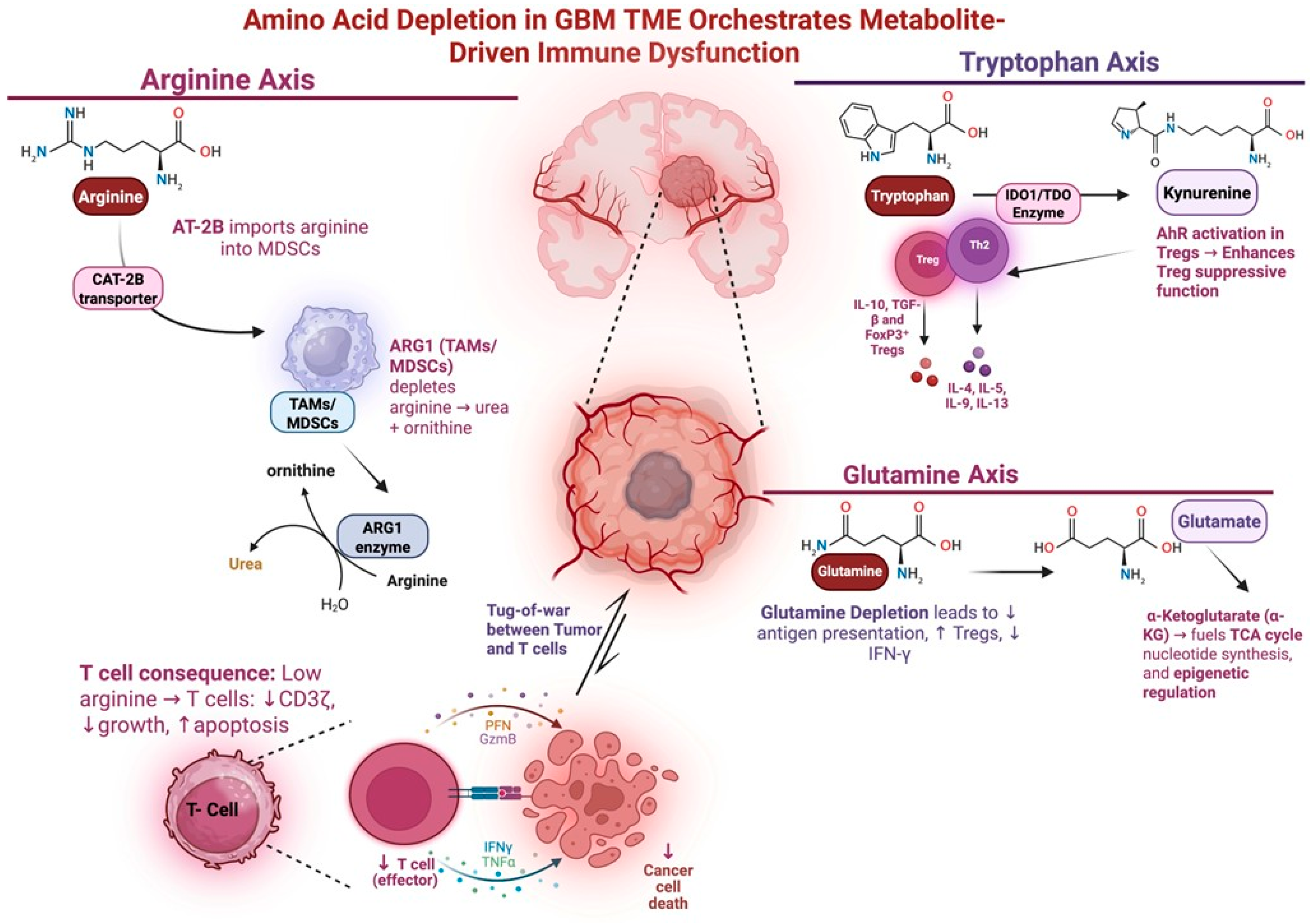
4. Amino Acid Metabolism and Immunosuppression
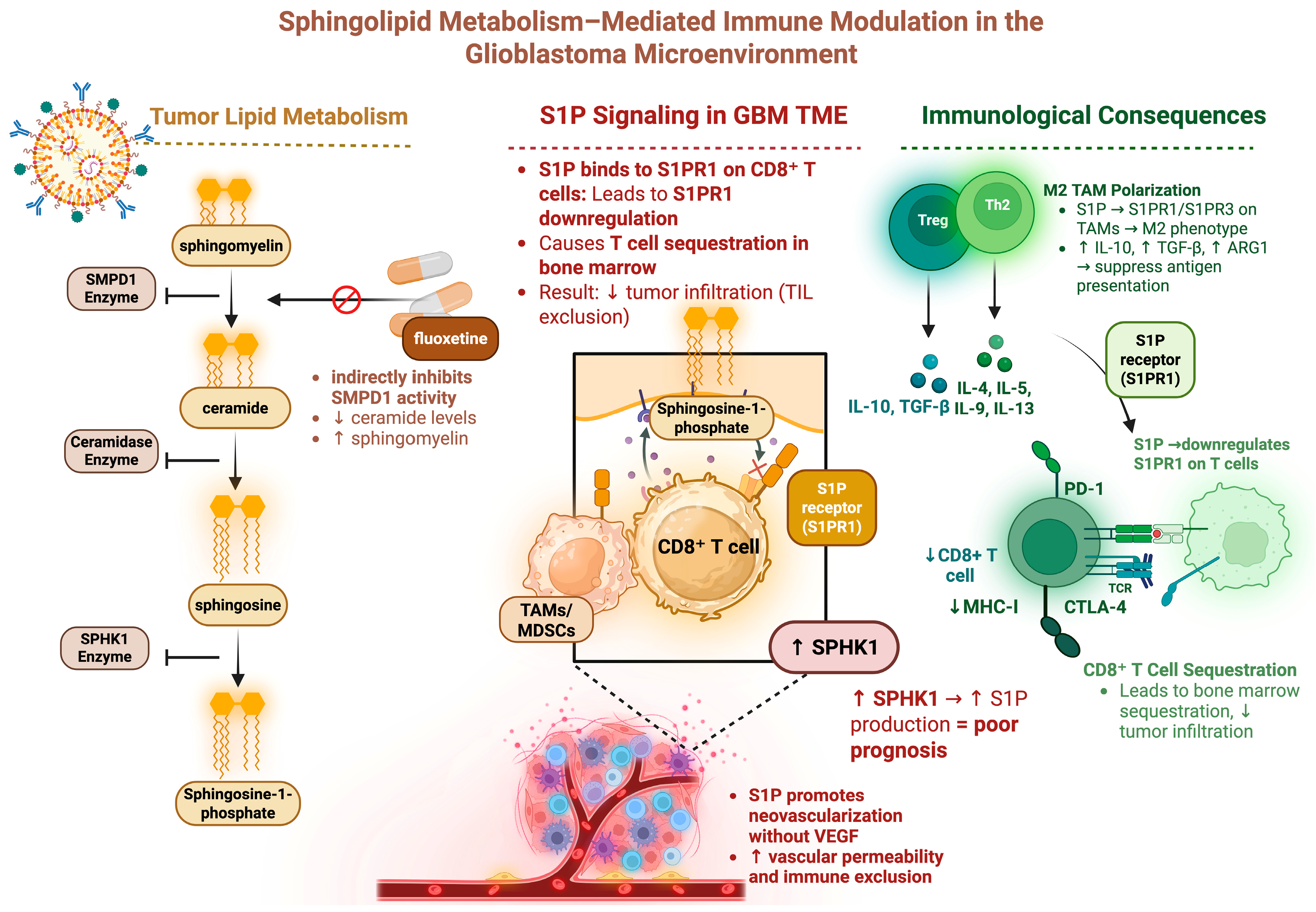
5. Sphingolipid Metabolism Affects Immune Cell Trafficking
6. Hypoxia in the GBM TME Contributes to Immune Resistance
7. The Relevance of Metabolic Alteration of the GBM TME on Immunotherapy Efficacy
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. Baltim. Md 1950 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Khagi, S.; Kotecha, R.; Gatson, N.T.N.; Jeyapalan, S.; Abdullah, H.I.; Avgeropoulos, N.G.; Batzianouli, E.T.; Giladi, M.; Lustgarten, L.; Goldlust, S.A. Recent Advances in Tumor Treating Fields (TTFields) Therapy for Glioblastoma. Oncologist 2025, 30, oyae227. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Le, S.B.; Hutchinson, T.E.; Calinescu, A.-A.; Sebastian, M.; Jin, D.; Liu, T.; Ghiaseddin, A.; Rahman, M.; Tran, D.D. Tumor Treating Fields Dually Activate STING and AIM2 Inflammasomes to Induce Adjuvant Immunity in Glioblastoma. J. Clin. Investig. 2022, 132, e149258. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III Trial of Chemoradiotherapy with Temozolomide plus Nivolumab or Placebo for Newly Diagnosed Glioblastoma with Methylated MGMT Promoter. Neuro-Oncology 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- Thomas, A.A.; Fisher, J.L.; Ernstoff, M.S.; Fadul, C.E. Vaccine-Based Immunotherapy for Glioblastoma. CNS Oncol. 2013, 2, 331–349. [Google Scholar] [CrossRef]
- Abikenari, M.A.; Enayati, I.; Fountain, D.M.; Leite, M.I. Navigating Glioblastoma Therapy: A Narrative Review of Emerging Immunotherapeutics and Small-Molecule Inhibitors. Microbes Immun. 2024, 5075. [Google Scholar] [CrossRef]
- Zhao, J.; Ma, X.; Gao, P.; Han, X.; Zhao, P.; Xie, F.; Liu, M. Advancing Glioblastoma Treatment by Targeting Metabolism. Neoplasia 2024, 51, 100985. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer Metabolism: A Therapeutic Perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, F.; D’Aprile, S.; Denaro, S.; Pavone, A.M.; Alberghina, C.; Zappalà, A.; Giuffrida, R.; Salvatorelli, L.; Broggi, G.; Magro, G.G.; et al. Epigenetics and Metabolism Reprogramming Interplay into Glioblastoma: Novel Insights on Immunosuppressive Mechanisms. Antioxidants 2023, 12, 220. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan Metabolism as a Common Therapeutic Target in Cancer, Neurodegeneration and Beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The Adenosine Pathway in Immuno-Oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of Antitumor T Cell Immunity by the Oncometabolite (R)-2-Hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Certo, M.; Tsai, C.-H.; Pucino, V.; Ho, P.-C.; Mauro, C. Lactate Modulation of Immune Responses in Inflammatory versus Tumour Microenvironments. Nat. Rev. Immunol. 2021, 21, 151–161. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of Immune Cells in Cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 Mediates Metabolic Responses to Intratumoral Hypoxia and Oncogenic Mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Miska, J.; Lee-Chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-Rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. HIF-1α Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019, 27, 226–237.e4. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological Mechanisms of the Antitumor Effects of Supplemental Oxygenation. Sci. Transl. Med. 2015, 7, 277ra30. [Google Scholar] [CrossRef] [PubMed]
- Adler, E.; Euler, H.V.; Günther, G.; Plass, M. isoCitric Dehydrogenase and Glutamic Acid Synthesis in Animal Tissues. Biochem. J. 1939, 33, 1028–1045. [Google Scholar] [CrossRef]
