
Identifying Candidate Gene Drivers Associated with Relapse in Pediatric T-Cell Acute Lymphoblastic Leukemia Using a Gene Co-Expression Network Approach
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. T-ALL Patient’s Samples
2.2. 10X Genomics Single-Cell Processing, Libraries Preparation, and Sequencing
2.3. scRNAseq Data Analysis
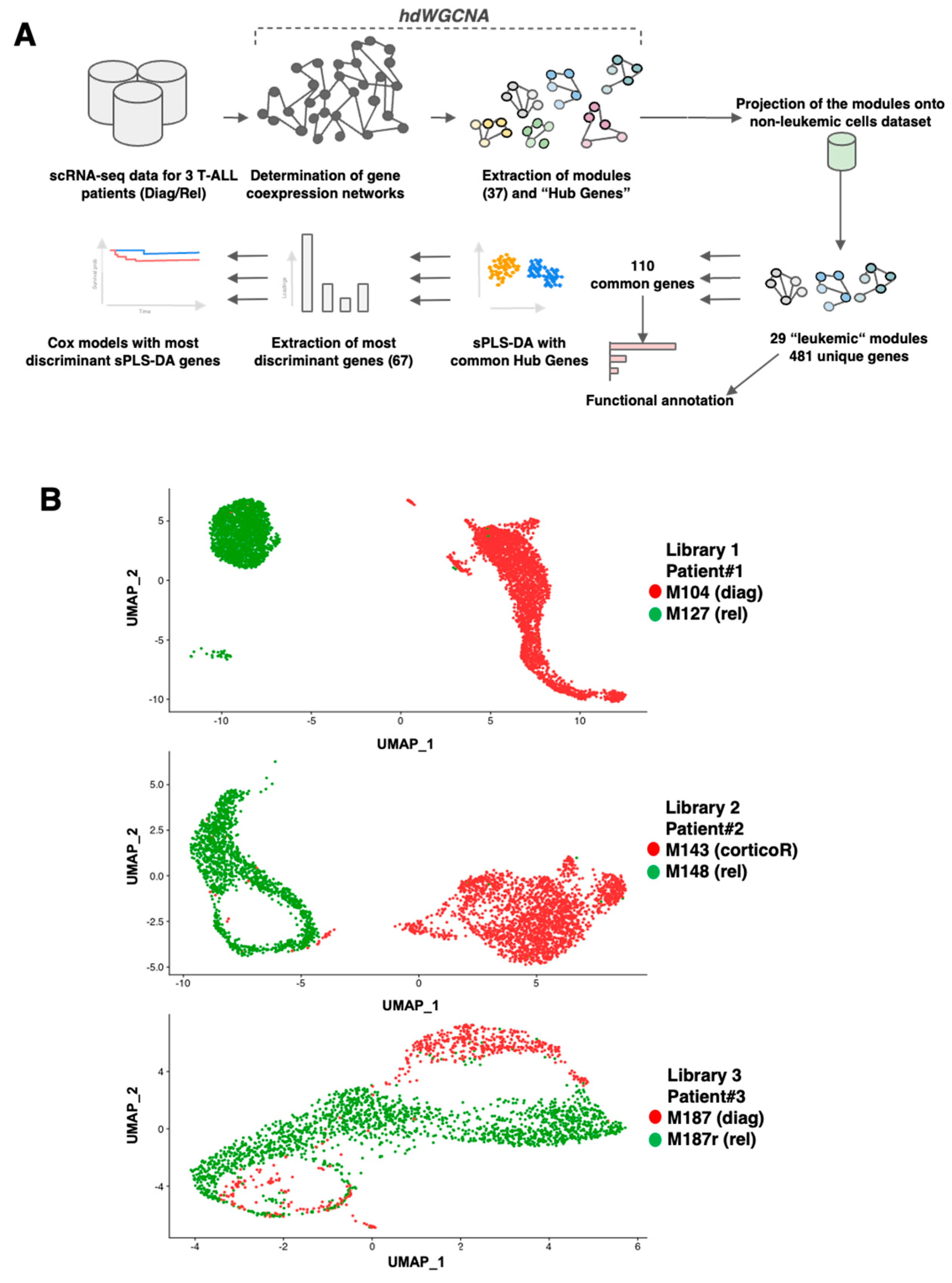
2.4. Gene Co-Expression Network Constructions
2.5. Module Projection on Healthy T-Cells Dataset
2.6. Module Preservation Statistics
2.7. Diagnosis–Relapse, Most Discriminating Genes
