
Integration of Computational Pipeline to Streamline Efficacious Drug Nomination and Biomarker Discovery in Glioblastoma

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. GBM Clinical Data Tested in Computational Modeling
2.2. GBM Mouse Avatar Model Utilized for Experimental Validation of Drug Candidates and Inferred Drug–Biomarker Relationships
2.3. Overview of Drug Discovery Pipeline
2.4. Overview of Biomarker Discovery Pipeline
2.5. Experimental Testing of Drug Candidates GBM Mouse Avatar Model
2.6. Experimental Testing of Inferred Drug–Biomarker Relationships in GBM Mouse Avatar Model
3. Results
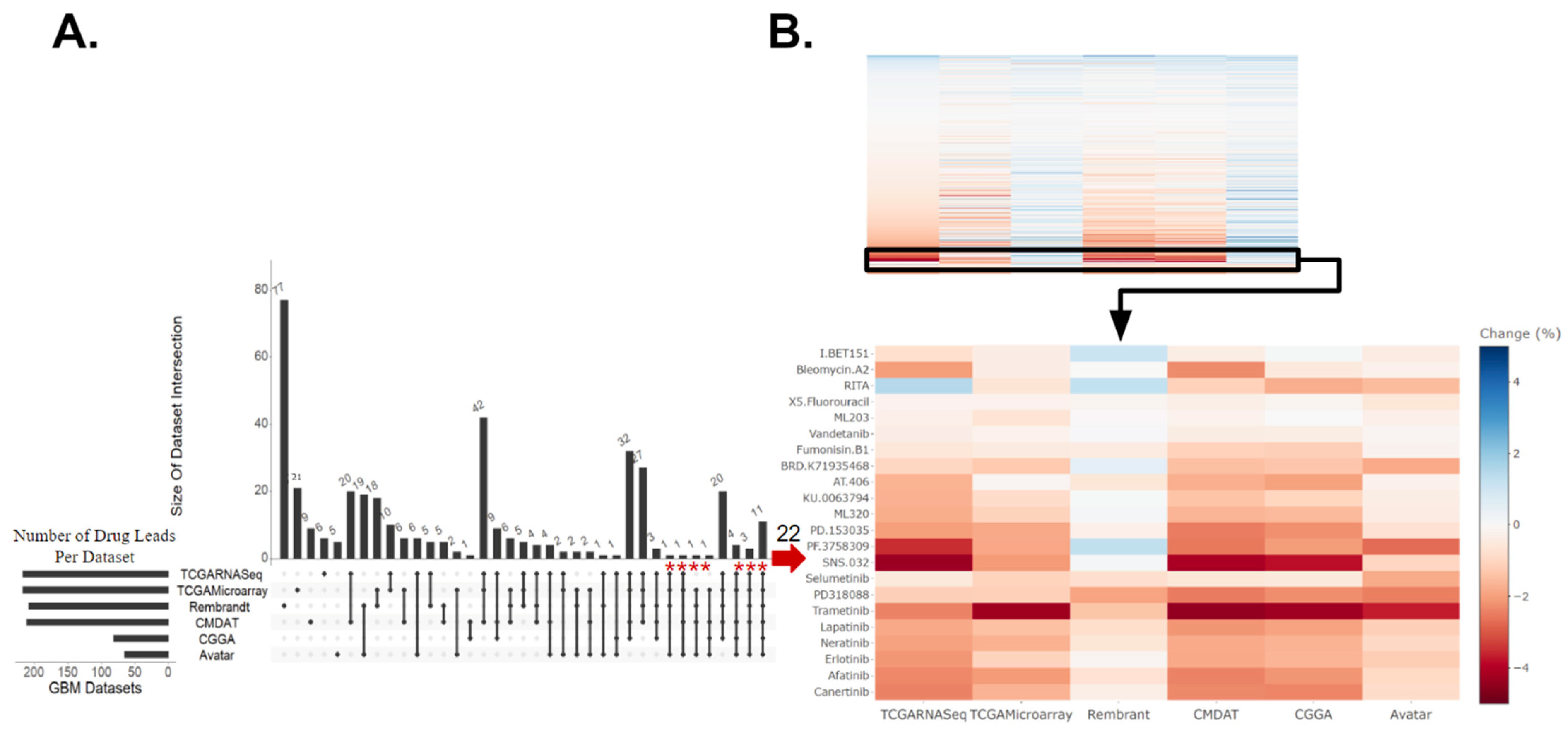
3.1. Identification of GBM Therapeutic Susceptibilities Utilizing Drug Response Prediction
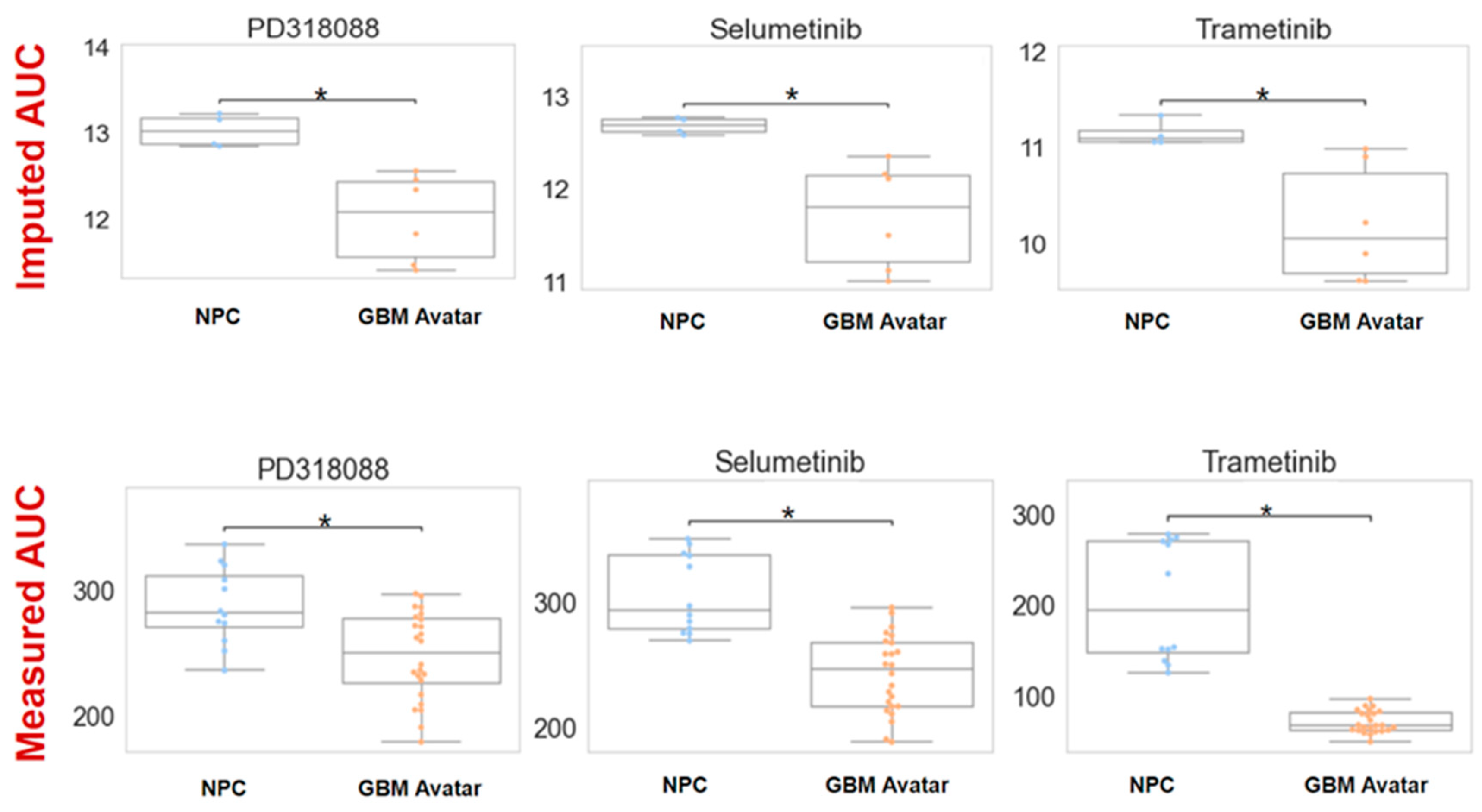
3.2. The Efficacy of MEKis Was Validated in GBM Avatar Model
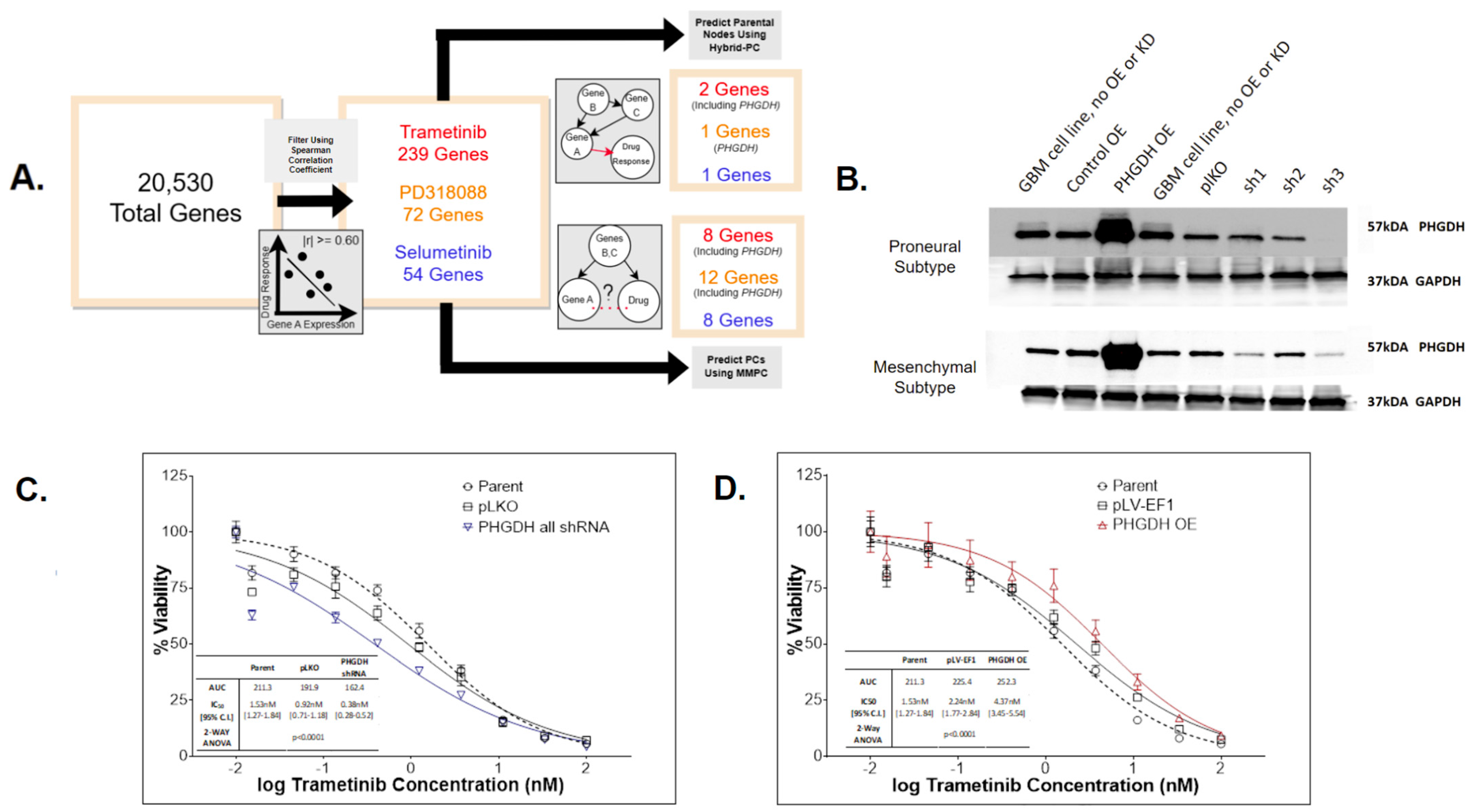
3.3. Application of Causal Inference to Identify Biomarkers Indicative of MEKi Response
3.4. PHGDH Expression Levels Help Inform MEKi Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smoll, N.R.; Schaller, K.; Gautschi, O.P. Long-term survival of patients with glioblastoma multiforme (GBM). J. Clin. Neurosci. 2013, 20, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Walid, M.S. Prognostic Factors for Long-Term Survival after Glioblastoma. Perm. J. 2008, 12, 45–48. [Google Scholar] [CrossRef]
- Krieg, S.M.; Sollmann, N.; Obermueller, T.; Sabih, J.; Bulubas, L.; Negwer, C.; Moser, T.; Droese, D.; Boeckh-Behrens, T.; Ringel, F.; et al. Changing the clinical course of glioma patients by preoperative motor mapping with navigated transcranial magnetic brain stimulation. BMC Cancer 2015, 15, 231. [Google Scholar] [CrossRef] [PubMed]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Briefings Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef] [PubMed]
