
Clonal Neoantigen: Emerging “Mechanism-based” Biomarker of Immunotherapy Response

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
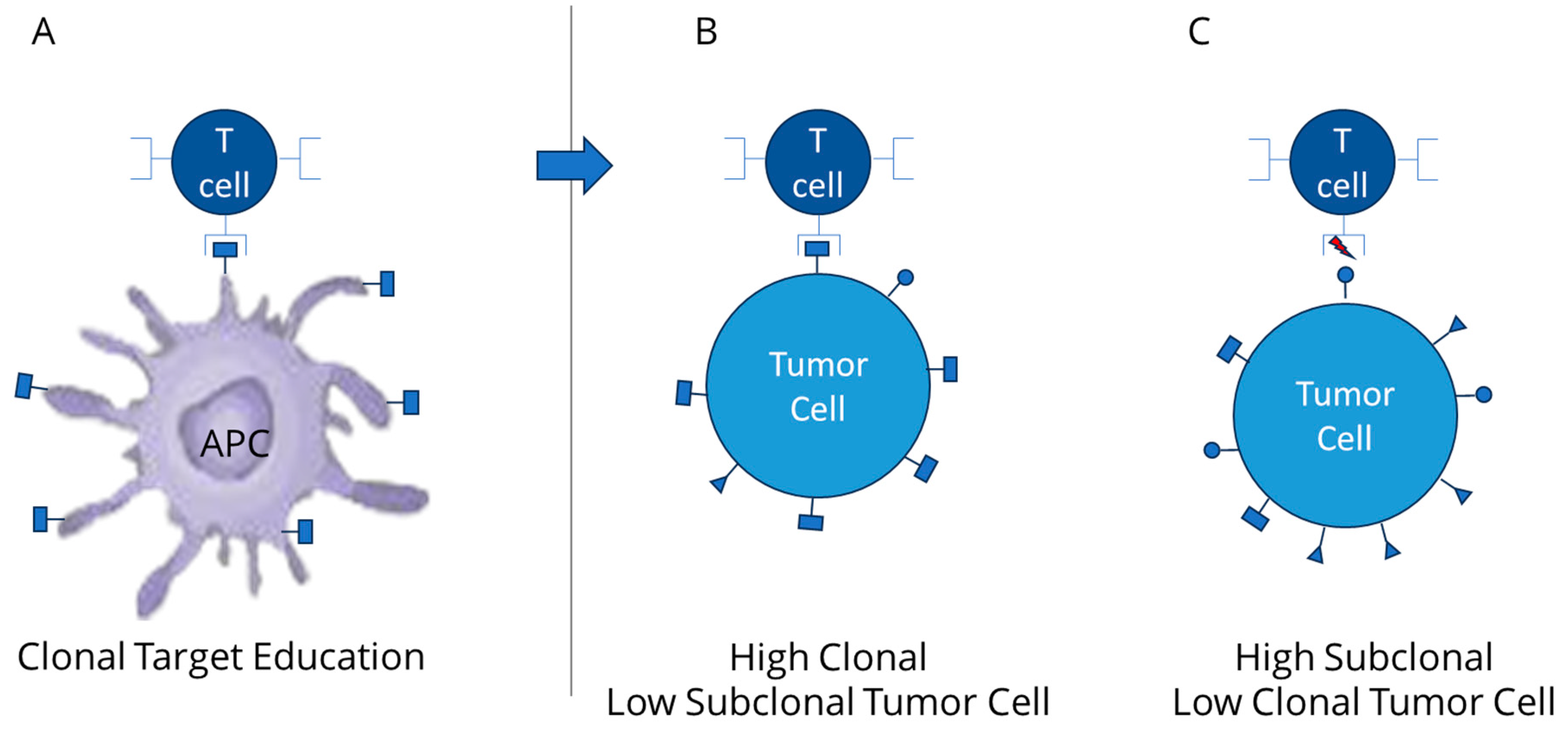
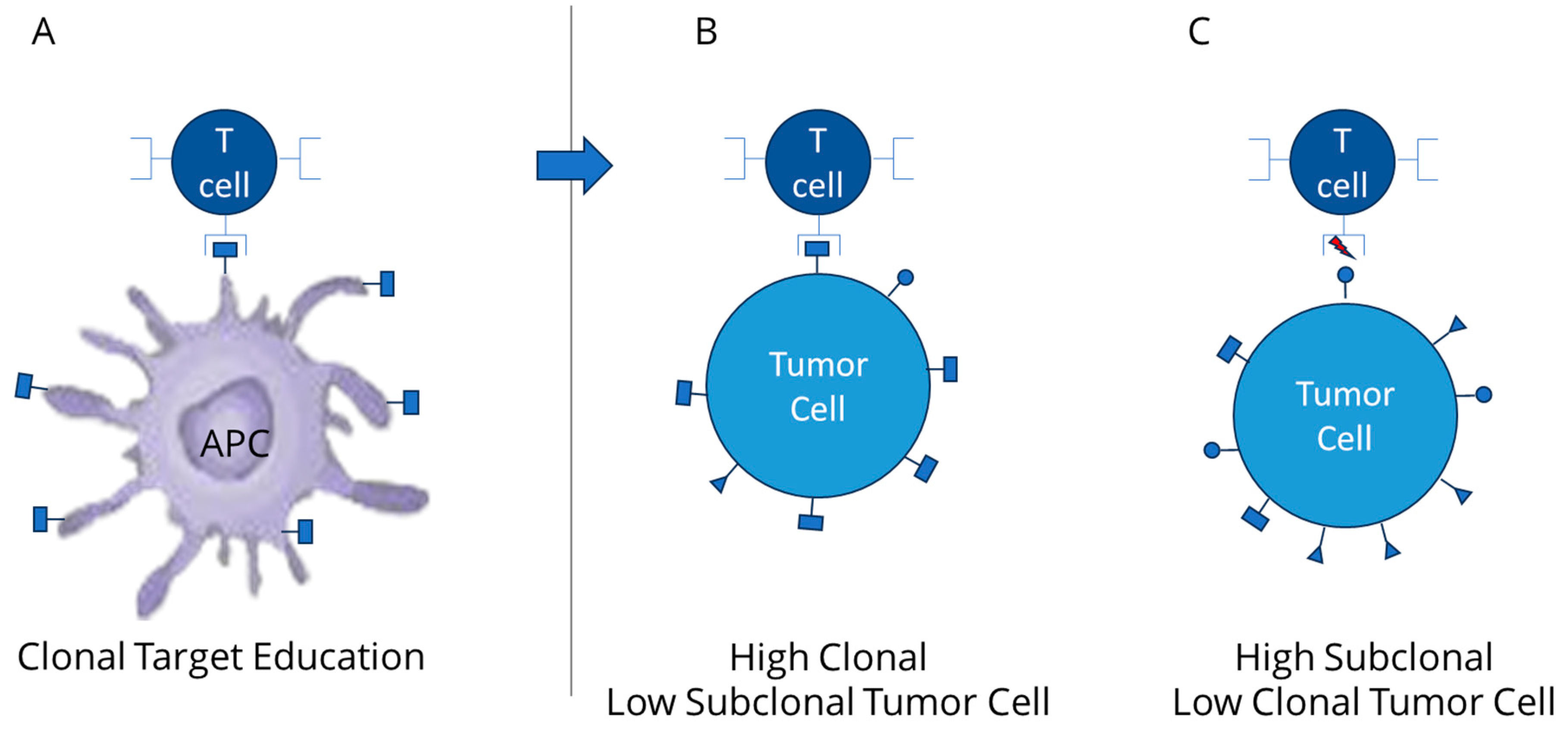
2. Relevance of Clonal Mutation Assessment
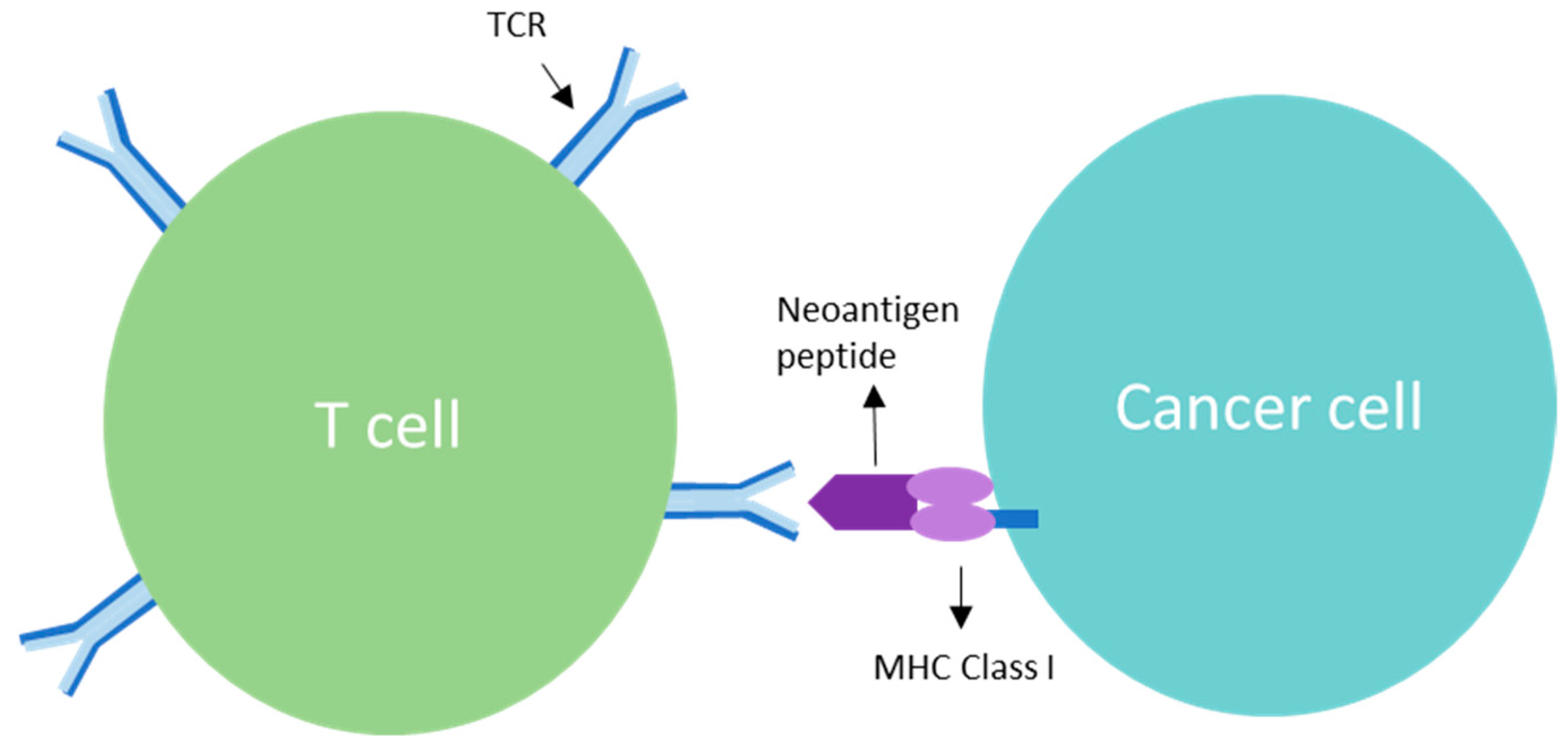
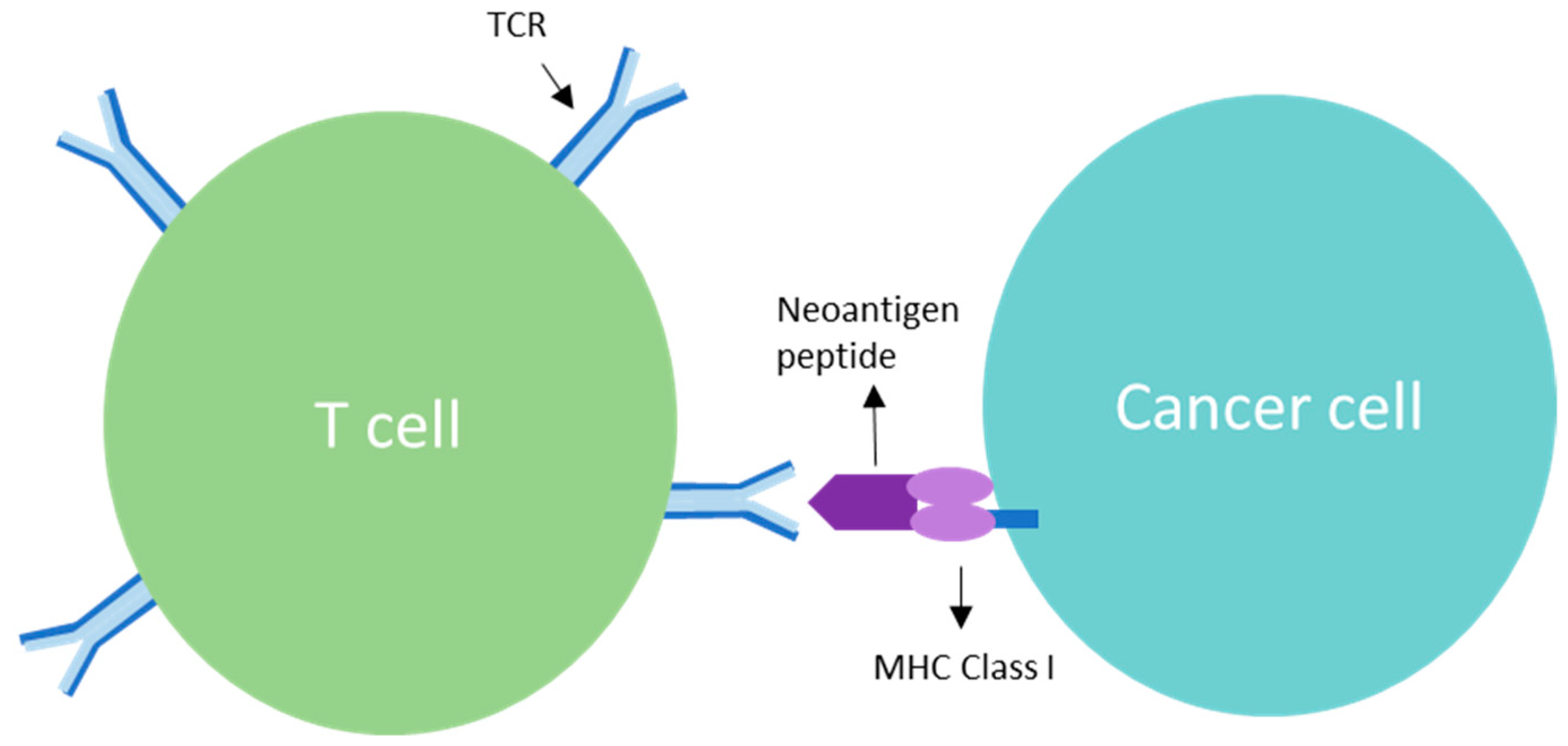
3. TCR Interaction and the Role of Clonal Neoantigens
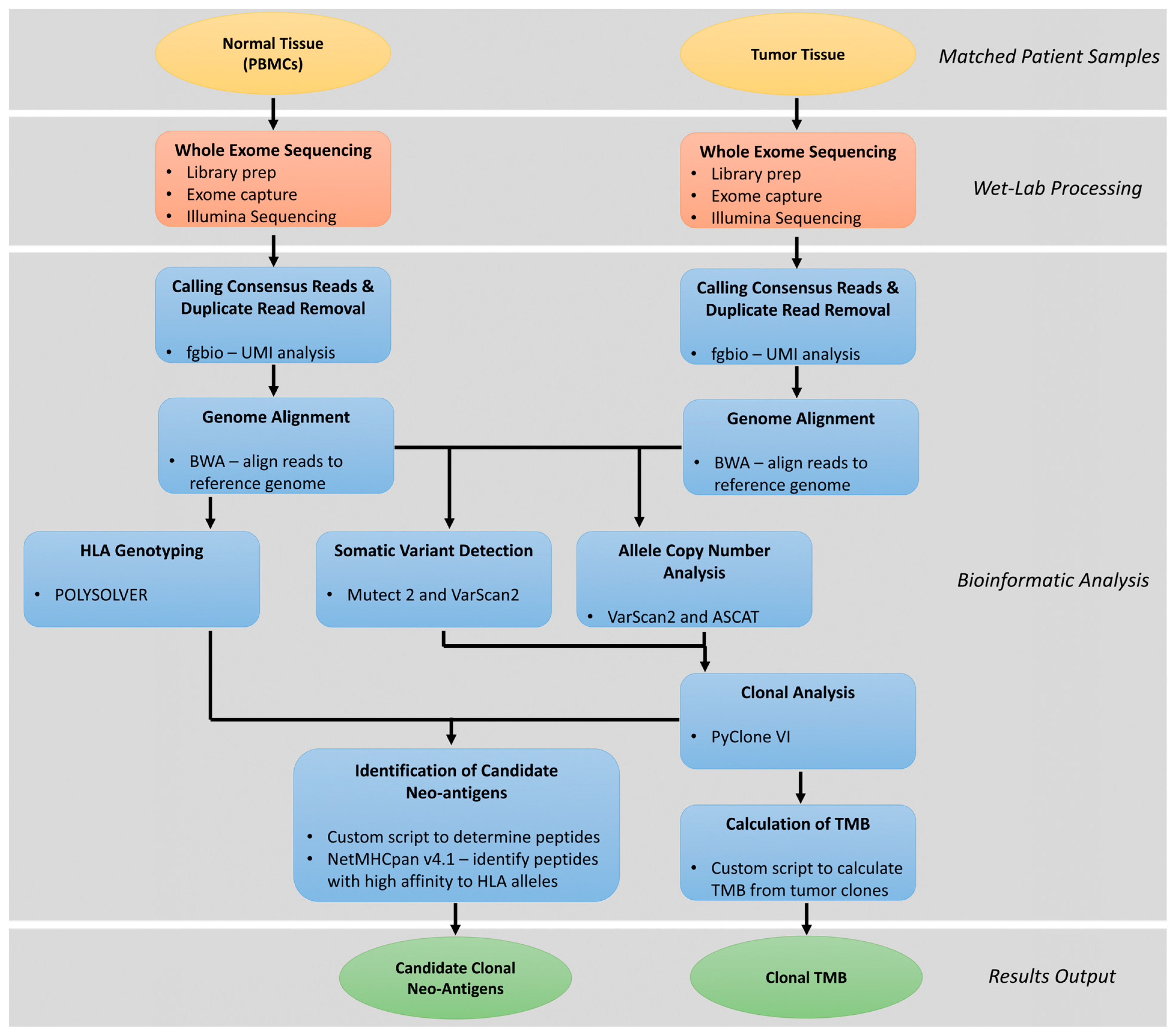
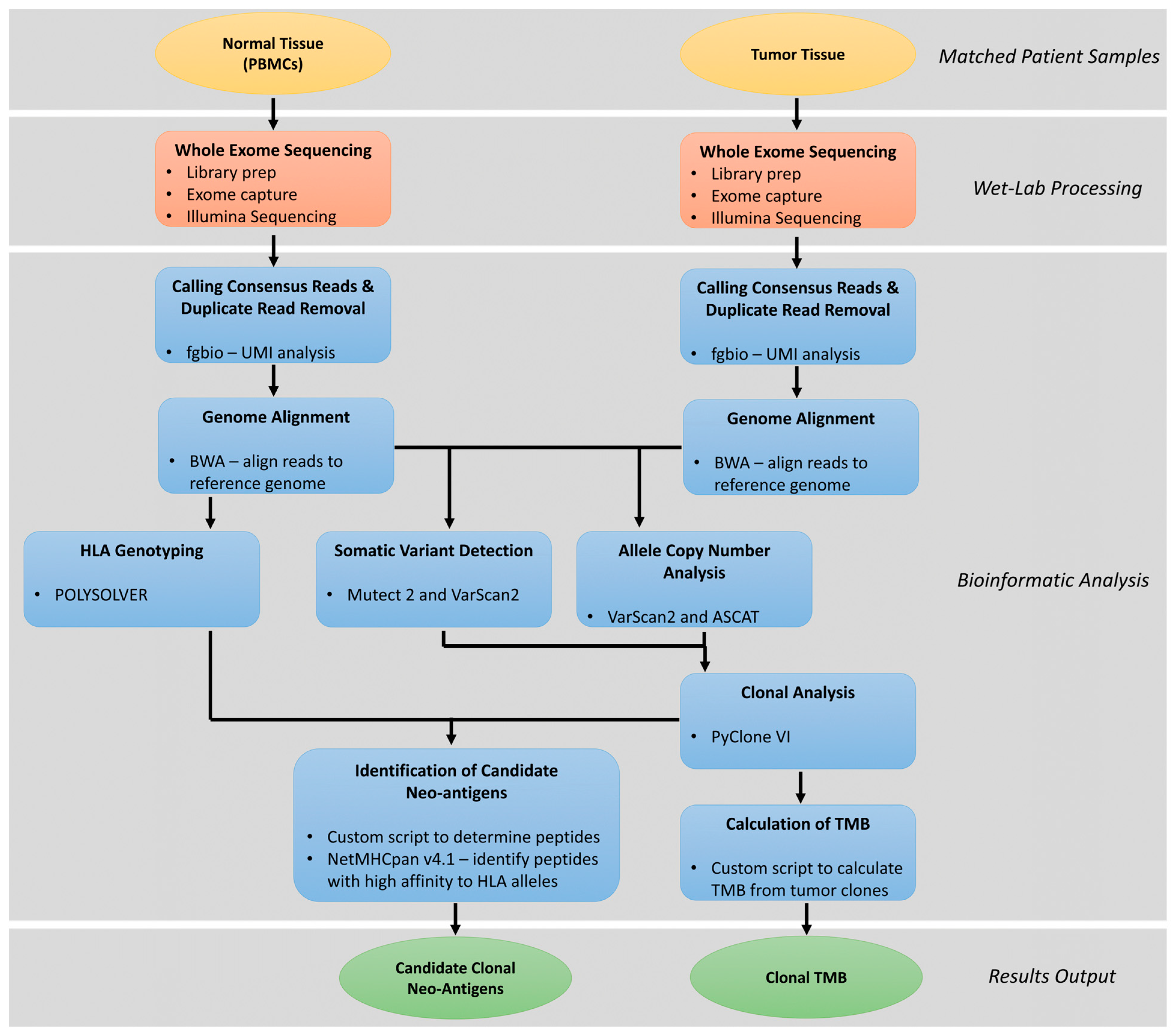
4. Clonal Neoantigen Identification and Analysis
5. Clinical Response to CPI Is Variable
6. Biomarkers of CPI Response
7. Limitations of CPI Based Therapy in Newly Diagnosed Advanced Ovarian Cancer
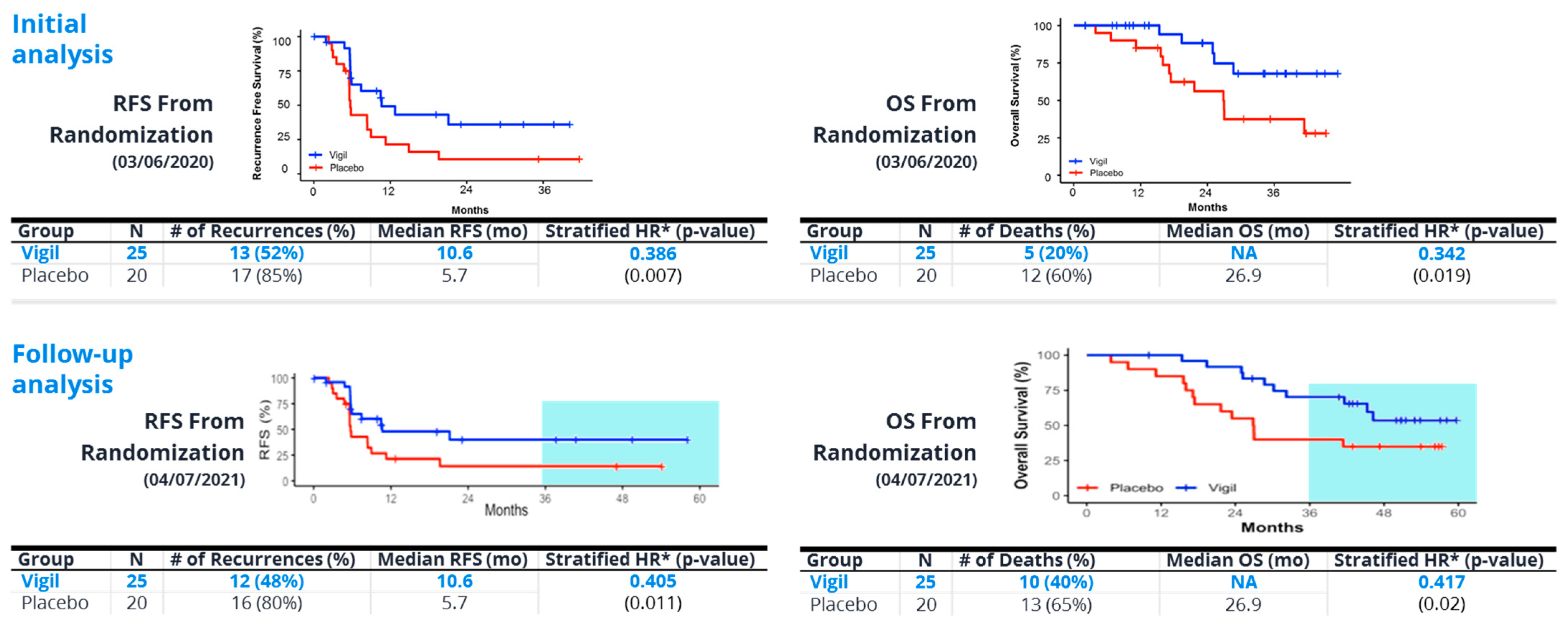
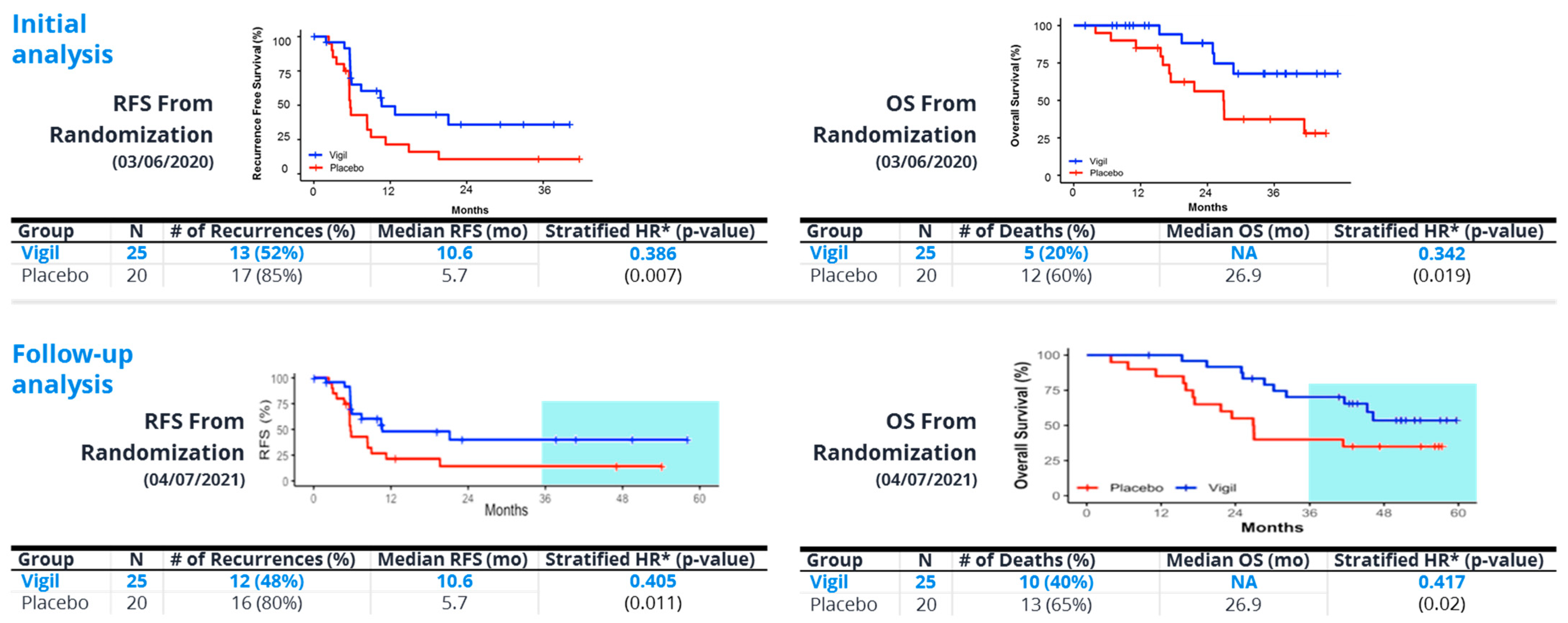
8. Vigil®—“Full Spectrum” Neoantigen Immune Training in a Permissive Environment
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fang, X.; Guo, Z.; Liang, J.; Wen, J.; Liu, Y.; Guan, X.; Li, H. Neoantigens and their potential applications in tumor immunotherapy. Oncol. Lett. 2022, 23, 88. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Wilmott, J.S.; Tembe, V.; Howle, J.R.; Sharma, R.; Thompson, J.F.; Rizos, H.; Lo, R.S.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Intratumoral molecular heterogeneity in a BRAF-mutant, BRAF inhibitor-resistant melanoma: A case illustrating the challenges for personalized medicine. Mol. Cancer Ther. 2012, 11, 2704–2708. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Yang, X.N.; Chen, Z.H.; Yang, J.J.; Zhou, Q.; Yan, H.H.; An, S.J.; et al. EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist 2012, 17, 978–985. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra117. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magri, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wolfel, C.; Huber, C.; Wolfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- DuPage, M.; Mazumdar, C.; Schmidt, L.M.; Cheung, A.F.; Jacks, T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012, 482, 405–409. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Ceglia, V.; Kelley, E.J.; Boyle, A.S.; Zurawski, S.; Mead, H.L.; Harms, C.E.; Blanck, J.P.; Flamar, A.L.; Kirschman, J.H.; Ogongo, P.; et al. A Framework to Identify Antigen-Expanded T Cell Receptor Clusters Within Complex Repertoires. Front. Immunol. 2021, 12, 735584. [Google Scholar] [CrossRef]
- Joshi, K.; Chain, B.M.; Peggs, K.S.; Quezada, S.A. The “Achilles’ Heel” of Cancer and Its Implications for the Development of Novel Immunotherapeutic Strategies. Cold Spring Harb. Perspect. Med. 2018, 8, a027086. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e514. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Wu, D.; Liu, Y.; Li, X.; Liu, Y.; Yang, Q.; Liu, Y.; Wu, J.; Tian, C.; Zeng, Y.; Zhao, Z.; et al. Identification of Clonal Neoantigens Derived From Driver Mutations in an EGFR-Mutated Lung Cancer Patient Benefitting From Anti-PD-1. Front. Immunol. 2020, 11, 1366. [Google Scholar] [CrossRef]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.A.; Riegel, K.; Pompaiah, M.; Juhl, H.; Rajalingam, K. Druggable genome and precision medicine in cancer: Current challenges. FEBS J. 2021, 288, 6142–6158. [Google Scholar] [CrossRef] [PubMed]
- Al Bakir, M.; Huebner, A.; Martinez-Ruiz, C.; Grigoriadis, K.; Watkins, T.B.K.; Pich, O.; Moore, D.A.; Veeriah, S.; Ward, S.; Laycock, J.; et al. The evolution of non-small cell lung cancer metastases in TRACERx. Nature 2023, 616, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Wu, Y.; Biswas, D.; Swanton, C. Impact of cancer evolution on immune surveillance and checkpoint inhibitor response. Semin. Cancer Biol. 2022, 84, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.A.; Carreno, L.J.; Coombs, D.; Mora, J.E.; Palmieri, E.; Goldstein, B.; Nathenson, S.G.; Kalergis, A.M. T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell. Proc. Natl. Acad. Sci. USA 2005, 102, 4824–4829. [Google Scholar] [CrossRef] [PubMed]
- Valitutti, S.; Muller, S.; Cella, M.; Padovan, E.; Lanzavecchia, A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature 1995, 375, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Boniface, J.J.; Reich, Z.; Lyons, D.; Hampl, J.; Arden, B.; Chien, Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998, 16, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Gejman, R.S.; Chang, A.Y.; Jones, H.F.; DiKun, K.; Hakimi, A.A.; Schietinger, A.; Scheinberg, D.A. Rejection of immunogenic tumor clones is limited by clonal fraction. Elife 2018, 7, e41090. [Google Scholar] [CrossRef]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jimenez-Sanchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235.e221. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef]
