
Heterocellular Adhesion in Cancer Invasion and Metastasis: Interactions between Cancer Cells and Cancer-Associated Fibroblasts



{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Cancer-Associated Fibroblasts in the Tumor Microenvironment
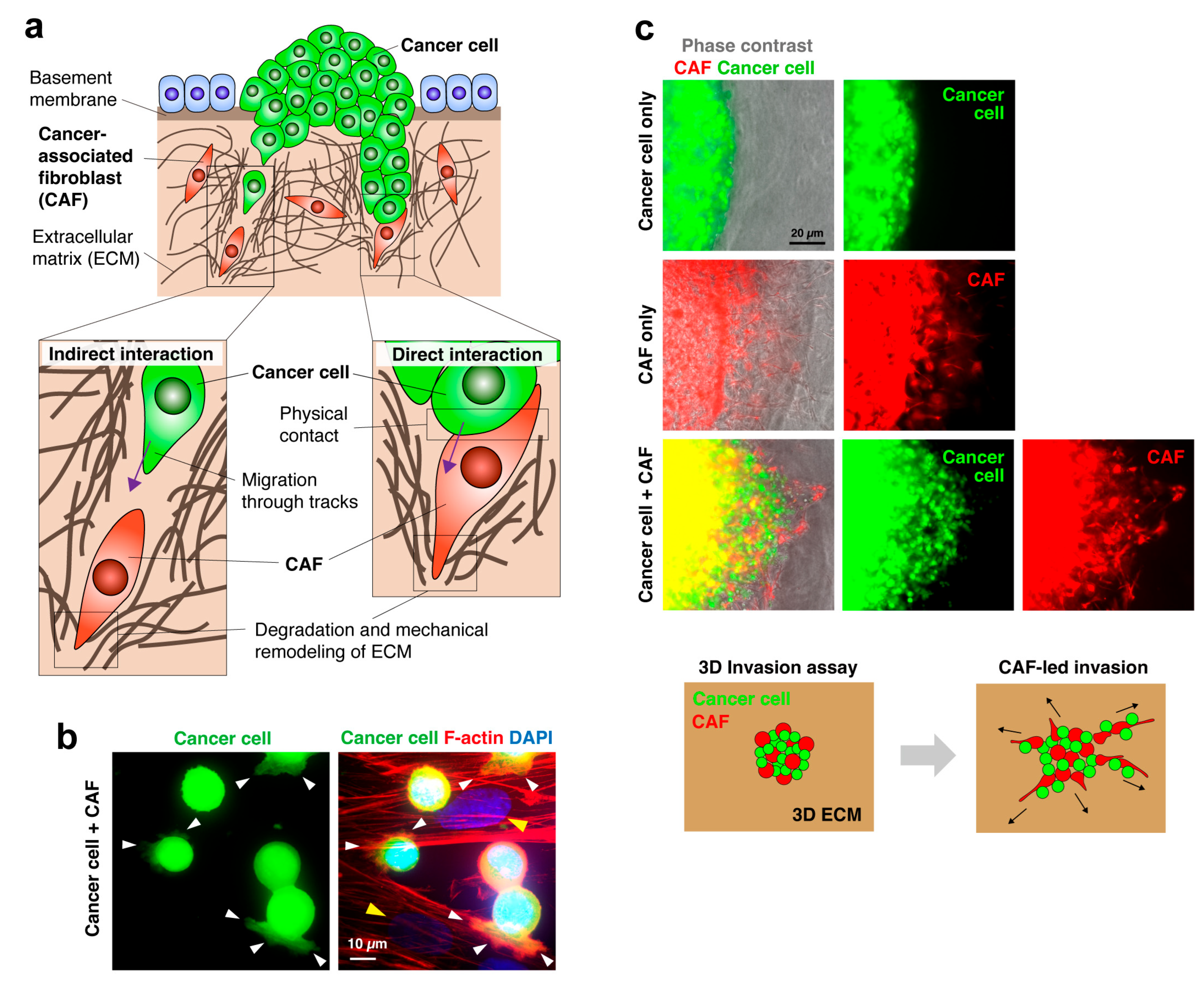
3. CAFs Lead Invasion of Cancer Cells
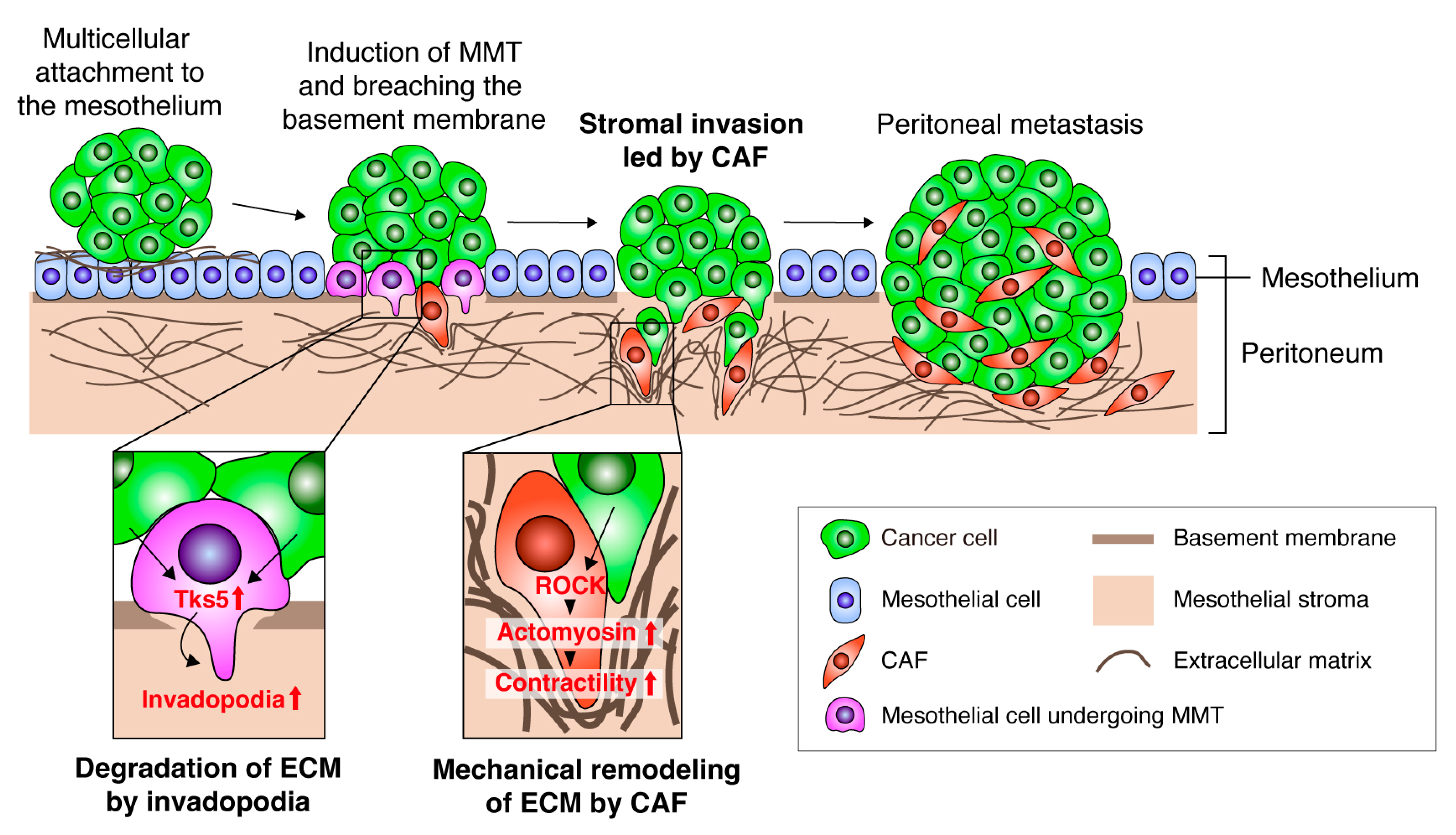
3.1. CAFs Generate the Path for Cancer Invasion by Remodeling the ECM
3.2. CAF-Led Invasion during Peritoneal Cancer Metastasis
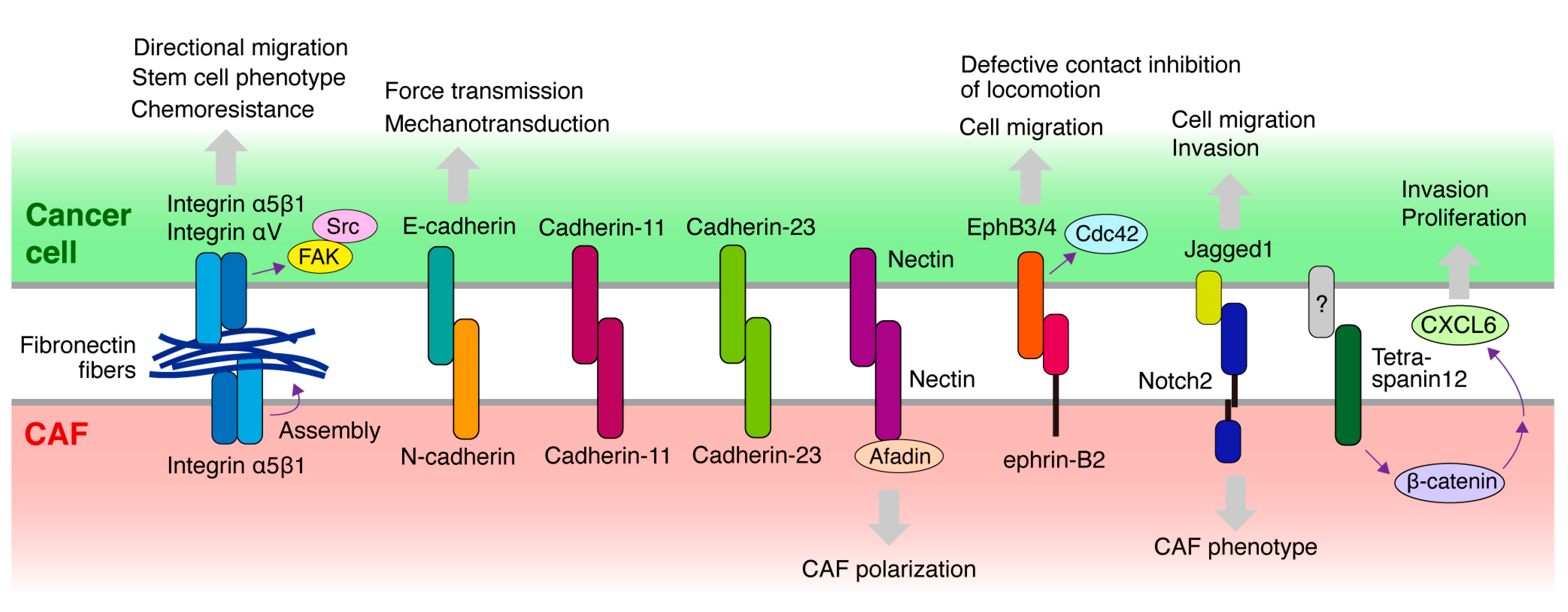
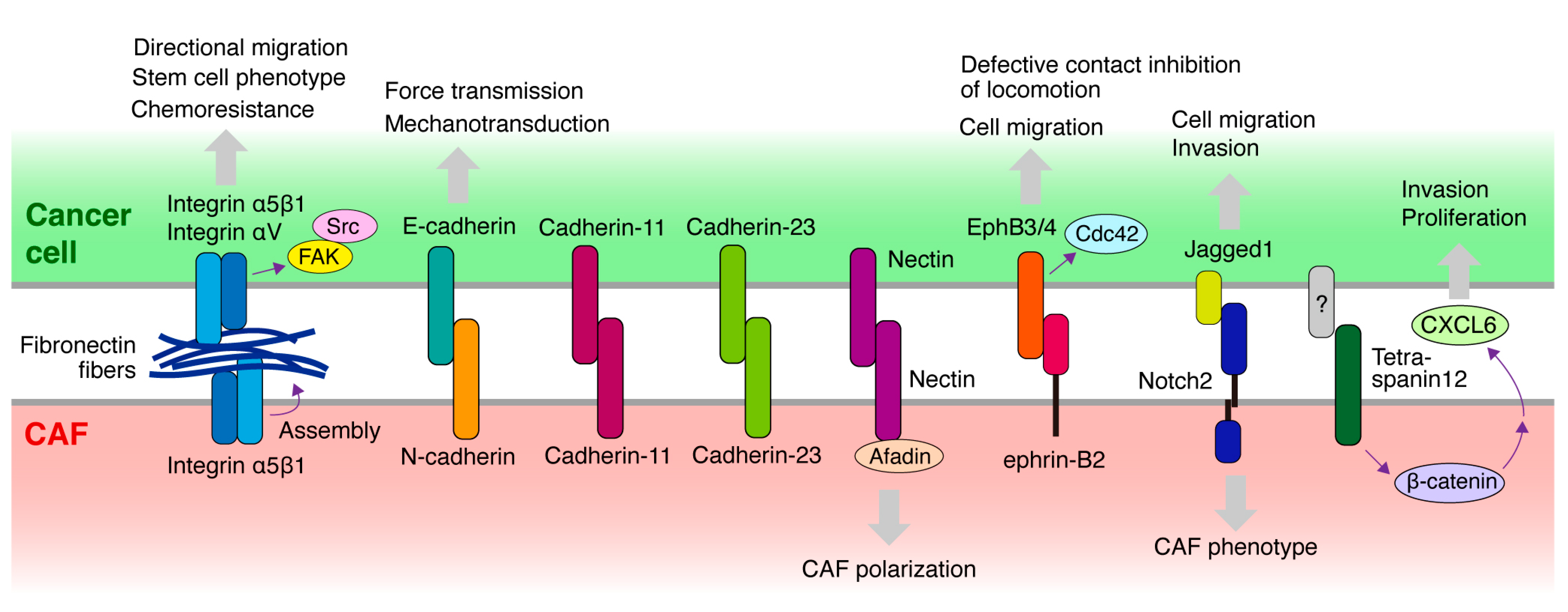
4. Molecules Mediating the Heterocellular Cancer Cell–CAF Adhesion and Downstream Signaling
4.1. Cadherin
4.2. Integrin and ECM
4.3. Eph/Ephrin
4.4. Other Molecules
5. Targeting Strategy for the Cancer Cell-CAF Adhesion for Cancer Therapy
6. Perspectives
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ganesh, K.; Massagué, J. Targeting Metastatic Cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Raudenská, M.; Petrláková, K.; Juriňáková, T.; Leischner Fialová, J.; Fojtů, M.; Jakubek, M.; Rösel, D.; Brábek, J.; Masařík, M. Engine Shutdown: Migrastatic Strategies and Prevention of Metastases. Trends Cancer 2023, 9, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Weiss, F.; Lauffenburger, D.; Friedl, P. Towards Targeting of Shared Mechanisms of Cancer Metastasis and Therapy Resistance. Nat. Rev. Cancer 2022, 22, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Beeghly, G.F.; Amofa, K.Y.; Fischbach, C.; Kumar, S. Regulation of Tumor Invasion by the Physical Microenvironment: Lessons from Breast and Brain Cancer. Annu. Rev. Biomed. Eng. 2022, 24, 29–59. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Joyce, J.A. The Evolving Tumor Microenvironment: From Cancer Initiation to Metastatic Outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, G.; Tuveson, D.A. Activated Fibroblasts in Cancer: Perspectives and Challenges. Cancer Cell 2023, 41, 434–449. [Google Scholar] [CrossRef]