- Adam, G.; Rampášek, L.; Safikhani, Z.; Smirnov, P.; Haibe-Kains, B.; Goldenberg, A. Machine learning approaches to drug response prediction: Challenges and recent progress. NPJ Precis. Oncol. 2020, 4, 19. [Google Scholar] [CrossRef]
- Zhang, W.; Lee, A.M.; Jena, S.; Huang, Y.; Ho, Y.; Tietz, K.T.; Miller, C.R.; Su, M.-C.; Mentzer, J.; Ling, A.L.; et al. Computational drug discovery for castration-resistant prostate cancers through in vitro drug response modeling. Proc. Natl. Acad. Sci. USA 2023, 120, e2218522120. [Google Scholar] [CrossRef]
- Gruener, R.F.; Ling, A.; Chang, Y.-F.; Morrison, G.; Geeleher, P.; Greene, G.L.; Huang, R.S. Facilitating Drug Discovery in Breast Cancer by Virtually Screening Patients Using In Vitro Drug Response Modeling. Cancers 2021, 13, 885. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Chaim, I.A.; Benitez, J.A.; Markmiller, S.; Parisian, A.D.; Hevner, R.F.; Turner, K.M.; Hessenauer, F.M.; D’Antonio, M.; Nguyen, N.D. Longitudinal assessment of tumor development using cancer avatars derived from genetically engineered pluripotent stem cells. Nat. Commun. 2020, 11, 550. [Google Scholar] [CrossRef]
- Sareen, H.; Ma, Y.; Becker, T.M.; Roberts, T.L.; de Souza, P.; Powter, B. Molecular Biomarkers in Glioblastoma: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2022, 23, 8835. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, K.-N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gusev, Y.; Bhuvaneshwar, K.; Song, L.; Zenklusen, J.-C.; Fine, H.; Madhavan, S. The REMBRANDT study, a large collection of genomic data from brain cancer patients. Sci. Data 2018, 5, 180158. [Google Scholar] [CrossRef] [PubMed]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An Interactive Resource to Identify Cancer Genetic and Lineage Dependencies Targeted by Small Molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Cell Line Encyclopedia Consortium; The Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015, 528, 84–87. [Google Scholar] [CrossRef]