- Andersen, M.H.; Fensterle, J.; Ugurel, S.; Reker, S.; Houben, R.; Guldberg, P.; Berger, T.G.; Schadendorf, D.; Trefzer, U.; Brocker, E.B.; et al. Immunogenicity of constitutively active V599EBRaf. Cancer Res. 2004, 64, 5456–5460. [Google Scholar] [CrossRef]
- Bowman, R.L.; Hennessey, R.C.; Weiss, T.J.; Tallman, D.A.; Crawford, E.R.; Murphy, B.M.; Webb, A.; Zhang, S.; La Perle, K.M.; Burd, C.J.; et al. UVB mutagenesis differs in Nras- and Braf-mutant mouse models of melanoma. Life Sci. Alliance 2021, 4, e202101135. [Google Scholar] [CrossRef]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Stanbery, L.; Walter, A.; Rocconi, R.P.; Stephens, P. Rationale of Homologous Recombination Proficient Molecular Profile as Biomarker for Therapeutic Targeting in Ovarian Cancer. Oncol. Rev. 2023, 17, 11471. [Google Scholar] [CrossRef] [PubMed]
- Rocconi, R.P.; Monk, B.J.; Walter, A.; Herzog, T.J.; Galanis, E.; Manning, L.; Bognar, E.; Wallraven, G.; Stanbery, L.; Aaron, P.; et al. Gemogenovatucel-T (Vigil) immunotherapy demonstrates clinical benefit in homologous recombination proficient (HRP) ovarian cancer. Gynecol. Oncol. 2021, 161, 676–680. [Google Scholar] [CrossRef]
- Rocconi, R.P.; Grosen, E.A.; Ghamande, S.A.; Chan, J.K.; Barve, M.A.; Oh, J.; Tewari, D.; Morris, P.C.; Stevens, E.E.; Bottsford-Miller, J.N.; et al. Gemogenovatucel-T (Vigil) immunotherapy as maintenance in frontline stage III/IV ovarian cancer (VITAL): A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Oncol. 2020, 21, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
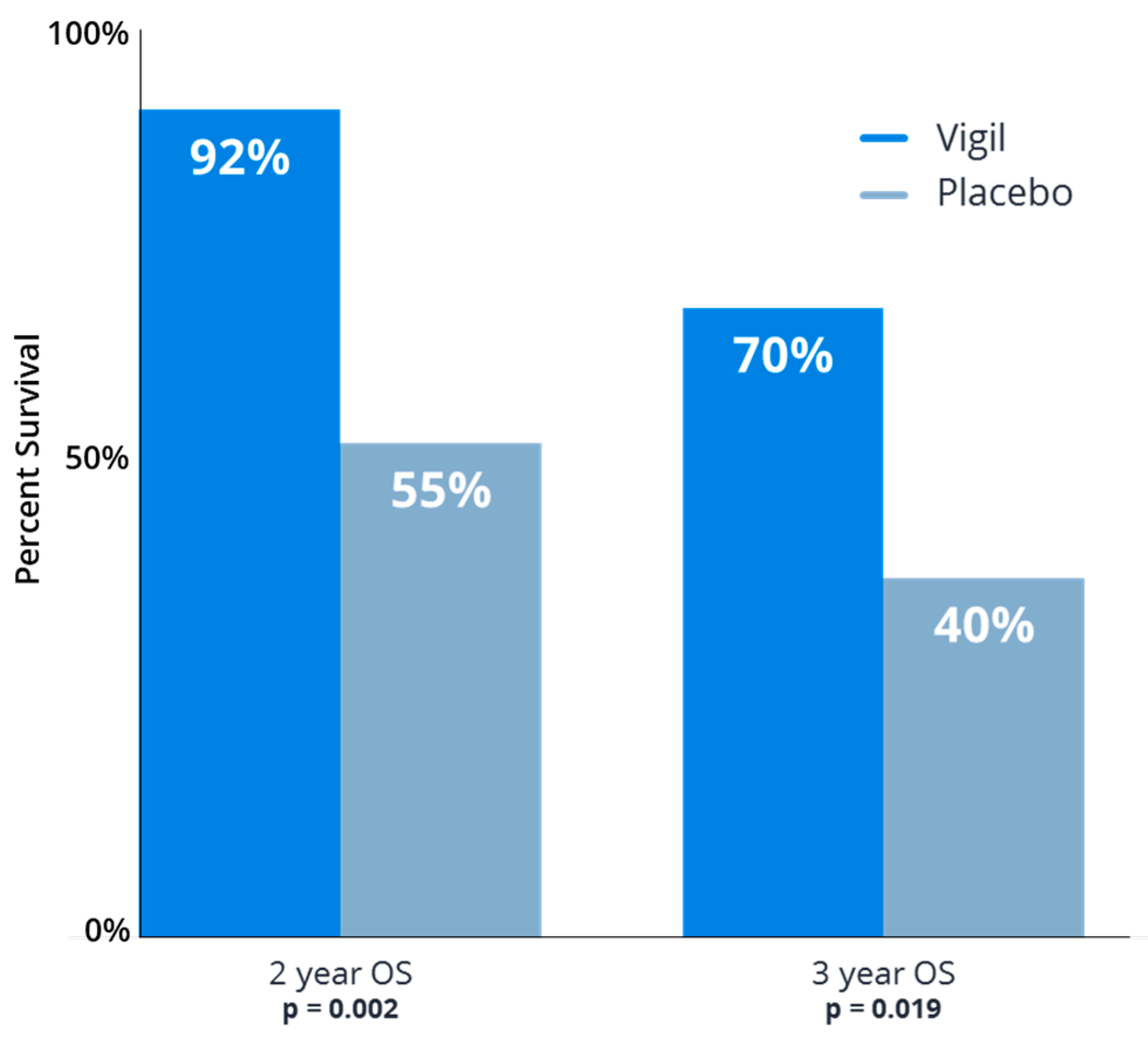
- Walter, A.; Rocconi, R.P.; Monk, B.J.; Herzog, T.J.; Manning, L.; Bognar, E.; Wallraven, G.; Aaron, P.; Horvath, S.; Tang, M.; et al. Gemogenovatucel-T (Vigil) maintenance immunotherapy: 3-year survival benefit in homologous recombination proficient (HRP) ovarian cancer. Gynecol. Oncol. 2021, 163, 459–464. [Google Scholar] [CrossRef]
- Lythe, G.; Callard, R.E.; Hoare, R.L.; Molina-Paris, C. How many TCR clonotypes does a body maintain? J. Theor. Biol. 2016, 389, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Wosen, J.E.; Mukhopadhyay, D.; Macaubas, C.; Mellins, E.D. Epithelial MHC Class II Expression and Its Role in Antigen Presentation in the Gastrointestinal and Respiratory Tracts. Front. Immunol. 2018, 9, 2144. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a Tumor-Infiltrating Lymphocyte Therapy, in Metastatic Melanoma. J. Clin. Oncol. 2021, 39, 2656–2666. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Peri, A.; Greenstein, E.; Alon, M.; Pai, J.A.; Dingjan, T.; Reich-Zeliger, S.; Barnea, E.; Barbolin, C.; Levy, R.; Arnedo-Pac, C.; et al. Combined presentation and immunogenicity analysis reveals a recurrent RAS.Q61K neoantigen in melanoma. J. Clin. Investig. 2021, 131, e129466. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Duitama, J.; Al Seesi, S.; Ayres, C.M.; Corcelli, S.A.; Pawashe, A.P.; Blanchard, T.; McMahon, D.; Sidney, J.; Sette, A.; et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med. 2014, 211, 2231–2248. [Google Scholar] [CrossRef] [PubMed]
- Spierings, E.; Brickner, A.G.; Caldwell, J.A.; Zegveld, S.; Tatsis, N.; Blokland, E.; Pool, J.; Pierce, R.A.; Mollah, S.; Shabanowitz, J.; et al. The minor histocompatibility antigen HA-3 arises from differential proteasome-mediated cleavage of the lymphoid blast crisis (Lbc) oncoprotein. Blood 2003, 102, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naive to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Chyuan, I.T.; Chu, C.L.; Hsu, P.N. Targeting the Tumor Microenvironment for Improving Therapeutic Effectiveness in Cancer Immunotherapy: Focusing on Immune Checkpoint Inhibitors and Combination Therapies. Cancers 2021, 13, 1188. [Google Scholar] [CrossRef]
- Petitprez, F.; Meylan, M.; de Reynies, A.; Sautes-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front Immunol 2020, 11, 784. [Google Scholar] [CrossRef]
- Zhang, J.; Endres, S.; Kobold, S. Enhancing tumor T cell infiltration to enable cancer immunotherapy. Immunotherapy 2019, 11, 201–213. [Google Scholar] [CrossRef]
- Melero, I.; Rouzaut, A.; Motz, G.T.; Coukos, G. T-cell and NK-cell infiltration into solid tumors: A key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014, 4, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef] [PubMed]