- Bernhard, C.; Reita, D.; Martin, S.; Entz-Werle, N.; Dontenwill, M. Glioblastoma Metabolism: Insights and Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 9137. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature 2010, 465, 966. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Kobayashi, A.; Cahill, D.P.; Wakimoto, H.; Tanaka, S. Molecular Biology and Novel Therapeutics for IDH Mutant Gliomas: The New Era of IDH Inhibitors. Biochim. Biophys. Acta BBA—Rev. Cancer 2024, 1879, 189102. [Google Scholar] [CrossRef]
- Choate, K.A.; Pratt, E.P.S.; Jennings, M.J.; Winn, R.J.; Mann, P.B. IDH Mutations in Glioma: Molecular, Cellular, Diagnostic, and Clinical Implications. Biology 2024, 13, 885. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Wang, N.; Yuan, Y.; Hu, T.; Xu, H.; Piao, H. Metabolism: An Important Player in Glioma Survival and Development. Discov. Oncol. 2024, 15, 577. [Google Scholar] [CrossRef]
- Velpula, K.K.; Bhasin, A.; Asuthkar, S.; Tsung, A.J. Combined Targeting of PDK1 and EGFR Triggers Regression of Glioblastoma by Reversing the Warburg Effect. Cancer Res. 2013, 73, 7277–7289. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Oda, Y.; Seino, Y.; Yamamoto, T.; Inagaki, N.; Yano, H.; Imura, H.; Shigemoto, R.; Kikuchi, H. Distribution of the Glucose Transporters in Human Brain Tumors. Cancer Res. 1992, 52, 3972–3979. [Google Scholar] [PubMed]
- Guda, M.R.; Labak, C.M.; Omar, S.I.; Asuthkar, S.; Airala, S.; Tuszynski, J.; Tsung, A.J.; Velpula, K.K. GLUT1 and TUBB4 in Glioblastoma Could Be Efficacious Targets. Cancers 2019, 11, 1308. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Scharping, N.E.; Goldrath, A.W. CD8+ T Cell Metabolism in Infection and Cancer. Nat. Rev. Immunol. 2021, 21, 718–738. [Google Scholar] [CrossRef]
- Guo, D.; Tong, Y.; Jiang, X.; Meng, Y.; Jiang, H.; Du, L.; Wu, Q.; Li, S.; Luo, S.; Li, M.; et al. Aerobic Glycolysis Promotes Tumor Immune Evasion by Hexokinase2-Mediated Phosphorylation of IκBα. Cell Metab. 2022, 34, 1312–1324.e6. [Google Scholar] [CrossRef]
- Liang, X.; Wang, Z.; Dai, Z.; Zhang, H.; Zhang, J.; Luo, P.; Liu, Z.; Liu, Z.; Yang, K.; Cheng, Q.; et al. Glioblastoma Glycolytic Signature Predicts Unfavorable Prognosis, Immunological Heterogeneity, and ENO1 Promotes Microglia M2 Polarization and Cancer Cell Malignancy. Cancer Gene Ther. 2023, 30, 481–496. [Google Scholar] [CrossRef]
- Geeraerts, X.; Bolli, E.; Fendt, S.-M.; Van Ginderachter, J.A. Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity. Front. Immunol. 2017, 8, 289. [Google Scholar] [CrossRef]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef]
- Jackson, C.M.; Pant, A.; Dinalankara, W.; Choi, J.; Jain, A.; Nitta, R.; Yazigi, E.; Saleh, L.; Zhao, L.; Nirschl, T.R.; et al. The Cytokine Meteorin-like Inhibits Anti-Tumor CD8+ T Cell Responses by Disrupting Mitochondrial Function. Immunity 2024, 57, 1864–1877.e9. [Google Scholar] [CrossRef]
- Waickman, A.T.; Powell, J.D. mTOR, Metabolism, and the Regulation of T-Cell Differentiation and Function. Immunol. Rev. 2012, 249, 43–58. [Google Scholar] [CrossRef]
- Gupta, S.; Roy, A.; Dwarakanath, B.S. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front. Oncol. 2017, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.H.; Jain, S.; Aghi, M.K. Metabolic Drivers of Invasion in Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 683276. [Google Scholar] [CrossRef] [PubMed]