2.8. Relationship between Gene Signatures and Survival
3. Results
3.1. scRNAseq Analysis of Three Paired Diagnosis–Relapse T-ALL Samples
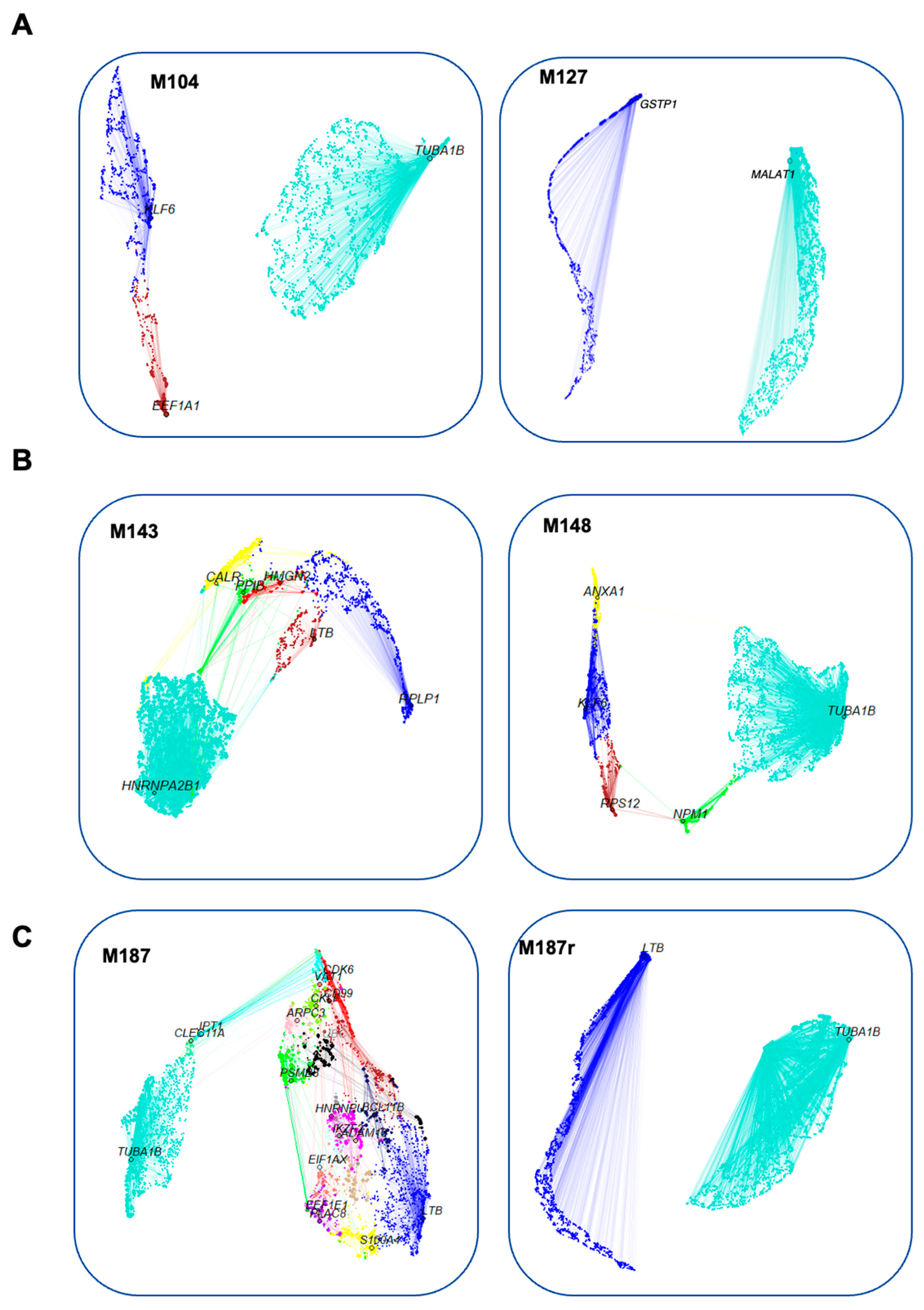
3.2. Construction of Gene Co-Expression Networks
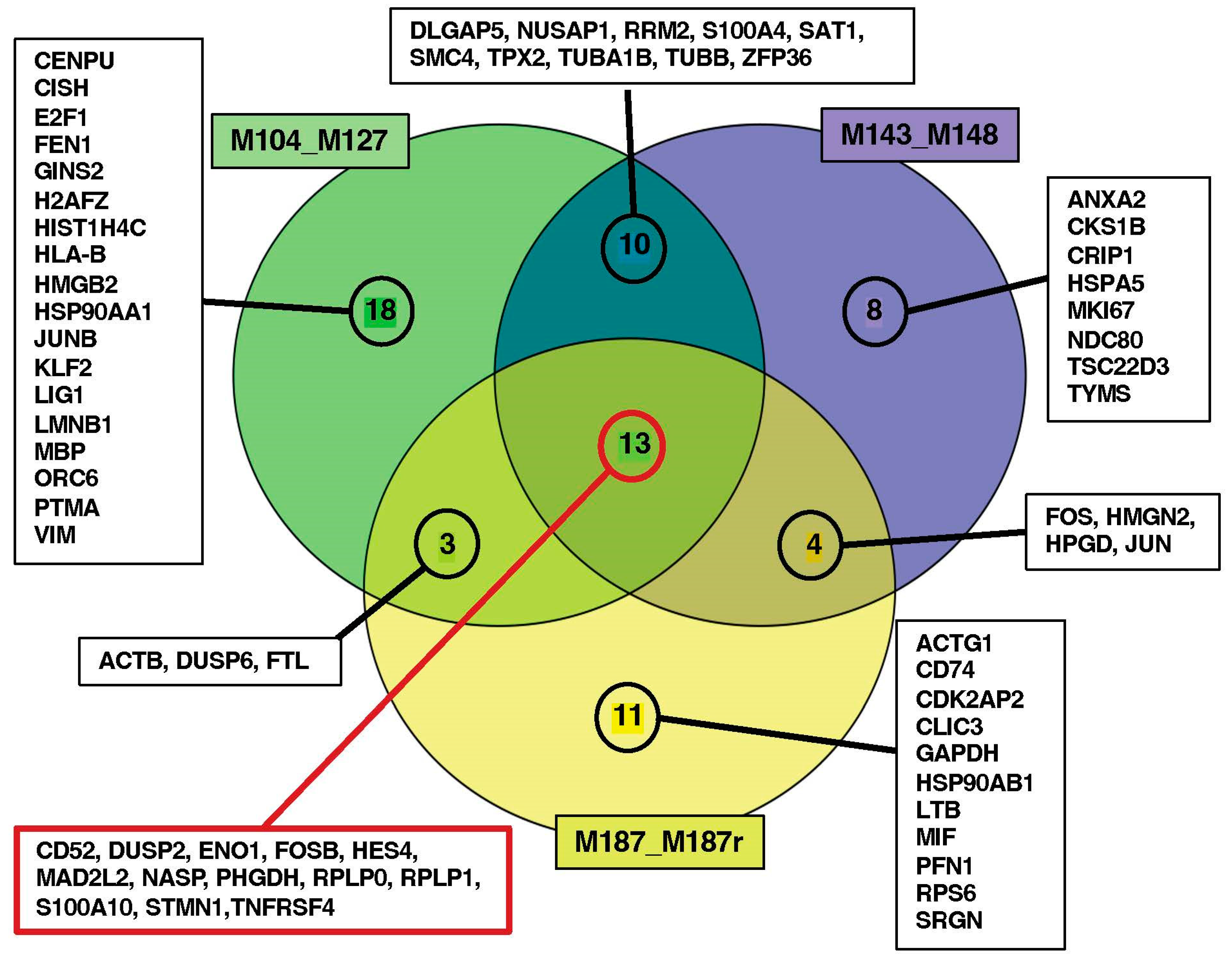
3.3. Search for Conserved Relapse-Associated Hub Genes
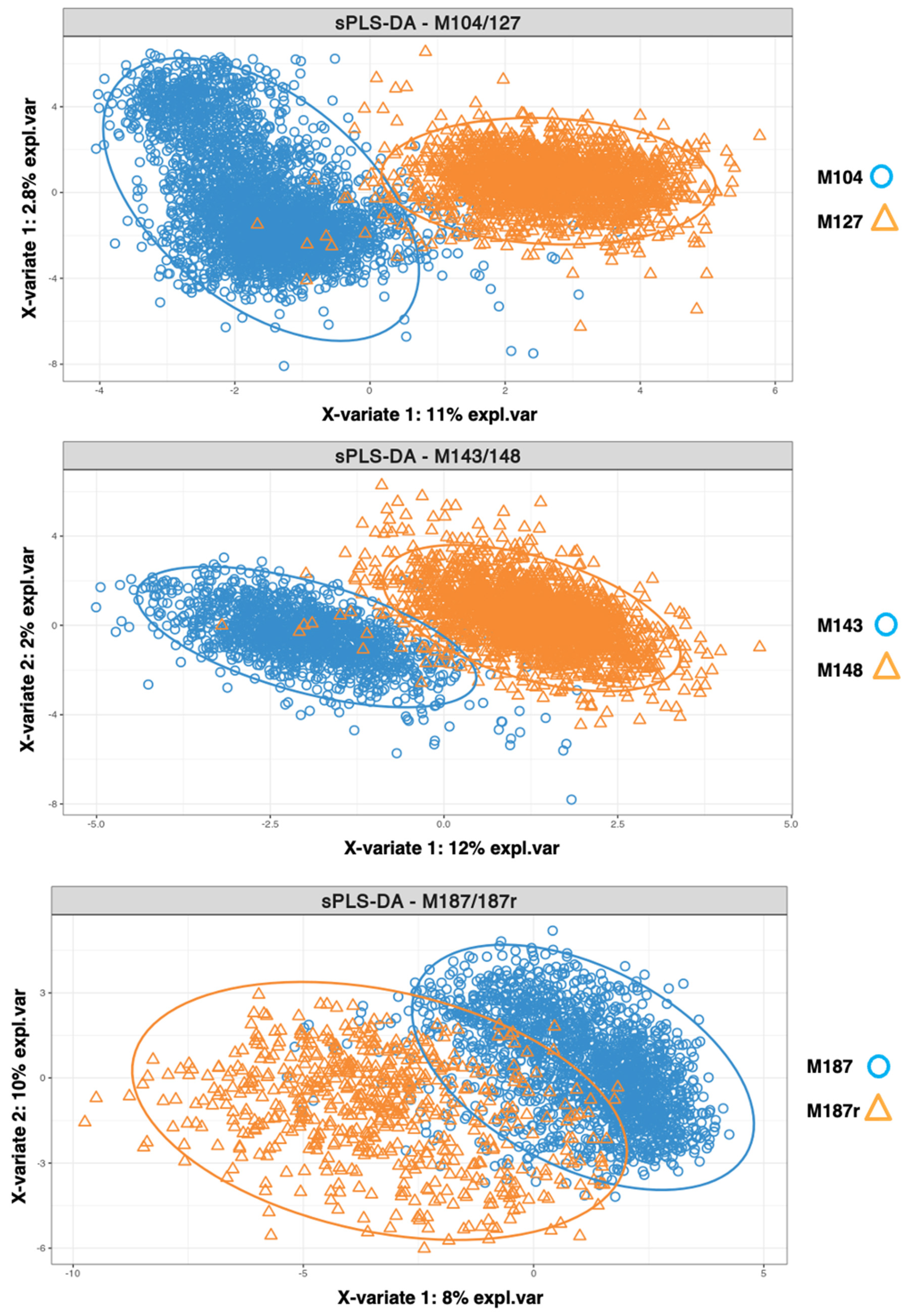
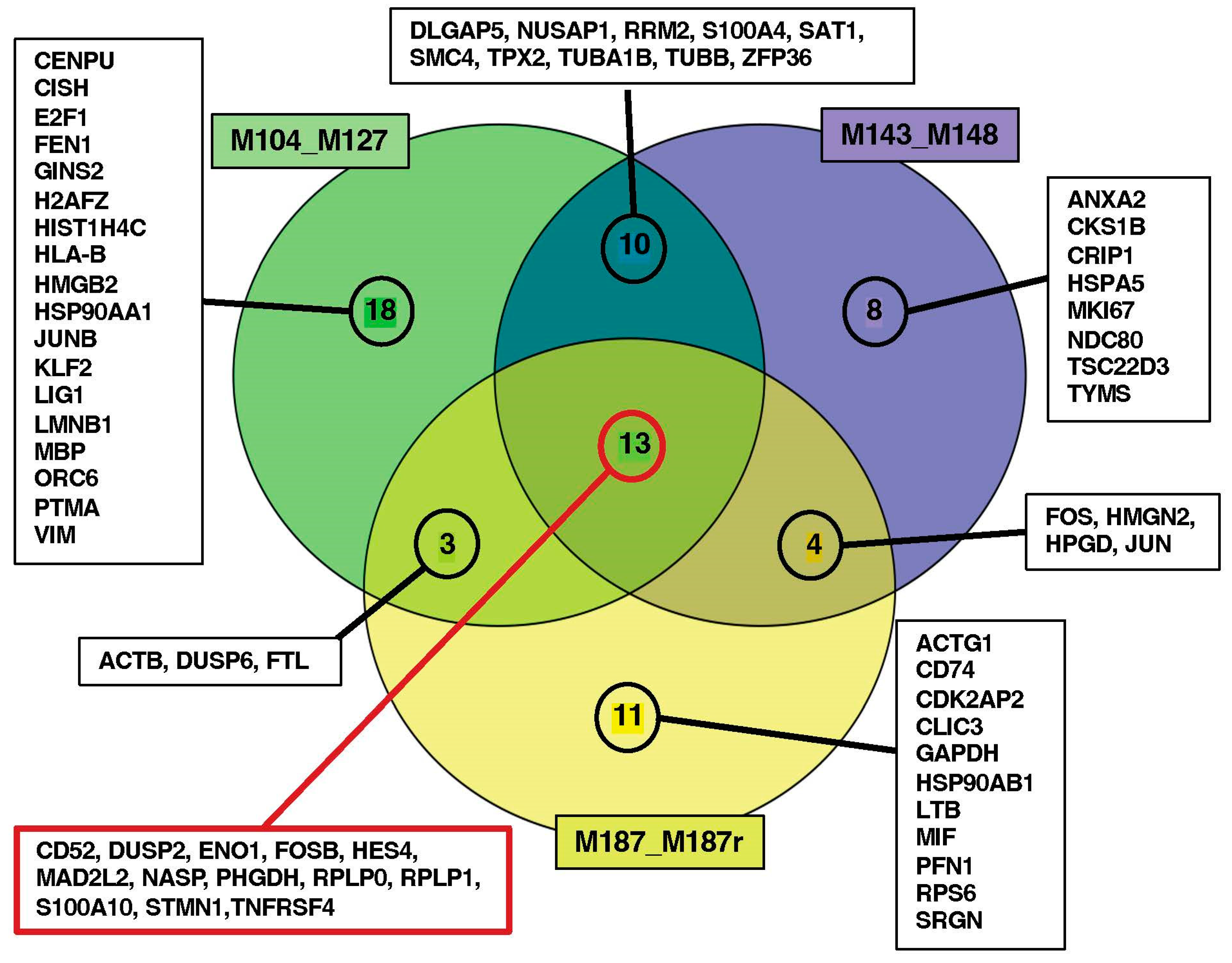
3.4. Identification of Diagnosis–Relapse Discriminating Genes
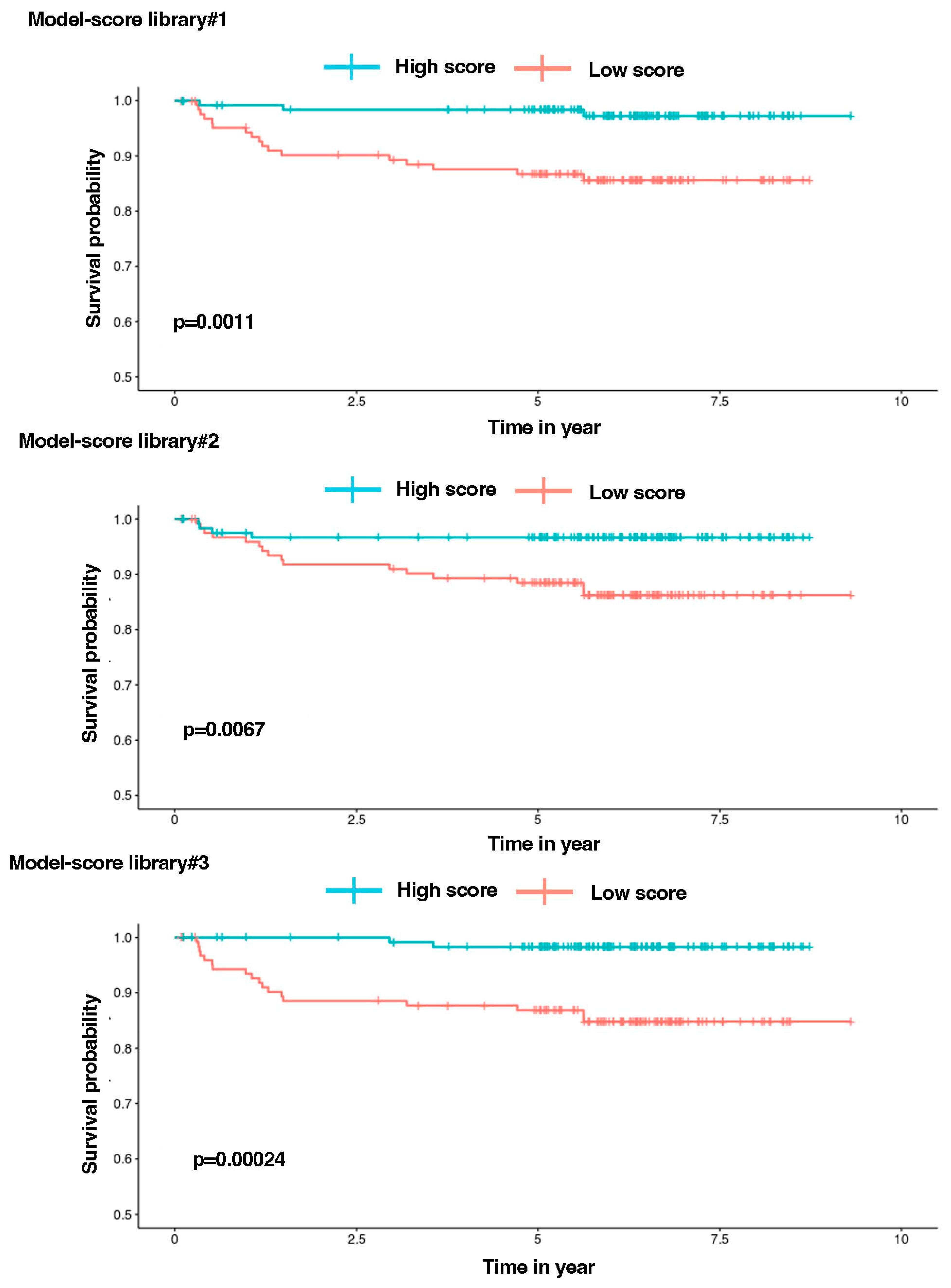
3.5. Establishment of Library Gene Signatures and Correlation with Patient’s Survival