- McKeithan, T.W. Kinetic proofreading in T-cell receptor signal transduction. Proc. Natl. Acad. Sci. USA 1995, 92, 5042–5046. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: A molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.A.; Boniface, J.J.; Davis, M.M. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity 1999, 10, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Kalergis, A.M.; Boucheron, N.; Doucey, M.A.; Palmieri, E.; Goyarts, E.C.; Vegh, Z.; Luescher, I.F.; Nathenson, S.G. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat. Immunol. 2001, 2, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Niknafs, N.; Balan, A.; Cherry, C.; Hummelink, K.; Monkhorst, K.; Shao, X.M.; Belcaid, Z.; Marrone, K.A.; Murray, J.; Smith, K.N.; et al. Persistent mutation burden drives sustained anti tumor immune responses. Nat. Med. 2023, 29, 440–449. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Boll, L.M.; Perera-Bel, J.; Rodriguez-Vida, A.; Arpi, O.; Rovira, A.; Juanpere, N.; Vazquez Montes de Oca, S.; Hernandez-Llodra, S.; Lloreta, J.; Alba, M.M.; et al. The impact of mutational clonality in predicting the response to immune checkpoint inhibitors in advanced urothelial cancer. Sci. Rep. 2023, 13, 15287. [Google Scholar] [CrossRef]
- Huang, J.; Teng, X. Expression of PD-L1 for predicting response to immune checkpoint inhibitors in metastatic urothelial carcinoma: A systematic review and meta-analysis. Curr. Oncol. 2020, 27, e656–e663. [Google Scholar] [CrossRef]
- So, W.V.; Dejardin, D.; Rossmann, E.; Charo, J. Predictive biomarkers for PD-1/PD-L1 checkpoint inhibitor response in NSCLC: An analysis of clinical trial and real-world data. J. Immunother. Cancer 2023, 11, e006464. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanic, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Parsons, D.W.; Peggs, K.S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Allison, J.P. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008, 68, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for Neoantigen Identification. Front. Immunol. 2019, 10, 1392. [Google Scholar] [CrossRef] [PubMed]
- Kalaora, S.; Samuels, Y. Cancer Exome-Based Identification of Tumor Neo-Antigens Using Mass Spectrometry. In Cancer Immunosurveillance: Methods and Protocols; López-Soto, A., Folgueras, A.R., Eds.; Springer: New York, NY, USA, 2019; pp. 203–214. [Google Scholar]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Coukos, G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 2016, 41, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Muller, M.; Huber, F.; Arnaud, M.; Kraemer, A.I.; Altimiras, E.R.; Michaux, J.; Taillandier-Coindard, M.; Chiffelle, J.; Murgues, B.; Gehret, T.; et al. Machine learning methods and harmonized datasets improve immunogenic neoantigen prediction. Immunity 2023, 56, 2650–2663. [Google Scholar] [CrossRef]
- Thuesen, N.H.; Klausen, M.S.; Gopalakrishnan, S.; Trolle, T.; Renaud, G. Benchmarking freely available HLA typing algorithms across varying genes, coverages and typing resolutions. Front. Immunol. 2022, 13, 987655. [Google Scholar] [CrossRef]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Côté, A.; Shah, S.P. PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Kote, S.; Pirog, A.; Bedran, G.; Alfaro, J.; Dapic, I. Mass spectrometry-based identification of MHC-associated peptides. Cancers 2020, 12, 535. [Google Scholar] [CrossRef] [PubMed]
- Haslem, D.S.; Van Norman, S.B.; Fulde, G.; Knighton, A.J.; Belnap, T.; Butler, A.M.; Rhagunath, S.; Newman, D.; Gilbert, H.; Tudor, B.P.; et al. A Retrospective Analysis of Precision Medicine Outcomes in Patients With Advanced Cancer Reveals Improved Progression-Free Survival Without Increased Health Care Costs. J. Oncol. Pract. 2017, 13, e108–e119. [Google Scholar] [CrossRef] [PubMed]
- Radovich, M.; Kiel, P.J.; Nance, S.M.; Niland, E.E.; Parsley, M.E.; Ferguson, M.E.; Jiang, G.; Ammakkanavar, N.R.; Einhorn, L.H.; Cheng, L.; et al. Clinical benefit of a precision medicine based approach for guiding treatment of refractory cancers. Oncotarget 2016, 7, 56491–56500. [Google Scholar] [CrossRef]
- Jardim, D.L.; Schwaederle, M.; Wei, C.; Lee, J.J.; Hong, D.S.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of a Biomarker-Based Strategy on Oncology Drug Development: A Meta-analysis of Clinical Trials Leading to FDA Approval. J. Natl. Cancer Inst. 2015, 107, djv253. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Lazar, V.; Leyland-Jones, B.; Schilsky, R.L.; Mendelsohn, J.; Kurzrock, R. Association of Biomarker-Based Treatment Strategies With Response Rates and Progression-Free Survival in Refractory Malignant Neoplasms: A Meta-analysis. JAMA Oncol. 2016, 2, 1452–1459. [Google Scholar] [CrossRef]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. 2015, 33, 3817–3825. [Google Scholar] [CrossRef]
- Dhir, M.; Choudry, H.A.; Holtzman, M.P.; Pingpank, J.F.; Ahrendt, S.A.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Zeh, H.J.; Singhi, A.D.; et al. Impact of genomic profiling on the treatment and outcomes of patients with advanced gastrointestinal malignancies. Cancer Med. 2017, 6, 195–206. [Google Scholar] [CrossRef]
- Chawla, A.; Janku, F.; Wheler, J.J.; Miller, V.A.; Ryan, J.; Anhorn, R.; Zhou, Z.; Signorovitch, J. Estimated Cost of Anticancer Therapy Directed by Comprehensive Genomic Profiling in a Single-Center Study. JCO Precis. Oncol. 2018, 2, 1–11. [Google Scholar] [CrossRef]