- Torrini, C.; Nguyen, T.T.T.; Shu, C.; Mela, A.; Humala, N.; Mahajan, A.; Seeley, E.H.; Zhang, G.; Westhoff, M.-A.; Karpel-Massler, G.; et al. Lactate Is an Epigenetic Metabolite That Drives Survival in Model Systems of Glioblastoma. Mol. Cell 2022, 82, 3061–3076.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dai, Z.; Zhang, H.; Liang, X.; Zhang, X.; Wen, Z.; Luo, P.; Zhang, J.; Liu, Z.; Zhang, M.; et al. Tumor-Secreted Lactate Contributes to an Immunosuppressive Microenvironment and Affects CD8 T-Cell Infiltration in Glioblastoma. Front. Immunol. 2023, 14, 894853. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Hortová-Kohoutková, M.; Filep, J.G.; Frič, J. Editorial: Lactate Metabolism and Regulation of the Immune Response. Front. Immunol. 2022, 13, 1103379. [Google Scholar] [CrossRef]
- Brown, T.P.; Ganapathy, V. Lactate/GPR81 Signaling and Proton Motive Force in Cancer: Role in Angiogenesis, Immune Escape, Nutrition, and Warburg Phenomenon. Pharmacol. Ther. 2020, 206, 107451. [Google Scholar] [CrossRef]
- Mehla, K.; Singh, P.K. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef]
- Cui, H.; Xie, N.; Banerjee, S.; Ge, J.; Jiang, D.; Dey, T.; Matthews, Q.L.; Liu, R.-M.; Liu, G. Lung Myofibroblasts Promote Macrophage Profibrotic Activity through Lactate-Induced Histone Lactylation. Am. J. Respir. Cell Mol. Biol. 2021, 64, 115–125. [Google Scholar] [CrossRef]
- Longhitano, L.; Vicario, N.; Tibullo, D.; Giallongo, C.; Broggi, G.; Caltabiano, R.; Barbagallo, G.M.V.; Altieri, R.; Baghini, M.; Di Rosa, M.; et al. Lactate Induces the Expressions of MCT1 and HCAR1 to Promote Tumor Growth and Progression in Glioblastoma. Front. Oncol. 2022, 12, 871798. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Arismendi-Morillo, G.; Zuccoli, G.; Lee, D.C.; Duraj, T.; Elsakka, A.M.; Maroon, J.C.; Mukherjee, P.; Ta, L.; Shelton, L.; et al. Metabolic Management of Microenvironment Acidity in Glioblastoma. Front. Oncol. 2022, 12, 968351. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of Microenvironment Acidity Reverses Anergy in Human and Murine Tumor-Infiltrating T Lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Cortes Ballen, A.I.; Amosu, M.; Ravinder, S.; Chan, J.; Derin, E.; Slika, H.; Tyler, B. Metabolic Reprogramming in Glioblastoma Multiforme: A Review of Pathways and Therapeutic Targets. Cells 2024, 13, 1574. [Google Scholar] [CrossRef] [PubMed]
- Maus, A.; Peters, G.J. Glutamate and α-Ketoglutarate: Key Players in Glioma Metabolism. Amino Acids 2017, 49, 21–32. [Google Scholar] [CrossRef]
- Srivastava, S.; Anbiaee, R.; Houshyari, M.; Laxmi; Sridhar, S.B.; Ashique, S.; Hussain, S.; Kumar, S.; Taj, T.; Akbarnejad, Z.; et al. Amino Acid Metabolism in Glioblastoma Pathogenesis, Immune Evasion, and Treatment Resistance. Cancer Cell Int. 2025, 25, 89. [Google Scholar] [CrossRef]
- Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 2020, 10, 429. [Google Scholar] [CrossRef]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. Baltim. Md 1950 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-Arginine Availability Regulates T-Lymphocyte Cell-Cycle Progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-Dioxygenase and Tumor-Induced Tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Nguyen, B.; Genet, M.; Kim, M.; Chen, P.; Mi, X.; et al. Tumor Cell IDO Enhances Immune Suppression and Decreases Survival Independent of Tryptophan Metabolism in Glioblastoma. Clin. Cancer Res. 2021, 27, 6514–6528. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T Cell Apoptosis by Tryptophan Catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Du, L.; Xing, Z.; Tao, B.; Li, T.; Yang, D.; Li, W.; Zheng, Y.; Kuang, C.; Yang, Q. Both IDO1 and TDO Contribute to the Malignancy of Gliomas via the Kyn–AhR–AQP4 Signaling Pathway. Signal Transduct. Target. Ther. 2020, 5, 10. [Google Scholar] [CrossRef]