- Bairoch, A. The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech. 2018, 29, 25–38. [Google Scholar] [CrossRef]
- Tang, K.; Ji, X.; Zhou, M.; Deng, Z.; Huang, Y.; Zheng, G.; Cao, Z. Rank-in: Enabling integrative analysis across microarray and RNA-seq for cancer. Nucleic Acids Res. 2021, 49, e99. [Google Scholar] [CrossRef]
- Hodges, J.L.; Lehmann, E.L. Estimates of Location Based on Rank Tests. Ann. Math. Stat. 1963, 34, 598–611. [Google Scholar] [CrossRef]
- Tsagris, M.; Tsamardinos, I. Feature selection with the R package MXM. F1000Research 2019, 7, 1505. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J.; et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16, 133. [Google Scholar] [CrossRef]
- Corchete, L.A.; Rojas, E.A.; Alonso-López, D.; Rivas, J.D.L.; Gutiérrez, N.C.; Burguillo, F.J. Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis. Sci. Rep. 2020, 10, 19737. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.H. Genetic wrappers for feature selection in decision tree induction and variable ordering in Bayesian network structure learning. Inf. Sci. 2004, 163, 103–122. [Google Scholar] [CrossRef]
- Irizarry, L.R.; Hambardzumyan, D.; Nakano, I.; Gladson, C.L.; Ahluwalia, M.S. Therapeutic targeting of VEGF in the treatment of glioblastoma. Expert Opin. Ther. Targets 2012, 16, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Zhou, Z.; Huang, X.; Chen, Z.; Zhang, L.; Zhang, J.; Hua, W.; Mao, Y. Use of Bevacizumab in recurrent glioblastoma: A scoping review and evidence map. BMC Cancer 2023, 23, 544. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Wakimoto, H.; Peters, C.W.; Antoszczyk, S.J.; Rabkin, S.D.; Martuza, R.L. Combinatorial Effects of VEGFR Kinase Inhibitor Axitinib and Oncolytic Virotherapy in Mouse and Human Glioblastoma Stem-Like Cell Models. Clin. Cancer Res. 2018, 24, 3409–3422. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zou, C.-L.; Zhang, Z.-M.; Lv, L.-J.; Qiao, H.-B.; Chen, X.-J. A multi-targeted tyrosine kinase inhibitor lenvatinib for the treatment of mice with advanced glioblastoma. Mol. Med. Rep. 2017, 16, 7105–7111. [Google Scholar] [CrossRef] [PubMed]
- Owen, S.; Alken, S.; Alshami, J.; Guiot, M.-C.; Kavan, P.; Reardon, D.A.; Muanza, T.; Gibson, N.; Pemberton, K.; Solca, F.; et al. Genomic Analysis of Tumors from Patients with Glioblastoma with Long-Term Response to Afatinib. OncoTargets Ther. 2022, 15, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Vengoji, R.; Macha, M.A.; Nimmakayala, R.K.; Rachagani, S.; Siddiqui, J.A.; Mallya, K.; Gorantla, S.; Jain, M.; Ponnusamy, M.P.; Batra, S.K. Afatinib and Temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 266. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.C.; Mercier, M.-C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Selvasaravanan, K.D.; Wiederspohn, N.; Hadzalic, A.; Strobel, H.; Payer, C.; Schuster, A.; Karpel-Massler, G.; Siegelin, M.D.; Halatsch, M.-E.; Debatin, K.-M.; et al. The limitations of targeting MEK signalling in Glioblastoma therapy. Sci. Rep. 2020, 10, 7401. [Google Scholar] [CrossRef]
- Fangusaro, J.; Onar-Thomas, A.; Poussaint, T.Y.; Wu, S.; Ligon, A.H.; Lindeman, N.; Campagne, O.; Banerjee, A.; Gururangan, S.; Kilburn, L.B.; et al. A phase II trial of selumetinib in children with recurrent optic pathway and hypothalamic low-grade glioma without NF1: A Pediatric Brain Tumor Consortium study. Neuro-Oncology 2021, 23, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [PubMed]