- Handorf, E.A.; McElligott, S.; Vachani, A.; Langer, C.J.; Bristol Demeter, M.; Armstrong, K.; Asch, D.A. Cost effectiveness of personalized therapy for first-line treatment of stage IV and recurrent incurable adenocarcinoma of the lung. J. Oncol. Pract. 2012, 8, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Pennell, N.A.; Mutebi, A.; Zhou, Z.Y.; Ricculli, M.L.; Tang, W.; Wang, H.; Guerin, A.; Arnhart, T.; Dalal, A.; Sasane, M.; et al. Economic Impact of Next-Generation Sequencing Versus Single-Gene Testing to Detect Genomic Alterations in Metastatic Non-Small-Cell Lung Cancer Using a Decision Analytic Model. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Signorovitch, J.; Zhou, Z.; Ryan, J.; Chawla, A. Comprehensive genomic profiling (CGP) versus conventional molecular diagnostic testing of patients with advanced non-small cell lung cancer (NSCLC): Overall survival (OS) and cost in a U.S. health plan population. J. Clin. Oncol. 2017, 35, 6599. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Pedersen, J.G.; Sokac, M.; Sorensen, B.S.; Luczak, A.A.; Aggerholm-Pedersen, N.; Birkbak, N.J.; Ollegaard, T.H.; Jakobsen, M.R. Increased Soluble PD-1 Predicts Response to Nivolumab plus Ipilimumab in Melanoma. Cancers 2022, 14, 3342. [Google Scholar] [CrossRef]
- Lenz, H.J.; Van Cutsem, E.; Luisa Limon, M.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; Garcia-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Behera, M.; Owonikoko, T.K.; Kamphorst, A.O.; Pakkala, S.; Belani, C.P.; Khuri, F.R.; Ahmed, R.; Ramalingam, S.S. Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non-small cell lung cancer: A systematic analysis of the literature. Cancer 2018, 124, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Carlino, M.S.; Long, G.V.; Schadendorf, D.; Robert, C.; Ribas, A.; Richtig, E.; Nyakas, M.; Caglevic, C.; Tarhini, A.; Blank, C.; et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006: A randomised clinical trial. Eur. J. Cancer 2018, 101, 236–243. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.G.; Dalle, S.; Haydon, A.M.; Meshcheryakov, A.; Khattak, A.; Carlino, M.S.; et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma (EORTC 1325-MG/KEYNOTE-054): Distant metastasis-free survival results from a double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 643–654. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Perez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Borghaei, H.; Langer, C.; Gadgeel, S.; Papadimitrakopoulou, V.; Patnaik, A.; Powell, S.; Gentzler, R.; Martins, R.; Stevenson, J.; Jalal, S.; et al. OA 17.01 Pemetrexed-Carboplatin Plus Pembrolizumab as First-Line Therapy for Advanced Nonsquamous NSCLC: KEYNOTE-021 Cohort G Update. J. Thorac. Oncol. 2017, 12, S1791. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Cay Senler, F.; Csoszi, T.; Fulop, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Ros, W.; Delord, J.P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, D.M.; Bariani, G.M.; Cassier, P.A.; Marabelle, A.; Hansen, A.R.; De Jesus Acosta, A.; Miller, W.H., Jr.; Safra, T.; Italiano, A.; Mileshkin, L.; et al. Pembrolizumab in Patients With Microsatellite Instability-High Advanced Endometrial Cancer: Results From the KEYNOTE-158 Study. J. Clin. Oncol. 2022, 40, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Colombo, N.; Dubot, C.; Lorusso, D.; Cáceres, V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Hoyos, E.; Yañez, E.; et al. LBA2 Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for persistent, recurrent, or metastatic cervical cancer: Randomized, double-blind, phase III KEYNOTE-826 study. Ann. Oncol. 2021, 32, S1307–S1308. [Google Scholar] [CrossRef]
- Santin, A.D.; Deng, W.; Frumovitz, M.; Buza, N.; Bellone, S.; Huh, W.; Khleif, S.; Lankes, H.A.; Ratner, E.S.; O’Cearbhaill, R.E.; et al. Phase II evaluation of nivolumab in the treatment of persistent or recurrent cervical cancer (NCT02257528/NRG-GY002). Gynecol. Oncol. 2020, 157, 161–166. [Google Scholar] [CrossRef]
- Naumann, R.W.; Hollebecque, A.; Meyer, T.; Devlin, M.J.; Oaknin, A.; Kerger, J.; Lopez-Picazo, J.M.; Machiels, J.P.; Delord, J.P.; Evans, T.R.J.; et al. Safety and Efficacy of Nivolumab Monotherapy in Recurrent or Metastatic Cervical, Vaginal, or Vulvar Carcinoma: Results From the Phase I/II CheckMate 358 Trial. J. Clin. Oncol. 2019, 37, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Tewari, K.S.; Monk, B.J.; Vergote, I.; Miller, A.; de Melo, A.C.; Kim, H.S.; Kim, Y.M.; Lisyanskaya, A.; Samouelian, V.; Lorusso, D.; et al. Survival with Cemiplimab in Recurrent Cervical Cancer. N. Engl. J. Med. 2022, 386, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Chase, D.M.; Slomovitz, B.M.; dePont Christensen, R.; Novak, Z.; Black, D.; Gilbert, L.; Sharma, S.; Valabrega, G.; Landrum, L.M.; et al. Dostarlimab for Primary Advanced or Recurrent Endometrial Cancer. N. Engl. J. Med. 2023, 388, 2145–2158. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbe, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Keilholz, U.; Mehnert, J.M.; Bauer, S.; Bourgeois, H.; Patel, M.R.; Gravenor, D.; Nemunaitis, J.J.; Taylor, M.H.; Wyrwicz, L.; Lee, K.W.; et al. Avelumab in patients with previously treated metastatic melanoma: Phase 1b results from the JAVELIN Solid Tumor trial. J. Immunother. Cancer 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Apolo, A.B.; Ellerton, J.A.; Infante, J.R.; Agrawal, M.; Gordon, M.S.; Aljumaily, R.; Gourdin, T.; Dirix, L.; Lee, K.W.; Taylor, M.H.; et al. Avelumab as second-line therapy for metastatic, platinum-treated urothelial carcinoma in the phase Ib JAVELIN Solid Tumor study: 2-year updated efficacy and safety analysis. J. Immunother. Cancer 2020, 8, e001246. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Massard, C.; Gordon, M.S.; Sharma, S.; Rafii, S.; Wainberg, Z.A.; Luke, J.; Curiel, T.J.; Colon-Otero, G.; Hamid, O.; Sanborn, R.E.; et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J. Clin. Oncol. 2016, 34, 3119–3125. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Mansfield, A.S.; Szczesna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Taylor, K.B.; Cohen, J.V.; Ayoubi, N.; Haugh, A.M.; Wang, D.Y.; Schlick, B.D.; Voorhees, A.L.; Gage, K.L.; Fintelmann, F.J.; et al. Anti-PD-1-Induced Pneumonitis Is Associated with Persistent Imaging Abnormalities in Melanoma Patients. Cancer Immunol. Res. 2019, 7, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Wang, X.; Alessi, J.V.; Rizvi, H.; Mahadevan, N.R.; Li, Y.Y.; Polio, A.; Lindsay, J.; Umeton, R.; Sinha, R.; et al. Association of High Tumor Mutation Burden in Non-Small Cell Lung Cancers With Increased Immune Infiltration and Improved Clinical Outcomes of PD-L1 Blockade Across PD-L1 Expression Levels. JAMA Oncol. 2022, 8, 1160–1168. [Google Scholar] [CrossRef]
- Rousseau, B.; Foote, M.B.; Maron, S.B.; Diplas, B.H.; Lu, S.; Argiles, G.; Cercek, A.; Diaz, L.A., Jr. The Spectrum of Benefit from Checkpoint Blockade in Hypermutated Tumors. N. Engl. J. Med. 2021, 384, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, P.; Leal, L.F.; Duval da Silva, V.; da Silva, E.C.A.; Cordeiro de Lima, V.C.; Reis, R.M. PD-L1 expression by Tumor Proportion Score (TPS) and Combined Positive Score (CPS) are similar in non-small cell lung cancer (NSCLC). J. Clin. Pathol. 2021, 74, 735–740. [Google Scholar] [CrossRef]
- Ito, T.; Okamoto, I.; Tokashiki, K.; Sato, H.; Okada, T.; Yamashita, G.; Nagao, T.; Hirai, H.; Saigusa, N.; Tsukahara, K. PD-L1 Expression and Survival Rates Using TPS and CPS for Nivolumab-treated Head-and-Neck Cancer. Anticancer Res. 2022, 42, 1547–1554. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Horiuchi, S.; Morooka, H.; Ibi, T.; Takahashi, N.; Ikeya, T.; Shimizu, Y.; Hoshi, E. Inter-tumor heterogeneity of PD-L1 expression in non-small cell lung cancer. J. Thorac. Dis. 2019, 11, 4982–4991. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, A.; Zhou, S.; Xu, Y.; Xiao, Y.; Bi, R.; Yang, W. Heterogeneity of PD-L1 expression in primary tumors and paired lymph node metastases of triple negative breast cancer. BMC Cancer 2018, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 Immunohistochemistry Assays for Lung Cancer: Results from Phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J. Thorac. Oncol. 2017, 12, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Choucair, K.; Morand, S.; Stanbery, L.; Edelman, G.; Dworkin, L.; Nemunaitis, J. TMB: A promising immune-response biomarker, and potential spearhead in advancing targeted therapy trials. Cancer Gene Ther. 2020, 27, 841–853. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef]
- Yamamoto, H.; Imai, K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin. Oncol. 2019, 46, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magri, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 71. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef]
- Anagnostopoulos, I.; Herbst, H.; Niedobitek, G.; Stein, H. Demonstration of monoclonal EBV genomes in Hodgkin’s disease and Ki-1-positive anaplastic large cell lymphoma by combined Southern blot and in situ hybridization. Blood 1989, 74, 810–816. [Google Scholar] [CrossRef]
- Sastre-Garau, X.; Peter, M.; Avril, M.F.; Laude, H.; Couturier, J.; Rozenberg, F.; Almeida, A.; Boitier, F.; Carlotti, A.; Couturaud, B.; et al. Merkel cell carcinoma of the skin: Pathological and molecular evidence for a causative role of MCV in oncogenesis. J. Pathol. 2009, 218, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Russell, J.; Lebbe, C.; Chmielowski, B.; Gambichler, T.; Grob, J.J.; Kiecker, F.; Rabinowits, G.; Terheyden, P.; Zwiener, I.; et al. Efficacy and Safety of First-line Avelumab Treatment in Patients With Stage IV Metastatic Merkel Cell Carcinoma: A Preplanned Interim Analysis of a Clinical Trial. JAMA Oncol. 2018, 4, e180077. [Google Scholar] [CrossRef] [PubMed]