- Sun, H.; Wang, X.; Wang, X.; Xu, M.; Sheng, W. The Role of Cancer-Associated Fibroblasts in Tumorigenesis of Gastric Cancer. Cell Death Dis. 2022, 13, 874. [Google Scholar] [CrossRef]
- Arima, Y.; Matsueda, S.; Saya, H. Significance of Cancer-Associated Fibroblasts in the Interactions of Cancer Cells with the Tumor Microenvironment of Heterogeneous Tumor Tissue. Cancers 2023, 15, 2536. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Ouyang, H. Intercellular Crosstalk between Cancer Cells and Cancer-Associated Fibroblasts via Exosomes in Gastrointestinal Tumors. Front. Oncol. 2024, 14, 1374742. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.; Ly, T.; Kriet, M.; Czirok, A.; Thomas, S.M. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers 2023, 15, 1899. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Chen, J.; Song, E. Targeting CAFs to Overcome Anticancer Therapeutic Resistance. Trends Cancer 2022, 8, 527–555. [Google Scholar] [CrossRef] [PubMed]
- Ansardamavandi, A.; Tafazzoli-Shadpour, M. The Functional Cross Talk between Cancer Cells and Cancer Associated Fibroblasts from a Cancer Mechanics Perspective. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2021, 1868, 119103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, J.; Huang, H.; Ye, M.; Li, X.; Wu, R.; Liu, H.; Song, Y. Metastasis-Associated Fibroblasts: An Emerging Target for Metastatic Cancer. Biomark. Res. 2021, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Asif, P.J.; Longobardi, C.; Hahne, M.; Medema, J.P. The Role of Cancer-Associated Fibroblasts in Cancer Invasion and Metastasis. Cancers 2021, 13, 4720. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Nagano, Y.; Miyazaki, M.; Nagamura, Y.; Sasaki, K.; Kawamura, T.; Yanagihara, K.; Imai, T.; Ohki, R.; Yashiro, M.; et al. Integrin A5 Mediates Cancer Cell-Fibroblast Adhesion and Peritoneal Dissemination of Diffuse-Type Gastric Carcinoma. Cancer Lett. 2022, 526, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-Led Collective Invasion of Carcinoma Cells with Differing Roles for RhoGTPases in Leading and Following Cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A Mechanically Active Heterotypic E-Cadherin/N-Cadherin Adhesion Enables Fibroblasts to Drive Cancer Cell Invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-Associated Fibroblasts Induce Metalloprotease-Independent Cancer Cell Invasion of the Basement Membrane. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef] [PubMed]
- Monster, J.L.; Kemp, L.J.S.; Gloerich, M.; van der Post, R.S. Diffuse Gastric Cancer: Emerging Mechanisms of Tumor Initiation and Progression. Biochim. Biophys. Acta BBA—Rev. Cancer 2022, 1877, 188719. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Yoshida, N.; Takanashi, M.; Ito, Y.; Fukami, K.; Yanagihara, K.; Yashiro, M.; Sakai, R. Stromal Fibroblasts Mediate Extracellular Matrix Remodeling and Invasion of Scirrhous Gastric Carcinoma Cells. PLoS ONE 2014, 9, e85485. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Yashiro, M.; Moyano-Galceran, L.; Sugimoto, A.; Ohira, M.; Lehti, K. Crosstalk Between Cancer Associated Fibroblasts and Cancer Cells in Scirrhous Type Gastric Cancer. Front. Oncol. 2020, 10, 568557. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Nakabo, A.; Nagano, Y.; Nagamura, Y.; Yanagihara, K.; Ohki, R.; Nakamura, Y.; Fukami, K.; Kawamoto, J.; Umayahara, K.; et al. Tissue Factor-Induced Fibrinogenesis Mediates Cancer Cell Clustering and Multiclonal Peritoneal Metastasis. Cancer Lett. 2023, 553, 215983. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M. Crosstalk of Tumor Stromal Cells Orchestrates Invasion and Spreading of Gastric Cancer. Pathol. Int. 2022, 72, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Cervero, P.; Eddy, R.; Condeelis, J. Mechanisms and Roles of Podosomes and Invadopodia. Nat. Rev. Mol. Cell Biol. 2023, 24, 86–106. [Google Scholar] [CrossRef] [PubMed]
- Satoyoshi, R.; Aiba, N.; Yanagihara, K.; Yashiro, M.; Tanaka, M. Tks5 Activation in Mesothelial Cells Creates Invasion Front of Peritoneal Carcinomatosis. Oncogene 2015, 34, 3176–3187. [Google Scholar] [CrossRef] [PubMed]
- Bootsma, S.; Bijlsma, M.F.; Vermeulen, L. The Molecular Biology of Peritoneal Metastatic Disease. EMBO Mol. Med. 2023, 15, e15914. [Google Scholar] [CrossRef]
- Avula, L.R.; Hagerty, B.; Alewine, C. Molecular Mediators of Peritoneal Metastasis in Pancreatic Cancer. Cancer Metastasis Rev. 2020, 39, 1223–1243. [Google Scholar] [CrossRef]
- Zajac, O.; Raingeaud, J.; Libanje, F.; Lefebvre, C.; Sabino, D.; Martins, I.; Roy, P.; Benatar, C.; Canet-Jourdan, C.; Azorin, P.; et al. Tumour Spheres with Inverted Polarity Drive the Formation of Peritoneal Metastases in Patients with Hypermethylated Colorectal Carcinomas. Nat. Cell Biol. 2018, 20, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Farsinejad, S.; Cattabiani, T.; Muranen, T.; Iwanicki, M. Ovarian Cancer Dissemination-A Cell Biologist’s Perspective. Cancers 2019, 11, 1957. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Yang, Z.; Xu, S.; Li, X.; Yang, X.; Jin, P.; Liu, Y.; Zhou, X.; Zhang, T.; Gong, C.; et al. Heterotypic CAF-Tumor Spheroids Promote Early Peritoneal Metastasis of Ovarian Cancer. J. Exp. Med. 2019, 216, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, M.; Kajiyama, H.; Yokoi, A.; Sugiyama, M.; Koya, Y.; Yamakita, Y.; Liu, W.; Nakamura, K.; Moriyama, Y.; Yasui, H.; et al. Ovarian Cancer-associated Mesothelial Cells Induce Acquired Platinum-resistance in Peritoneal Metastasis via the FN1/Akt Signaling Pathway. Int. J. Cancer 2020, 146, 2268–2280. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Sakai, R. Direct Interaction between Carcinoma Cells and Cancer Associated Fibroblasts for the Regulation of Cancer Invasion. Cancers Basel 2015, 7, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, M. Cell Sorting in Vitro and in Vivo: How Are Cadherins Involved? Semin. Cell Dev. Biol. 2023, 147, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Omelchenko, T.; Fetisova, E.; Ivanova, O.; Bonder, E.M.; Feder, H.; Vasiliev, J.M.; Gelfand, I.M. Contact Interactions between Epitheliocytes and Fibroblasts: Formation of Heterotypic Cadherin-Containing Adhesion Sites Is Accompanied by Local Cytoskeletal Reorganization. Proc. Natl. Acad. Sci. USA 2001, 98, 8632–8637. [Google Scholar] [CrossRef]
- Kang, W.; Fan, Y.; Du, Y.; Tonkova, E.A.; Hsu, Y.-H.; Tan, K.V.; Alexander, S.; Wong, B.S.; Yang, H.; Luo, J.; et al. Post-EMT: Cadherin-11 Mediates Cancer Hijacking Fibroblasts. eLife 2023, 12, RP87423. [Google Scholar] [CrossRef]
- Apostolopoulou, M.; Ligon, L. Cadherin-23 Mediates Heterotypic Cell-Cell Adhesion between Breast Cancer Epithelial Cells and Fibroblasts. PLoS ONE 2012, 7, e33289. [Google Scholar] [CrossRef]
- Miyazaki, K.; Oyanagi, J.; Hoshino, D.; Togo, S.; Kumagai, H.; Miyagi, Y. Cancer Cell Migration on Elongate Protrusions of Fibroblasts in Collagen Matrix. Sci. Rep. 2019, 9, 292. [Google Scholar] [CrossRef]