4. Discussion
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Pui, C.H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019, 20, e142–e154. [Google Scholar] [CrossRef]
- Ferrando, A. Can one target T-cell ALL. Best. Pract. Res. Clin. Haematol. 2018, 31, 361–366. [Google Scholar] [CrossRef]
- Brady, S.W.; Roberts, K.G.; Gu, Z.; Shi, L.; Pounds, S.; Pei, D.; Cheng, C.; Dai, Y.; Devidas, M.; Qu, C.; et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat. Genet. 2022, 54, 1376–1389. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Langfelder, P.; Fuller, T.; Dong, J.; Li, A.; Hovarth, S. Weighted gene coexpression network analysis: State of the art. J. Biopharm. Stat. 2010, 20, 281–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Cahu, X.; Calvo, J.; Poglio, S.; Prade, N.; Colsch, B.; Arcangeli, M.-L.; Leblanc, T.; Petit, A.; Baleydier, F.; Baruchel, A.; et al. Bone marrow sites differently imprint dormancy and chemoresistance to T-cell acute lymphoblastic leukemia. Blood Adv. 2017, 1, 1760–1772. [Google Scholar] [CrossRef]
- Available online: https://cran.r-project.org/web/packages/Seurat/citation.html (accessed on 20 April 2024).
- Chung, N.C.; Storey, J.D. Statistical significance of variables driving systematic variation in high-dimensional data. Bioinformatics 2015, 31, 545–554. [Google Scholar] [CrossRef]
- Van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene–disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Fionda, V. Networks in Biology. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 915–921. [Google Scholar]
