
Hypomethylation of ATP1A1 Is Associated with Poor Prognosis and Cancer Progression in Triple-Negative Breast Cancer
 and
and {kind=link}
{kind=link}
{kind=link}
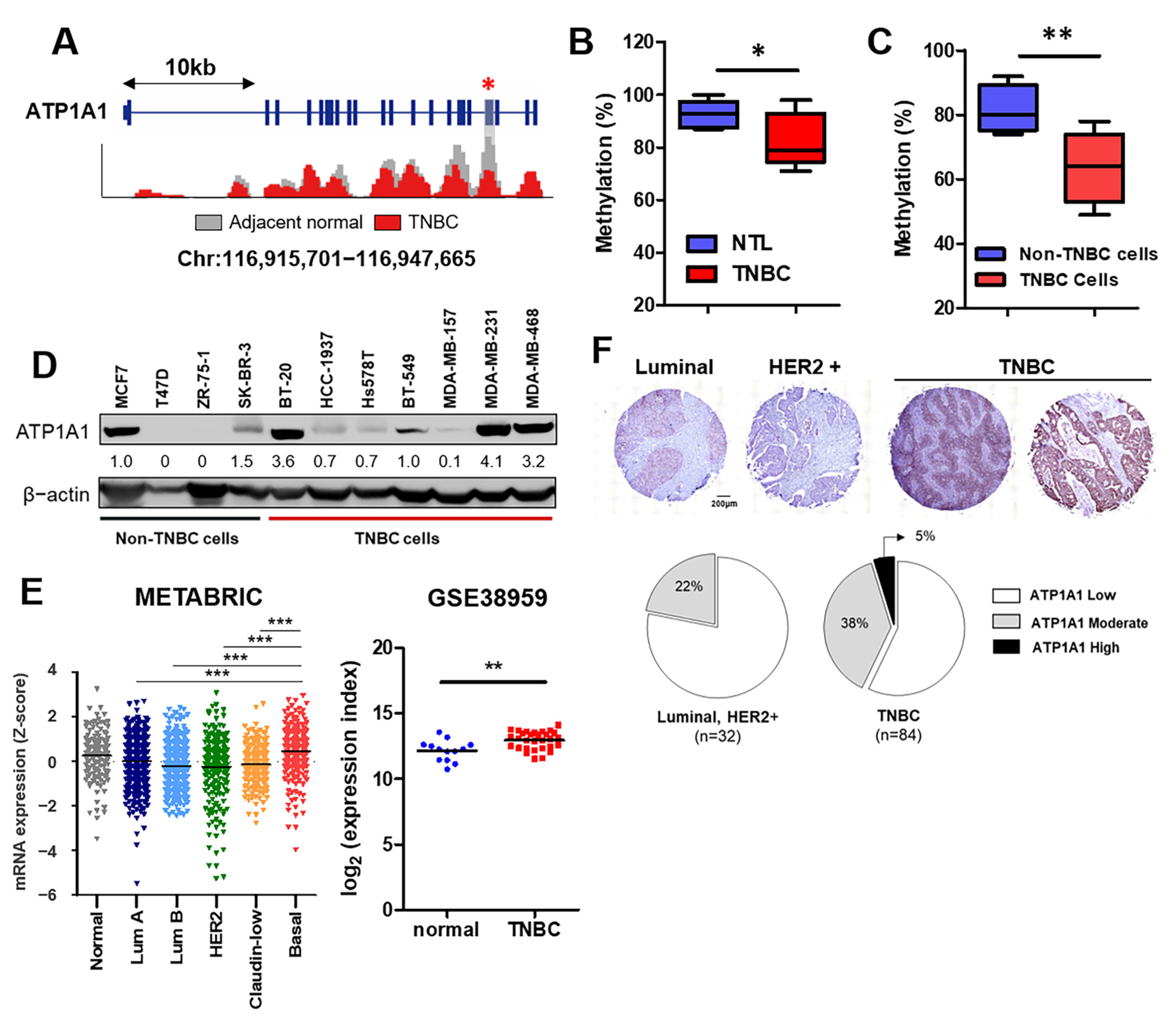{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Breast Cancer Tissue
2.2. MBD-Seq Library Preparation and Next-Generation Sequencing
2.3. Data Analysis of MBD-Seq
2.4. RNA Preparation and mRNA Microarray
2.5. Gene Ontology (GO) Analysis
2.6. Acquisition of TCGA, GEO Data, and Analysis of Survival and Its Association with Molecular Subtype
2.7. Bisulfite Treatment and MS-HRM Analysis
2.8. Cell Culture and Transfection
2.9. Western Blotting Analysis
2.10. Immunohistochemical Analysis of Tissue Microarrays (TMAs)
2.11. Cell Viability Assay
2.12. Mammosphere Formation
2.13. Colony Formation Assay
2.14. Apoptosis Analysis
2.15. Statistical Analysis
3. Results
3.1. Identification of TNBC-Specific DNA Methylation
3.2. Integrative Analysis of DNA Methylation and Gene Expression in TNBC
3.3. Strong Association between Hypomethylation/High Expression of ATP1A1 and Poor Survival
3.4. Validation of Methylation Status and Expression Levels of ATP1A1 in Patients with TNBC
3.5. Inhibition of Tumor Aggressiveness by Depletion of ATP1A1 in TNBC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harris, L.; Fritsche, H.; Mennel, R.; Norton, L.; Ravdin, P.; Taube, S.; Somerfield, M.R.; Hayes, D.F.; Bast, R.C., Jr. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 5287–5312. [Google Scholar] [CrossRef] [PubMed]
- Cleator, S.; Heller, W.; Coombes, R.C. Triple-negative breast cancer: Therapeutic options. Lancet Oncol. 2007, 8, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Blows, F.M.; Driver, K.E.; Schmidt, M.K.; Broeks, A.; van Leeuwen, F.E.; Wesseling, J.; Cheang, M.C.; Gelmon, K.; Nielsen, T.O.; Blomqvist, C.; et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: A collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010, 7, e1000279. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Martienssen, R. A blueprint for a Human Epigenome Project: The AACR Human Epigenome Workshop. Cancer Res. 2005, 65, 11241–11246. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Schuebel, K.E. Genomic biology: The epigenomic era opens. Nature 2007, 448, 548–549. [Google Scholar] [CrossRef] [PubMed][Green Version]
- The American Association for Cancer Research Human Epigenome Task Force; European Union, Network of Excellence, Scientific Advisory Board. Moving AHEAD with an international human epigenome project. Nature 2008, 454, 711–715. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef]