- Li, A.; Barsoumian, H.B.; Schoenhals, J.E.; Caetano, M.S.; Wang, X.; Menon, H.; Valdecanas, D.R.; Niknam, S.; Younes, A.I.; Cortez, M.A.; et al. IDO1 Inhibition Overcomes Radiation-Induced “Rebound Immune Suppression” by Reducing Numbers of IDO1-Expressing Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Int. J. Radiat. Oncol. 2019, 104, 903–912. [Google Scholar] [CrossRef]
- Ladomersky, E.; Zhai, L.; Lenzen, A.; Lauing, K.L.; Qian, J.; Scholtens, D.M.; Gritsina, G.; Sun, X.; Liu, Y.; Yu, F.; et al. IDO1 Inhibition Synergizes with Radiation and PD-1 Blockade to Durably Increase Survival against Advanced Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2559–2573. [Google Scholar] [CrossRef]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.-J.; Hellmann, M.D.; Cervantes, A.; Ochoa de Olza, M.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef]
- Lukas, R.; Sachdev, S.; Kumthekar, P.; Dixit, K.; Grimm, S.; Gondi, V.; Sharp, L.; Lezon, R.; James, D.; Lesniak, M.; et al. CTIM-12. A phase 1 trial of immunoradiotherapy with the ido enzyme inhibitor (BMS-986205) and nivolumab in patients with newly diagnosed mgmt promoter unmethylated IDHwt glioblastoma. Neuro-Oncology 2021, 23, vi51–vi52. [Google Scholar] [CrossRef]
- Bomalaski, J.S.; Chen, K.-T.; Chuang, M.-J.; Liau, C.-T.; Peng, M.-T.; Chen, P.-Y.; Lee, C.-C.; Johnston, A.; Liu, H.-F.; Huang, Y.-L.S.; et al. Phase IB Trial of Pegylated Arginine Deiminase (ADI-PEG 20) plus Radiotherapy and Temozolomide in Patients with Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2022, 40, 2057. [Google Scholar] [CrossRef]
- Hou, X.; Chen, S.; Zhang, P.; Guo, D.; Wang, B. Targeted Arginine Metabolism Therapy: A Dilemma in Glioma Treatment. Front. Oncol. 2022, 12, 938847. [Google Scholar] [CrossRef] [PubMed]
- Hajji, N.; Garcia-Revilla, J.; Soto, M.S.; Perryman, R.; Symington, J.; Quarles, C.C.; Healey, D.R.; Guo, Y.; Orta-Vázquez, M.L.; Mateos-Cordero, S.; et al. Arginine Deprivation Alters Microglial Polarity and Synergizes with Radiation to Eradicate Non-Arginine-Auxotrophic Glioblastoma Tumors. J. Clin. Investig. 2022, 132, e142137. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zuo, M.; Li, W.; Chen, S.; Wang, Z.; Yuan, Y.; Yang, Y.; Liu, Y. A Novel Score System Based on Arginine Metabolism-Related Genes to Predict Prognosis, Characterize Immune Microenvironment, and Forecast Response to Immunotherapy in IDH-Wildtype Glioblastoma. Front. Pharmacol. 2023, 14, 1145828. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Naing, A.; Papadopoulos, K.P.; Pishvaian, M.J.; Rahma, O.; Hanna, G.J.; Garralda, E.; Saavedra, O.; Gogov, S.; Kallender, H.; Cheng, L.; et al. First-in-Human Phase 1 Study of the Arginase Inhibitor INCB001158 Alone or Combined with Pembrolizumab in Patients with Advanced or Metastatic Solid Tumours. BMJ Oncol. 2024, 3, e000249. [Google Scholar] [CrossRef]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.-R.; Ho, P.-C. Metabolic and Epigenetic Regulation of T-Cell Exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef]
- Das, A.; Hoare, M.; Davies, N.; Lopes, A.R.; Dunn, C.; Kennedy, P.T.F.; Alexander, G.; Finney, H.; Lawson, A.; Plunkett, F.J.; et al. Functional Skewing of the Global CD8 T Cell Population in Chronic Hepatitis B Virus Infection. J. Exp. Med. 2008, 205, 2111–2124. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Ernstoff, M.S.; Hernandez, C.; Atkins, M.; Zabaleta, J.; Sierra, R.; Ochoa, A.C. Arginase I–Producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma Are a Subpopulation of Activated Granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.S.; Braganhol, E.; Whiteside, T.L. Arginase-1+ Exosomes from Reprogrammed Macrophages Promote Glioblastoma Progression. Int. J. Mol. Sci. 2020, 21, 3990. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Zea, A.H.; DeSalvo, J.; Culotta, K.S.; Zabaleta, J.; Quiceno, D.G.; Ochoa, J.B.; Ochoa, A.C. L-Arginine Consumption by Macrophages Modulates the Expression of CD3 Zeta Chain in T Lymphocytes. J. Immunol. Baltim. Md 1950 2003, 171, 1232–1239. [Google Scholar] [CrossRef]
- Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules 2020, 10, 1357. [Google Scholar] [CrossRef] [PubMed]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A Metabolic Shift Favoring Sphingosine 1-Phosphate at the Expense of Ceramide Controls Glioblastoma Angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef]
- Song, Z.; Zhao, Z.; Liu, X.; Song, Y.; Zhu, S.; Jia, Z.; Li, Y.; Wang, Z.; Sun, B.; Jin, Q.; et al. Sphingosine Kinase 1 Promotes M2 Macrophage Infiltration and Enhances Glioma Cell Migration via the JAK2/STAT3 Pathway. Sci. Rep. 2025, 15, 4152. [Google Scholar] [CrossRef]
- Li, W.; Cai, H.; Ren, L.; Yang, Y.; Yang, H.; Liu, J.; Li, S.; Zhang, Y.; Zheng, X.; Tan, W.; et al. Sphingosine Kinase 1 Promotes Growth of Glioblastoma by Increasing Inflammation Mediated by the NF-κB/IL-6/STAT3 and JNK/PTX3 Pathways. Acta Pharm. Sin. B 2022, 12, 4390–4406. [Google Scholar] [CrossRef]
- Bi, J.; Khan, A.; Tang, J.; Armando, A.M.; Wu, S.; Zhang, W.; Gimple, R.C.; Reed, A.; Jing, H.; Koga, T.; et al. Targeting Glioblastoma Signaling and Metabolism with a Re-Purposed Brain-Penetrant Drug. Cell Rep. 2021, 37, 109957. [Google Scholar] [CrossRef]
- Sousa, N.; Geiß, C.; Bindila, L.; Lieberwirth, I.; Kim, E.; Régnier-Vigouroux, A. Targeting Sphingolipid Metabolism with the Sphingosine Kinase Inhibitor SKI-II Overcomes Hypoxia-Induced Chemotherapy Resistance in Glioblastoma Cells: Effects on Cell Death, Self-Renewal, and Invasion. BMC Cancer 2023, 23, 762. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T Cells in Bone Marrow in the Setting of Glioblastoma and Other Intracranial Tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Guo, X.-D.; Ji, J.; Xue, T.-F.; Sun, Y.-Q.; Guo, R.-B.; Cheng, H.; Sun, X.-L. FTY720 Exerts Anti-Glioma Effects by Regulating the Glioma Microenvironment Through Increased CXCR4 Internalization by Glioma-Associated Microglia. Front. Immunol. 2020, 11, 178. [Google Scholar] [CrossRef]
- Feldman, L. Hypoxia within the Glioblastoma Tumor Microenvironment: A Master Saboteur of Novel Treatments. Front. Immunol. 2024, 15, 1384249. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, A.C.; Darnell, N.G.; Hoefflin, R.; Simkin, D.; Mount, C.W.; Gonzalez Castro, L.N.; Harnik, Y.; Dumont, S.; Hirsch, D.; Nomura, M.; et al. Integrative Spatial Analysis Reveals a Multi-Layered Organization of Glioblastoma. Cell 2024, 187, 2485–2501.e26. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-Inducible Factor 1 and VEGF Upregulate CXCR4 in Glioblastoma: Implications for Angiogenesis and Glioma Cell Invasion. Lab. Investig. J. Tech. Methods Pathol. 2006, 86, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, R.C.; Brennan, C.E.; Kingsmore, K.M.; Munson, J.M. Convective Forces Increase CXCR4-Dependent Glioblastoma Cell Invasion in GL261 Murine Model. Sci. Rep. 2018, 8, 17057. [Google Scholar] [CrossRef]
- Wu, A.; Maxwell, R.; Xia, Y.; Cardarelli, P.; Oyasu, M.; Belcaid, Z.; Kim, E.; Hung, A.; Luksik, A.S.; Garzon-Muvdi, T.; et al. Combination Anti-CXCR4 and Anti-PD-1 Immunotherapy Provides Survival Benefit in Glioblastoma through Immune Cell Modulation of Tumor Microenvironment. J. Neurooncol. 2019, 143, 241–249. [Google Scholar] [CrossRef]