- Morabito, S.; Reese, F.; Rahimzadeh, N.; Miyoshi, E.; Swarup, V. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell Rep. Methods 2023, 3, 100498. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Hasankhani, A.; Bahrami, A.; Sheybani, N.; Aria, B.; Hemati, B.; Fatehi, F.; Farahani, H.G.M.; Javanmard, G.; Rezaee, M.; Kastelic, J.P.; et al. Differential Co-Expression Network Analysis Reveals Key Hub-High Traffic Genes as Potential Therapeutic Targets for COVID-19 Pandemic. Front. Immunol. 2021, 12, 789317. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Luo, R.; Oldham, M.C.; Horvath, S. Is My Network Module Preserved and Reproducible? PLoS Comput. Biol. 2011, 7, e1001057. [Google Scholar] [CrossRef]
- Ding, M.; Li, F.; Wang, B.; Chi, G.; Liu, H. A comprehensive analysis of WGCNA and serum metabolomics manifests the lung cancer-associated disordered glucose metabolism. J. Cell. Biochem. 2019, 120, 10855–10863. [Google Scholar] [CrossRef]
- Lê Cao, K.A.; Welham, Z.M. mixOmics: Omics Data Integration Project. Bioconductor 3.17. Available online: https://www.bioconductor.org/packages/release/bioc/html/mixOmics.html (accessed on 20 April 2024).