- AbdulJabbar, K.; Raza, S.E.A.; Rosenthal, R.; Jamal-Hanjani, M.; Veeriah, S.; Akarca, A.; Lund, T.; Moore, D.A.; Salgado, R.; Al Bakir, M.; et al. Geospatial immune variability illuminates differential evolution of lung adenocarcinoma. Nat. Med. 2020, 26, 1054–1062. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Liu, Y.T.; Sun, Z.J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Zabransky, D.J.; Yarchoan, M.; Jaffee, E.M. Strategies for Heating Up Cold Tumors to Boost Immunotherapies. Annu. Rev. Cancer Biol. 2023, 7, 149–170. [Google Scholar] [CrossRef]
- Benoit, A.; Vogin, G.; Duhem, C.; Berchem, G.; Janji, B. Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy. Cells 2023, 12, 1787. [Google Scholar] [CrossRef]
- Moore, K.N.; Bookman, M.; Sehouli, J.; Miller, A.; Anderson, C.; Scambia, G.; Myers, T.; Taskiran, C.; Robison, K.; Maenpaa, J.; et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). J. Clin. Oncol. 2021, 39, 1842–1855. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Colombo, N.; Oza, A.M.; Fujiwara, K.; Birrer, M.J.; Randall, L.M.; Poddubskaya, E.V.; Scambia, G.; Shparyk, Y.V.; Lim, M.C.; et al. LBA 25—Scientific Plenary: Avelumab in combination with and/or following chemotherapy vs chemotherapy alone in patients with previously untreated epithelial ovarian cancer: Results from the phase 3 javelin ovarian 100 trial. Gynecol. Oncol. 2020, 159, 13–14. [Google Scholar] [CrossRef]
- Monk, B.J.; Colombo, N.; Oza, A.M.; Fujiwara, K.; Birrer, M.J.; Randall, L.; Poddubskaya, E.V.; Scambia, G.; Shparyk, Y.V.; Lim, M.C.; et al. Chemotherapy with or without avelumab followed by avelumab maintenance versus chemotherapy alone in patients with previously untreated epithelial ovarian cancer (JAVELIN Ovarian 100): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Colombo, N.; Oza, A.; Fujiwara, K.; Birrer, M.; Randall, L.; Poddubskaya, E.; Scambia, G.; Shparyk, Y.; Lim, M.; et al. 1 Avelumab in combination with and/or following chemotherapy vs chemotherapy in treatment-naive patients with ovarian cancer: Biomarker analyses from the phase 3 JAVELIN Ovarian 100 trial. Int. J. Gynecol. Cancer 2020, 30, A1. [Google Scholar] [CrossRef]
- Moore, K.N.; Okamoto, A.; Wu, F.; Lin, Y.G.; Pignata, S. IMagyn050/GOG3015/ENGOT-ov39: A randomized, double-blind, phase III study of atezolizumab vs placebo combined with chemotherapy + bevacizumab in stage III-IV ovarian, fallopian tube & peritoneal cancers (OC). Ann. Oncol. 2017, 28, V350–V351. [Google Scholar]
- Yu, H.; Boyle, T.A.; Zhou, C.; Rimm, D.L.; Hirsch, F.R. PD-L1 Expression in Lung Cancer. J. Thorac. Oncol. 2016, 11, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Riviere, P.; Goodman, A.M.; Okamura, R.; Barkauskas, D.A.; Whitchurch, T.J.; Lee, S.; Khalid, N.; Collier, R.; Mareboina, M.; Frampton, G.M.; et al. High Tumor Mutational Burden Correlates with Longer Survival in Immunotherapy-Naive Patients with Diverse Cancers. Mol. Cancer Ther. 2020, 19, 2139–2145. [Google Scholar] [CrossRef]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef]
- Martin, S.D.; Brown, S.D.; Wick, D.A.; Nielsen, J.S.; Kroeger, D.R.; Twumasi-Boateng, K.; Holt, R.A.; Nelson, B.H. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PLoS ONE 2016, 11, e0155189. [Google Scholar] [CrossRef]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef]
- Fan, S.; Gao, X.; Qin, Q.; Li, H.; Yuan, Z.; Zhao, S. Association between tumor mutation burden and immune infiltration in ovarian cancer. Int. Immunopharmacol. 2020, 89, 107126. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Chen, R.; Bai, Y.; Lu, X. The prognostic value of tumor-infiltrating T lymphocytes in ovarian cancer. Oncotarget 2017, 8, 15621–15631. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef]
- Rocconi, R.P.; Stevens, E.E.; Bottsford-Miller, J.N.; Ghamande, S.A.; Elder, J.; DeMars, L.L.; Munkarah, A.; Aaron, P.; Stanbery, L.; Wallraven, G.; et al. Proof of principle study of sequential combination atezolizumab and Vigil in relapsed ovarian cancer. Cancer Gene Ther. 2022, 29, 369–382. [Google Scholar] [CrossRef]
- Barve, M.; Aaron, P.; Manning, L.; Bognar, E.; Wallraven, G.; Horvath, S.; Stanbery, L.; Nemunaitis, J. Pilot Study of Combination Gemogenovatucel-T (Vigil) and Durvalumab in Women With Relapsed BRCA-wt Triple-Negative Breast or Ovarian Cancer. Clin. Med. Insights Oncol. 2022, 16, 11795549221110501. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Senzer, N.; Barve, M.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Lazar, M.; Lifshitz, S.; Magee, M.; Oh, J.; Mill, S.W.; et al. Phase I trial of “bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol. Ther. 2012, 20, 679–686. [Google Scholar] [CrossRef]