- Laget, S.; Joulie, M.; Le Masson, F.; Sasai, N.; Christians, E.; Pradhan, S.; Roberts, R.J.; Defossez, P.A. The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS ONE 2010, 5, e11982. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Cai, T. Na+-K+--ATPase-mediated signal transduction: From protein interaction to cellular function. Mol. Interv. 2003, 3, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Bublitz, M.; Morth, J.P.; Nissen, P. P-type ATPases at a glance. J. Cell Sci. 2011, 124, 2515–2519. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, A.; Moiseenko, F.; Dubina, M. Abnormal expression of ATP1A1 and ATP1A2 in breast cancer. F1000Res 2017, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Prassas, I.; Diamandis, E.P. Novel therapeutic applications of cardiac glycosides. Nat. Rev. Drug Discov. 2008, 7, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Magpusao, A.N.; Omolloh, G.; Johnson, J.; Gascon, J.; Peczuh, M.W.; Fenteany, G. Cardiac glycoside activities link Na(+)/K(+) ATPase ion-transport to breast cancer cell migration via correlative SAR. ACS Chem. Biol. 2015, 10, 561–569. [Google Scholar] [CrossRef]
- Kim, N.; Yim, H.Y.; He, N.; Lee, C.J.; Kim, J.H.; Choi, J.S.; Lee, H.S.; Kim, S.; Jeong, E.; Song, M.; et al. Cardiac glycosides display selective efficacy for STK11 mutant lung cancer. Sci. Rep. 2016, 6, 29721. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Lienhard, M.; Grimm, C.; Morkel, M.; Herwig, R.; Chavez, L. MEDIPS: Genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 2014, 30, 284–286. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000; p. xiii. 350p. [Google Scholar]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. IHC Profiler: An open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE 2014, 9, e96801. [Google Scholar] [CrossRef]
- Komatsu, M.; Yoshimaru, T.; Matsuo, T.; Kiyotani, K.; Miyoshi, Y.; Tanahashi, T.; Rokutan, K.; Yamaguchi, R.; Saito, A.; Imoto, S.; et al. Molecular features of triple negative breast cancer cells by genome-wide gene expression profiling analysis. Int. J. Oncol. 2013, 42, 478–506. [Google Scholar] [CrossRef]
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Rajasekaran, A.K.; Rajasekaran, S.A. Role of Na-K-ATPase in the assembly of tight junctions. Am. J. Physiol. Ren. Physiol. 2003, 285, F388–F396. [Google Scholar] [CrossRef]
- Contreras, R.G.; Shoshani, L.; Flores-Maldonado, C.; Lazaro, A.; Cereijido, M. Relationship between Na+,K+-ATPase and cell attachment. J. Cell Sci. 1999, 112 Pt 23, 4223–4232. [Google Scholar] [CrossRef]