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Cao, Z.; Sun, X.; Icli, B.; Wara, A.K.; Feinberg, M.W. Role of Kruppel-like factors in leukocyte development, function, and disease. Blood 2010, 116, 4404–4414. [Google Scholar] [CrossRef]
- Narla, G.; Heath, K.E.; Reeves, H.L.; Li, D.; Giono, L.E.; Kimmelman, A.C.; Glucksman, M.J.; Narla, J.; Eng, F.J.; Chan, A.M.; et al. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science 2001, 294, 2563–2566. [Google Scholar] [CrossRef] [PubMed]
- Hatami, R.; Sieuwerts, A.M.; Izadmehr, S.; Yao, Z.; Qiao, R.F.; Papa, L.; Look, M.P.; Smid, M.; Ohlssen, J.; Levine, A.C.; et al. KLF6-SV1 drives breast cancer metastasis and is associated with poor survival. Sci. Transl. Med. 2013, 5, 169ra12. [Google Scholar] [CrossRef] [PubMed]
- Warke, V.G.; Nambiar, M.P.; Krishnan, S.; Tenbrock, K.; Geller, D.A.; Koritschoner, N.P.; Atkins, J.L.; Farber, D.L.; Tsokos, G.C. Transcriptional activation of the human inducible nitric-oxide synthase promoter by Kruppel-like factor 6. J. Biol. Chem. 2003, 278, 14812–14819. [Google Scholar] [CrossRef] [PubMed]
- DeKelver, R.C.; Lewin, B.; Lam, K.; Komeno, Y.; Yan, M.; Rundle, C.; Lo, M.C.; Zhang, D.E. Cooperation between RUNX1-ETO9a and novel transcriptional partner KLF6 in upregulation of Alox5 in acute myeloid leukemia. PLoS Genet. 2013, 9, e1003765. [Google Scholar] [CrossRef] [PubMed]
- Sharaf-Eldein, M.; Elghannam, D.; Elderiny, W.; Abdel-Malak, C. Prognostic Implication of MIF Gene Expression in Childhood Acute Lymphoblastic Leukemia. Clin. Lab. 2018, 64, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Shalek, A.K.; Benson, M. Single-cell analyses to tailor treatments. Sci. Transl. Med. 2017, 9, eaan4730. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kypraios, A.; Bennour, J.; Imbert, V.; David, L.; Calvo, J.; Pflumio, F.; Bonnet, R.; Couralet, M.; Magnone, V.; Lebrigand, K.; et al. Identifying Candidate Gene Drivers Associated with Relapse in Pediatric T-Cell Acute Lymphoblastic Leukemia Using a Gene Co-Expression Network Approach. Cancers 2024, 16, 1667. https://doi.org/10.3390/cancers16091667
Kypraios A, Bennour J, Imbert V, David L, Calvo J, Pflumio F, Bonnet R, Couralet M, Magnone V, Lebrigand K, et al. Identifying Candidate Gene Drivers Associated with Relapse in Pediatric T-Cell Acute Lymphoblastic Leukemia Using a Gene Co-Expression Network Approach. Cancers. 2024; 16(9):1667. https://doi.org/10.3390/cancers16091667
Chicago/Turabian StyleKypraios, Anthony, Juba Bennour, Véronique Imbert, Léa David, Julien Calvo, Françoise Pflumio, Raphaël Bonnet, Marie Couralet, Virginie Magnone, Kevin Lebrigand, and et al. 2024. "Identifying Candidate Gene Drivers Associated with Relapse in Pediatric T-Cell Acute Lymphoblastic Leukemia Using a Gene Co-Expression Network Approach" Cancers 16, no. 9: 1667. https://doi.org/10.3390/cancers16091667
APA StyleKypraios, A., Bennour, J., Imbert, V., David, L., Calvo, J., Pflumio, F., Bonnet, R., Couralet, M., Magnone, V., Lebrigand, K., Barbry, P., Rohrlich, P. S., & Peyron, J.-F. (2024). Identifying Candidate Gene Drivers Associated with Relapse in Pediatric T-Cell Acute Lymphoblastic Leukemia Using a Gene Co-Expression Network Approach. Cancers, 16(9), 1667. https://doi.org/10.3390/cancers16091667