- Doepner, M.; Lee, I.Y.; Ridky, T.W. Drug Resistant Melanoma May Be Vulnerable to Inhibitors of Serine Synthesis. J. Investig. Dermatol. 2020, 140, 2114–2116. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.Q.; Teh, J.L.; Purwin, T.J.; Chervoneva, I.; Davies, M.A.; Nathanson, K.L.; Cheng, P.F.; Levesque, M.P.; Dummer, R.; Aplin, A.E. Targeting PHGDH Upregulation Reduces Glutathione Levels and Resensitizes Resistant NRAS-Mutant Melanoma to MAPK Kinase Inhibition. J. Investig. Dermatol. 2020, 140, 2242–2252.e7. [Google Scholar] [CrossRef]
- Cai, S.; Lu, J.X.; Wang, Y.P.; Shi, C.J.; Yuan, T.; Wang, X.P. SH2B3, Transcribed by STAT1, Promotes Glioblastoma Progression Through Transducing IL-6/gp130 Signaling to Activate STAT3 Signaling. Front. Cell Dev. Biol. 2021, 9, 606527. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeser, D.; Gruener, R.F.; Galvin, R.; Lee, A.; Koga, T.; Grigore, F.-N.; Suzuki, Y.; Furnari, F.B.; Chen, C.; Huang, R.S. Integration of Computational Pipeline to Streamline Efficacious Drug Nomination and Biomarker Discovery in Glioblastoma. Cancers 2024, 16, 1723. https://doi.org/10.3390/cancers16091723
Maeser D, Gruener RF, Galvin R, Lee A, Koga T, Grigore F-N, Suzuki Y, Furnari FB, Chen C, Huang RS. Integration of Computational Pipeline to Streamline Efficacious Drug Nomination and Biomarker Discovery in Glioblastoma. Cancers. 2024; 16(9):1723. https://doi.org/10.3390/cancers16091723
Chicago/Turabian StyleMaeser, Danielle, Robert F. Gruener, Robert Galvin, Adam Lee, Tomoyuki Koga, Florina-Nicoleta Grigore, Yuta Suzuki, Frank B. Furnari, Clark Chen, and R. Stephanie Huang. 2024. "Integration of Computational Pipeline to Streamline Efficacious Drug Nomination and Biomarker Discovery in Glioblastoma" Cancers 16, no. 9: 1723. https://doi.org/10.3390/cancers16091723
APA StyleMaeser, D., Gruener, R. F., Galvin, R., Lee, A., Koga, T., Grigore, F.-N., Suzuki, Y., Furnari, F. B., Chen, C., & Huang, R. S. (2024). Integration of Computational Pipeline to Streamline Efficacious Drug Nomination and Biomarker Discovery in Glioblastoma. Cancers, 16(9), 1723. https://doi.org/10.3390/cancers16091723