- Rajasekaran, S.A.; Palmer, L.G.; Quan, K.; Harper, J.F.; Ball, W.J., Jr.; Bander, N.H.; Peralta Soler, A.; Rajasekaran, A.K. Na,K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol. Biol. Cell 2001, 12, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Askari, A. Na(+)/K(+)-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Van Quaquebeke, E.; Delest, B.; Debeir, O.; Darro, F.; Kiss, R. Cardiotonic steroids on the road to anti-cancer therapy. Biochim. Et Biophys. Acta 2007, 1776, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.K.; Kim, K.; Chae, H.; Evans, J.; Yan, P.; Zhang, B.T.; Gray, J.; Spellman, P.; Huang, T.H.; Nephew, K.P.; et al. Integrated analysis of genome-wide DNA methylation and gene expression profiles in molecular subtypes of breast cancer. Nucleic. Acids Res. 2013, 41, 8464–8474. [Google Scholar] [CrossRef] [PubMed]
- Ruike, Y.; Imanaka, Y.; Sato, F.; Shimizu, K.; Tsujimoto, G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genom. 2010, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Fackler, M.J.; Umbricht, C.B.; Williams, D.; Argani, P.; Cruz, L.A.; Merino, V.F.; Teo, W.W.; Zhang, Z.; Huang, P.; Visvananthan, K.; et al. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Res. 2011, 71, 6195–6207. [Google Scholar] [CrossRef] [PubMed]
- Locke, W.J.; Zotenko, E.; Stirzaker, C.; Robinson, M.D.; Hinshelwood, R.A.; Stone, A.; Reddel, R.R.; Huschtscha, L.I.; Clark, S.J. Coordinated epigenetic remodelling of transcriptional networks occurs during early breast carcinogenesis. Clin. Epigenetics 2015, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.J.; Stone, A.; Armstong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6, 5899. [Google Scholar] [CrossRef]
- Kawazu, M.; Kojima, S.; Ueno, T.; Totoki, Y.; Nakamura, H.; Kunita, A.; Qu, W.; Yoshimura, J.; Soda, M.; Yasuda, T.; et al. Integrative analysis of genomic alterations in triple-negative breast cancer in association with homologous recombination deficiency. PLoS Genet. 2017, 13, e1006853. [Google Scholar] [CrossRef]
- Fang, F.; Turcan, S.; Rimner, A.; Kaufman, A.; Giri, D.; Morris, L.G.; Shen, R.; Seshan, V.; Mo, Q.; Heguy, A.; et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci. Transl. Med. 2011, 3, 75ra25. [Google Scholar] [CrossRef]