- Senzer, N.; Barve, M.; Nemunaitis, J.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Magee, M.; Oh, J.; Bedell, C.; Kumar, P.; et al. Long Term Follow Up: Phase I Trial of “bi-shRNA furin/GMCSF DNA/Autologous Tumor Cell” Immunotherapy (FANG™) in Advanced Cancer. J. Vaccines Vaccin. 2013, 4, 209. [Google Scholar]
- Oh, J.; Barve, M.; Matthews, C.M.; Koon, E.C.; Heffernan, T.P.; Fine, B.; Grosen, E.; Bergman, M.K.; Fleming, E.L.; DeMars, L.R.; et al. Phase II study of Vigil(R) DNA engineered immunotherapy as maintenance in advanced stage ovarian cancer. Gynecol. Oncol. 2016, 143, 504–510. [Google Scholar] [CrossRef]
- Oh, J.; Barve, M.; Senzer, N.; Aaron, P.; Manning, L.; Wallraven, G.; Bognar, E.; Stanbery, L.; Horvath, S.; Manley, M.; et al. Long-term follow-up of Phase 2A trial results involving advanced ovarian cancer patients treated with Vigil(R) in frontline maintenance. Gynecol. Oncol. Rep. 2020, 34, 100648. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Kochupurakkal, B.; Izarzugaza, J.M.; Eklund, A.C.; Li, Y.; Liu, J.; Szallasi, Z.; Matulonis, U.A.; Richardson, A.L.; Iglehart, J.D.; et al. Tumor mutation burden forecasts outcome in ovarian cancer with BRCA1 or BRCA2 mutations. PLoS ONE 2013, 8, e80023. [Google Scholar] [CrossRef] [PubMed]
- Sliheet, E.; Robinson, M.; Morand, S.; Choucair, K.; Willoughby, D.; Stanbery, L.; Aaron, P.; Bognar, E.; Nemunaitis, J. Network based analysis identifies TP53m-BRCA1/2wt-homologous recombination proficient (HRP) population with enhanced susceptibility to Vigil immunotherapy. Cancer Gene Ther. 2022, 29, 993–1000. [Google Scholar] [CrossRef]
- Rocconi, R.P.; Stanbery, L.; Tang, M.; Walter, A.; Monk, B.J.; Herzog, T.J.; Coleman, R.L.; Manning, L.; Wallraven, G.; Horvath, S.; et al. ENTPD1/CD39 as a predictive marker of treatment response to gemogenovatucel-T as maintenance therapy in newly diagnosed ovarian cancer. Commun. Med. 2022, 2, 106. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPI (Target) | Indication/Population | Study (Arms) | Response | Ref. |
|---|---|---|---|---|
| Pembrolizumab (anti-PD-1) | Unresectable or metastatic BRAFV600E mutated metastatic melanoma, refractory to CTLA-4 therapy and BRAF inhibitor | NCT01295827 (Pembrolizumab) | ORR: 24% | [104] |
| Unresectable or metastatic, untreated melanoma regardless or BRAF status | Keynote-006 (Pembrolizumab vs. Ipilimumab) | 1-year OS: 74.1% (vs. 58.2%; p = 0.0005); 1-year PFS: 47.3% (vs. 26.5%; p < 0.001); PD-L1 ≥1%: 24 months OS and PFS: 58% and 33% vs. 45% and 13%, respectively | [105,106] | |
| Stage III melanoma/adjuvant | Keynote-054 (pembrolizumab vs. placebo) | RFS: 65.33% vs. 49.4%; p < 0.0001 | [107] | |
| Previously treated, PD-L1 positive, advanced NSCLC | Keynote-010 (pembrolizumab vs. docetaxel) | OS: 10.4 vs. 8.5 months; p = 0.0008 | [12,108] | |
| Untreated, PD-L1 ≥50 advanced NSCLC | Keynote-024 (pembrolizumab vs. chemotherapy) | 6 months OS: 80.2% vs. 72.4%; p = 0.005 Median PFS: 10.3 vs. 6.0 months; p < 0.001 ORR: 44.8% vs. 27.8% | [109] | |
| Untreated, non-squamous NSCLC without sensitizing mutations, regardless of PD-L1 level | Keynote-021 (pembrolizumab plus chemotherapy vs. chemotherapy | Median PFS: 19.0 vs. 8.9 months (HR: 0.53; p = 0.0049) ORR: 56.7% vs. 26.4%; p = 0.0016 Note: data from Keynote-189 and 407 revealed consistent PFS and OS advantage for pembrolizumab | [110,111,112] | |
| Recurrent/metastatic head and neck squamous cancer; 2nd line or 1st line in CPS ≥1 | Keynote-012 (pembrolizumab) | ORR: 16% | [113,114] | |
| Unresectable/metastatic urothelial cancer; 2nd line | Keynote-045 (pembrolizumab) | Median OS: 10.3 vs. 7.4 months (p = 0.002) | [115] | |
| Unresectable/metastatic urothelial cancer, cisplatin-ineligible; 1st line in CPS ≥10 | Keynote-052 (pembrolizumab) | ORR: 38% | [116] | |
| MSI-H, dMMR or TMB-H solid tumors | Keynote-012, 016, 028, 158 * and 164 (pembrolizumab) | MSI-H or dMMR studies: ORRs ~ 40% TMB-H: ORR: 29% (vs. 6% TMB-L) Cervical cancer cohort/MSI-H or dMMR: ORR: 12.2% Endometrial cancer cohort/MSI-H or dMMR: ORR: 48% | [117,118,119] | |
| Persistent/recurrent or metastatic cervical cancer with PD-L1 CPS ≥ 1 | Keynote-826 (pembrolizumab vs. placebo + paclitaxel/platinum +/− bevacizumab | Median OS: not reached vs. 16.3 months (p = 0.0001) Median PFS: 10.4 vs. 8.2 months (p < 0.0001) ORR: 68% vs. 50%; median DOR: 18 vs. 10.4 months | [120] | |
| Nivolumab (anti-PD-1) | Persistent/recurrent cervical cancer | NRG-GY002 (nivolumab) | 6 months PFS: 16%; 6 months OS: 78% Median duration of SD: 5.7 months | [121] |
| Persistent/recurrent cervical cancer | CheckMate-358 (nivolumab) | ORR: 26.3% Median OS: 21.9 months | [122] | |