- Miyazaki, K.; Togo, S.; Okamoto, R.; Idiris, A.; Kumagai, H.; Miyagi, Y. Collective Cancer Cell Invasion in Contact with Fibroblasts through Integrin-α5β1/Fibronectin Interaction in Collagen Matrix. Cancer Sci. 2020, 111, 4381–4392. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-Associated Fibroblasts Promote Directional Cancer Cell Migration by Aligning Fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital Imaging Reveals How BRAF Inhibition Generates Drug-Tolerant Microenvironments with High Integrin Β1/FAK Signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; McMillan, R.H.; Chang, Y.-T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct Interactions with Cancer-Associated Fibroblasts Lead to Enhanced Pancreatic Cancer Stem Cell Function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef]
- Astin, J.W.; Batson, J.; Kadir, S.; Charlet, J.; Persad, R.A.; Gillatt, D.; Oxley, J.D.; Nobes, C.D. Competition amongst Eph Receptors Regulates Contact Inhibition of Locomotion and Invasiveness in Prostate Cancer Cells. Nat. Cell Biol. 2010, 12, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Eph Receptors and Ephrins in Cancer Progression. Nat. Rev. Cancer 2024, 24, 5–27. [Google Scholar] [CrossRef]
- Kakarla, M.; ChallaSivaKanaka, S.; Dufficy, M.F.; Gil, V.; Filipovich, Y.; Vickman, R.; Crawford, S.E.; Hayward, S.W.; Franco, O.E. Ephrin B Activate Src Family Kinases in Fibroblasts Inducing Stromal Remodeling in Prostate Cancer. Cancers 2022, 14, 2336. [Google Scholar] [CrossRef] [PubMed]
- Duraivelan, K.; Samanta, D. Emerging Roles of the Nectin Family of Cell Adhesion Molecules in Tumour-Associated Pathways. Biochim. Biophys. Acta BBA—Rev. Cancer 2021, 1876, 188589. [Google Scholar] [CrossRef] [PubMed]