- Wei, R.; Li, J.; Lin, W.; Pang, X.; Yang, H.; Lai, S.; Wei, X.; Jiang, X.; Yuan, Y.; Yang, R. Nanoparticle-Mediated Blockade of CXCL12/CXCR4 Signaling Enhances Glioblastoma Immunotherapy: Monitoring Early Responses with MRI Radiomics. Acta Biomater. 2024, 177, 414–430. [Google Scholar] [CrossRef]
- Xue, H.; Yuan, G.; Guo, X.; Liu, Q.; Zhang, J.; Gao, X.; Guo, X.; Xu, S.; Li, T.; Shao, Q.; et al. A Novel Tumor-Promoting Mechanism of IL6 and the Therapeutic Efficacy of Tocilizumab: Hypoxia-Induced IL6 Is a Potent Autophagy Initiator in Glioblastoma via the p-STAT3-MIR155-3p-CREBRF Pathway. Autophagy 2016, 12, 1129–1152. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, L.; Gong, S.; Xiong, S.; Wang, J.; Zou, D.; Pan, J.; Deng, Y.; Yan, Q.; Wu, N.; et al. HIF1α/HIF2α–Sox2/Klf4 Promotes the Malignant Progression of Glioblastoma via the EGFR–PI3K/AKT Signalling Pathway with Positive Feedback under Hypoxia. Cell Death Dis. 2021, 12, 312. [Google Scholar] [CrossRef]
- He, X.-C.; Wang, J.; Shi, M.-Y.; Liu, C.-M.; Teng, Z.-Q. Hypoxia-Induced One-Carbon Metabolic Reprogramming in Glioma Stem-like Cells. Life Med. 2023, 2, lnad048. [Google Scholar] [CrossRef]
- Wu, C.Y.-J.; Chen, Y.; Lin, Y.-J.; Wei, K.-C.; Chang, K.-Y.; Feng, L.-Y.; Chen, K.-T.; Li, G.; Ren, A.L.; Nitta, R.T.; et al. Tumor-Associated Microglia Secrete Extracellular ATP to Support Glioblastoma Progression. Cancer Res. 2024, 84, 4017–4030. [Google Scholar] [CrossRef]
- Xia, C.; Yin, S.; To, K.K.W.; Fu, L. CD39/CD73/A2AR Pathway and Cancer Immunotherapy. Mol. Cancer 2023, 22, 44. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A Adenosine Receptor Protects Tumors from Antitumor T Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, H.; Xu, J.; Lu, Y.; Ji, X.; Yao, Y.; Chao, H.; Zhang, J.; Zhang, X.; Yao, S.; et al. Different T-Cell Subsets in Glioblastoma Multiforme and Targeted Immunotherapy. Cancer Lett. 2021, 496, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting Immunosuppressive Adenosine in Cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune Profiling of Human Tumors Identifies CD73 as a Combinatorial Target in Glioblastoma. Nat. Med. 2020, 26, 39–46. [Google Scholar] [CrossRef]
- Bendell, J.; LoRusso, P.; Overman, M.; Noonan, A.M.; Kim, D.-W.; Strickler, J.H.; Kim, S.-W.; Clarke, S.; George, T.J.; Grimison, P.S.; et al. First-in-Human Study of Oleclumab, a Potent, Selective Anti-CD73 Monoclonal Antibody, Alone or in Combination with Durvalumab in Patients with Advanced Solid Tumors. Cancer Immunol. Immunother. CII 2023, 72, 2443–2458. [Google Scholar] [CrossRef]
- Guha, P.; Heatherton, K.R.; O’Connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10, 655. [Google Scholar] [CrossRef]
- Maccari, M.; Baek, C.; Caccese, M.; Mandruzzato, S.; Fiorentino, A.; Internò, V.; Bosio, A.; Cerretti, G.; Padovan, M.; Idbaih, A.; et al. Present and Future of Immunotherapy in Patients With Glioblastoma: Limitations and Opportunities. The Oncologist 2024, 29, 289–302. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, F.; Ali, H.; Lathia, J.D.; Chen, P. Immunotherapy for Glioblastoma: Current State, Challenges, and Future Perspectives. Cell. Mol. Immunol. 2024, 21, 1354–1375. [Google Scholar] [CrossRef]