| Cemiplimab (anti-PD-1) | Recurrent or metastatic cervical cancer after platinum-based chemotherapy, regardless of PD-L1 | NCT03257267 (cemiplimab vs. investigator-choice chemotherapy) | ORR: 16.4% vs. 6.3% Median OS: 12.0 vs. 8.5 months (p < 0.001) Median PFS: HR 0.75, p < 0.001 | [123] |
| Dostarlimab-gxly (anti-PD-1) | Primary advanced or recurrent dMMR or MSI-H endometrial cancer | RUBY (dostarlimab-gxly vs. placebo + carboplatin-paclitaxel, followed by dostarlimab or placebo | Median PFS: 30.3 vs. 7.7 months (p < 0.0001) | [124] |
| Avelumab (anti-PD-L1) | Chemo-refractory, metastatic Merkel cell carcinoma, regardless of PD-L1 | NCT02155647 (Avelumab) | ORR: 31.8% (28/88); CR: 8/88 and PR: 20/88 | [125] |
| Advanced urothelial carcinoma, 2nd line setting | JAVELIN Solid Tumor (Avelumab) | ORR: 16.5%; CR: 4.1%; PR: 12.4% Median DoR: 20.5 months Median PFS: 1.6 months Median OS: 7.0 months; 24-month OS: 20.1% | [126,127] | |
| Advanced renal cell carcinoma (1st line in combination with Axitinib | JAVELIN Renal (Avelumab + Axitinib vs. sunitinib) | PD-L1+: median PFS: 13.8 vs. 7.2 months (p < 0.001); ORR: 55.2% (vs. 25.5%) Overall population: median PFS: 13.8 vs. 8.4 (p < 0.001) | [128] | |
| Durvalumab (anti-PD-L1) | Advanced/metastatic urothelial carcinoma, platinum-refractory | NCT01693562 (Durvalumab) | ORR: 31% (all patients), 46.4% (PD-L1 positive), 0% (PD-L1 negative) | [129] |
| Stage III NSCLC, maintenance therapy, post-chemo-radiation | PACIFIC Trial (Durvalumab vs. placebo) | Median PFS: 16.8 vs. 5.6 months (p < 0.001) ORR: 28.4% vs. 16% (p < 0.001) | [130] | |
| Atezolizumab (anti-PD-L1) | Metastatic urothelial carcinoma, platinum ineligible or refractory, regardless of PD-L1 | IMvigor210 (Atezolizumab) | ORR: 23% Median PFS: 2.7 months Median OS: 15.9 months | [131] |
| OK | Metastatic NSCLC, 2nd line post platinum-based chemotherapy, regardless of PD-L1 | OAK/POPLAR (Atezolizumab vs. docetaxel) | Median OS: 12.6–13.3 vs. 9.7–9.8 months (p = 0.05) | [132,133] |
| Extensive stage SCLC, 1st line setting | IMpower 133 (Chemo/atezolizumab vs. chemotherapy/ placebo) | Median OS: 12.3 vs. 10.3 months (p = 0.007) Median PFS: 5.2 vs. 4.3 months (p = 0.02) | [134] |
| Syndrome | Affected Maintenance Mechanism | Main Type of Genome Instability | Major Cancer Predisposition |
|---|---|---|---|
| Xeroderma pigmentosum | NER (±Transcription coupled repair) | Point mutations | UV-induced skin cancer |
| Cockayne syndrome | Transcription coupled repair | Point mutations | None * |
| Trichothiodystrophy | NER/Transcription coupled repair | Point mutations | None * |
| Ataxia telangiectasia (AT) | DSB response/repair | Chromosome aberrations | Lymphomas |
| AT-like disorder | DSB response/repair | Chromosome aberrations | Lymphomas |
| Nijmegen breakage syndrome | DSB response/repair | Chromosome aberrations | Lymphomas |
| BRCA 1/BRCA 2 | Homologous recombination | Chromosome aberrations | Breast (ovarian) cancer |
| Werner syndrome | Homologous recombination/TLS | Chromosome aberrations | Various cancers |
| Bloom syndrome | Homologous recombination | Chromosome aberrations (SCE↑) | Leukemia, lymphoma, others |
| Rothmund-Thomson syndrome | Homologous recombination | Chromosome aberrations | Osteosarcoma |
| Ligase IV deficiency † | EJ | Recombination fidelity | Leukemia |
| HNPCC | MMR | Point mutations | Colorectal cancer |
| Xeroderma pigmentosum variant | TLS ‡ | Point mutations | UV-induced skin cancer |
| ERCC6L2 deficiency | NER | Point mutations | hematologic |
| Constitutional mismatch repair disorder | MMR | Point mutations and insertion/deletions | Hematologic, brain and intestinal tract |
| Fanconi Anemia | FA | Chromosomal aberrations | SCC, AML, MDS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemunaitis, J.; Stanbery, L.; Willoughby, D.; Bognar, E.; Brun, S.; Walter, A.; Monk, B.J.; Rocconi, R.P.; Choucair, K.; Coleman, R.L. Clonal Neoantigen: Emerging “Mechanism-based” Biomarker of Immunotherapy Response. Cancers 2023, 15, 5616. https://doi.org/10.3390/cancers15235616
Nemunaitis J, Stanbery L, Willoughby D, Bognar E, Brun S, Walter A, Monk BJ, Rocconi RP, Choucair K, Coleman RL. Clonal Neoantigen: Emerging “Mechanism-based” Biomarker of Immunotherapy Response. Cancers. 2023; 15(23):5616. https://doi.org/10.3390/cancers15235616
Chicago/Turabian StyleNemunaitis, John, Laura Stanbery, David Willoughby, Ernest Bognar, Scott Brun, Adam Walter, Bradley J. Monk, Rodney P. Rocconi, Khalil Choucair, and Robert L. Coleman. 2023. "Clonal Neoantigen: Emerging “Mechanism-based” Biomarker of Immunotherapy Response" Cancers 15, no. 23: 5616. https://doi.org/10.3390/cancers15235616
APA StyleNemunaitis, J., Stanbery, L., Willoughby, D., Bognar, E., Brun, S., Walter, A., Monk, B. J., Rocconi, R. P., Choucair, K., & Coleman, R. L. (2023). Clonal Neoantigen: Emerging “Mechanism-based” Biomarker of Immunotherapy Response. Cancers, 15(23), 5616. https://doi.org/10.3390/cancers15235616