- Kulis, M.; Heath, S.; Bibikova, M.; Queiros, A.C.; Navarro, A.; Clot, G.; Martinez-Trillos, A.; Castellano, G.; Brun-Heath, I.; Pinyol, M.; et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Shiozaki, A.; Kosuga, T.; Shimizu, H.; Kudou, M.; Ohashi, T.; Arita, T.; Konishi, H.; Komatsu, S.; Kubota, T.; et al. The expression of the alpha1 subunit of Na+/K+-ATPase is related to tumor development and clinical outcomes in gastric cancer. Gastric. Cancer 2021, 24, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Xu, L.; Wang, P.; Jiang, Y.; Yong, P.; Zhang, C.; Zhang, H.; Meng, Z.; Yang, P. Na+/K+-ATPase alpha1 subunit, a novel therapeutic target for hepatocellular carcinoma. Oncotarget 2015, 6, 28183–28193. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, K.; Winnicka, K.; Bielawska, A. Inhibition of DNA topoisomerases I and II, and growth inhibition of breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A. Biol. Pharm. Bull. 2006, 29, 1493–1497. [Google Scholar] [CrossRef]
- Lawrence, T.S. Ouabain sensitizes tumor cells but not normal cells to radiation. Int. J. Radiat. Oncol. Biol. Phys. 1988, 15, 953–958. [Google Scholar] [CrossRef]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef]
- Eskiocak, U.; Ramesh, V.; Gill, J.G.; Zhao, Z.; Yuan, S.W.; Wang, M.; Vandergriff, T.; Shackleton, M.; Quintana, E.; Johnson, T.M.; et al. Synergistic effects of ion transporter and MAP kinase pathway inhibitors in melanoma. Nat. Commun. 2016, 7, 12336. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Ko, J.Y.; Kong, H.K.; Lee, M.; Chung, W.; Lim, S.; Son, D.; Oh, S.; Park, J.W.; Kim, D.Y.; et al. Hypomethylation of ATP1A1 Is Associated with Poor Prognosis and Cancer Progression in Triple-Negative Breast Cancer. Cancers 2024, 16, 1666. https://doi.org/10.3390/cancers16091666
Kim Y, Ko JY, Kong HK, Lee M, Chung W, Lim S, Son D, Oh S, Park JW, Kim DY, et al. Hypomethylation of ATP1A1 Is Associated with Poor Prognosis and Cancer Progression in Triple-Negative Breast Cancer. Cancers. 2024; 16(9):1666. https://doi.org/10.3390/cancers16091666
Chicago/Turabian StyleKim, Yesol, Je Yeong Ko, Hyun Kyung Kong, Minyoung Lee, Woosung Chung, Sera Lim, Dasom Son, Sumin Oh, Jee Won Park, Do Yeon Kim, and et al. 2024. "Hypomethylation of ATP1A1 Is Associated with Poor Prognosis and Cancer Progression in Triple-Negative Breast Cancer" Cancers 16, no. 9: 1666. https://doi.org/10.3390/cancers16091666
APA StyleKim, Y., Ko, J. Y., Kong, H. K., Lee, M., Chung, W., Lim, S., Son, D., Oh, S., Park, J. W., Kim, D. Y., Lee, M., Han, W., Park, W.-Y., Yoo, K. H., & Park, J. H. (2024). Hypomethylation of ATP1A1 Is Associated with Poor Prognosis and Cancer Progression in Triple-Negative Breast Cancer. Cancers, 16(9), 1666. https://doi.org/10.3390/cancers16091666