- Choudhary, N.; Osorio, R.C.; Oh, J.Y.; Aghi, M.K. Metabolic Barriers to Glioblastoma Immunotherapy. Cancers 2023, 15, 1519. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, M.; Wang, G.; Lai, W.; Liao, S.; Chen, Y.; Ning, Q.; Tang, S. Metabolic Cross-Talk between Glioblastoma and Glioblastoma-Associated Microglia/Macrophages: From Basic Insights to Therapeutic Strategies. Crit. Rev. Oncol. Hematol. 2025, 208, 104649. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Greenberg, P.D. Tolerance and Exhaustion: Defining Mechanisms of T Cell Dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs. Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bähr, O.; et al. Radiotherapy Combined with Nivolumab or Temozolomide for Newly Diagnosed Glioblastoma with Unmethylated MGMT Promoter: An International Randomized Phase III Trial. Neuro-Oncology 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin States Define Tumour-Specific T Cell Dysfunction and Reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with Temozolomide for Patients with Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral Oncolytic Herpes Virus G47∆ for Residual or Recurrent Glioblastoma: A Phase 2 Trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-Term Outcomes Following CAR T Cell Therapy: What We Know so Far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Yin, Y.; Rodriguez, J.L.; Li, N.; Thokala, R.; Nasrallah, M.P.; Hu, L.; Zhang, L.; Zhang, J.V.; Logun, M.T.; Kainth, D.; et al. Locally Secreted BiTEs Complement CAR T Cells by Enhancing Killing of Antigen Heterogeneous Solid Tumors. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 2537–2553. [Google Scholar] [CrossRef]
- Choi, B.D.; Gerstner, E.R.; Frigault, M.J.; Leick, M.B.; Mount, C.W.; Balaj, L.; Nikiforow, S.; Carter, B.S.; Curry, W.T.; Gallagher, K.; et al. Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N. Engl. J. Med. 2024, 390, 1290–1298. [Google Scholar] [CrossRef]
- Bagley, S.J.; Logun, M.; Fraietta, J.A.; Wang, X.; Desai, A.S.; Bagley, L.J.; Nabavizadeh, A.; Jarocha, D.; Martins, R.; Maloney, E.; et al. Intrathecal Bivalent CAR T Cells Targeting EGFR and IL13Rα2 in Recurrent Glioblastoma: Phase 1 Trial Interim Results. Nat. Med. 2024, 30, 1320–1329. [Google Scholar] [CrossRef]
- Baxter, M.E.; Miller, H.A.; Chen, J.; Williams, B.J.; Frieboes, H.B. Metabolomic Differentiation of Tumor Core versus Edge in Glioma. Neurosurg. Focus 2023, 54, E4. [Google Scholar] [CrossRef]
- Rahman, M.; Abbatematteo, J.; De Leo, E.K.; Kubilis, P.S.; Vaziri, S.; Bova, F.; Sayour, E.; Mitchell, D.; Quinones-Hinojosa, A. The Effects of New or Worsened Postoperative Neurological Deficits on Survival of Patients with Glioblastoma. J. Neurosurg. 2017, 127, 123–131. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Sensor/Pathway | Trigger in GBM TME | Immune Effects | Therapeutic Implications |
|---|---|---|---|
| METRNL | Secreted by checkpoint-expressing tumor-infiltrating T cells | Bioenergetic failure of T cells and diminished effector function | METRNL inhibition may rescue anti-tumor T cell function |
| mTOR | Glucose deprivation, chronic stress | Impaired effector function, metabolic arrest | Consider mTOR agonists or T-cell metabolic reprogramming |
| Lactic acid | Key byproduct of glycolysis | Promotes M2 polarization of TAMs, Treg expansion | Alleviating lactate-driven acidity may improve T cell function and immunotherapy efficacy |
| Trial ID | Study Start Date | Phase | Target | Other Results |
|---|---|---|---|---|
| NCT05106296 | 02/08/2022 | 1 (Recruiting) | IDO inhibitor | |
| NCT04049669 | 10/02/2019 | 2 (Recruiting) | IDO inhibitor | |
| NCT04047706 | 08/13/2019 | 1 (Active, not recruiting) | IDO1 inhibitor, anti-PD-1 | Dose limiting toxicity of the combination is reversible hepatic transaminitis, phase 2/3 trial is approved [70] |
| NCT03707457 | 03/22/2019 | 1 (Terminated) | IDO1 inhibitor, anti-PD-1, anti-CTLA-4, anti-GITR | |
| NCT04587830 | 09/14/2020 | 2 (Recruiting) | ADI-PEG 20 (arginine depletion) | In newly diagnosed GBM subjects, peripheral arginine levels were suppressed with reciprocally elevated citrulline for 4–6 weeks, and preliminary OS was reported to be encouraging [71] |
| Immune Cell Type | Metabolic Profile | Functional State in GBM | Immunological Role | Metabolic Dependencies |
|---|---|---|---|---|
| CD8+ Effector T cells | Aerobic glycolysis (Warburg effect) | Exhausted, hyporesponsive | Cytotoxicity, IFN-γ secretion | Glucose, mTOR, OXPHOS backup |
| Regulatory T cells (Tregs) | Fatty acid oxidation (FAO), OXPHOS | Expanded, suppressive | IL-10/TGF-β production, immune suppression | CPT1a, PPARγ, adenosine |
| M2-like TAMs | OXPHOS, lipid metabolism | Pro-tumoral, immunosuppressive | Angiogenesis, Treg recruitment, phagocytosis | FAO, lactate, AhR signaling |
| M1-like TAMs | Glycolytic, NO-driven metabolism | Rare, immunostimulatory | Antigen presentation, TNF/IL-12 production | Glycolysis, HIF-1α |
| Dendritic Cells (DCs) | Mixed; rely on glutamine for activation | Tolerogenic, immature | Poor antigen presentation, low costimulation | Glutamine, α-KG, mTOR |
| Trial ID | Immune Checkpoint Target | Additional Treatments | Primary vs. Recurrent | Clinical Trial Phase |
|---|---|---|---|---|
| NCT06896110 | Anti-PD-1 | Azacitidine: disrupts DNA methylation, RNA processing, and protein synthesis | Recurrent | Phase I |
| NCT04145115 | Anti-PD-1, Anti-CTLA-4 | None | Recurrent | Phase II |
| NCT06325683 | Anti-PD-1, Anti-LAG-3 | Lomustine | Recurrent | Phase II |
| NCT03174197 | Anti-PD-L1 | Temozolomide and radiation | Primary | Phase I/II |
| NCT04977375 | Anti-PD-1 | Stereotactic radiation | Recurrent | Phase Ib/II |
| NCT02287428 | Anti-PD-1 | Neoantigen-based vaccine, radiation, temozolomide | Primary | Phase I |
| NCT05465954 | Anti-PD-1 | Recombinant IL-7 | Recurrent | Phase II |
| NCT06558214 | Anti-PD-1 | TTFs, MRI-guided laser ablation | Recurrent | Phase II |
| NCT05084430 | Anti-PD-1 | Herpes oncolytic virus | Primary or Recurrent | Phase I/II |
| NCT05743595 | Anti-PD-1 | Neoantigen based personalized DNA vaccine | Primary | Phase I |
| NCT06672575 | Bispecific antibody targeting PD-1 and VEGF | None | Recurrent | Phase I/II |
| NCT05039281 | Anti-PD-L1 | Tyrosine Kinase Inhibitor | Recurrent | Phase I/II |
| NCT04201873 | Anti-PD-1 | Dendritic cell vaccination | Recurrent | Phase I |
| NCT04003649 | Anti-PD-1, Anti-CTLA-4 | IL13Rα2-CAR T cells | Recurrent | Phase I |
| NCT06556563 | Anti-PD-1 | TTFs, Temozolomide | Primary | Phase III |
| NCT03277638 | Anti-PD-1 | Laser interstitial thermal therapy | Recurrent | Phase I/II |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medikonda, R.; Abikenari, M.; Schonfeld, E.; Lim, M. The Metabolic Orchestration of Immune Evasion in Glioblastoma: From Molecular Perspectives to Therapeutic Vulnerabilities. Cancers 2025, 17, 1881. https://doi.org/10.3390/cancers17111881
Medikonda R, Abikenari M, Schonfeld E, Lim M. The Metabolic Orchestration of Immune Evasion in Glioblastoma: From Molecular Perspectives to Therapeutic Vulnerabilities. Cancers. 2025; 17(11):1881. https://doi.org/10.3390/cancers17111881
Chicago/Turabian StyleMedikonda, Ravi, Matthew Abikenari, Ethan Schonfeld, and Michael Lim. 2025. "The Metabolic Orchestration of Immune Evasion in Glioblastoma: From Molecular Perspectives to Therapeutic Vulnerabilities" Cancers 17, no. 11: 1881. https://doi.org/10.3390/cancers17111881
APA StyleMedikonda, R., Abikenari, M., Schonfeld, E., & Lim, M. (2025). The Metabolic Orchestration of Immune Evasion in Glioblastoma: From Molecular Perspectives to Therapeutic Vulnerabilities. Cancers, 17(11), 1881. https://doi.org/10.3390/cancers17111881