- Strell, C.; Paulsson, J.; Jin, S.-B.; Tobin, N.P.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M.; et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Körner, C.; Ben-Baruch, A. Tumor-Stroma-Inflammation Networks Promote Pro-Metastatic Chemokines and Aggressiveness Characteristics in Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 3513. [Google Scholar] [CrossRef]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Morein, D.; Rubinstein-Achiasaf, L.; Sprinzak, D.; Wiemann, S.; Körner, C.; Ehrlich, M.; Ben-Baruch, A. Notch-Mediated Tumor-Stroma-Inflammation Networks Promote Invasive Properties and CXCL8 Expression in Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 804. [Google Scholar] [CrossRef]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.G.; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell–Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef]
- Otomo, R.; Otsubo, C.; Matsushima-Hibiya, Y.; Miyazaki, M.; Tashiro, F.; Ichikawa, H.; Kohno, T.; Ochiya, T.; Yokota, J.; Nakagama, H.; et al. TSPAN12 Is a Critical Factor for Cancer-Fibroblast Cell Contact-Mediated Cancer Invasion. Proc. Natl. Acad. Sci. USA 2014, 111, 18691–18696. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kasashima, H.; Fukui, Y.; Tsujio, G.; Yashiro, M.; Maeda, K. The Heterogeneity of Cancer-associated Fibroblast Subpopulations: Their Origins, Biomarkers, and Roles in the Tumor Microenvironment. Cancer Sci. 2023, 114, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-Associated Fibroblasts That Restrain Cancer Progression: Hypotheses and Perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and Therapeutic Relevance of Cancer-Associated Fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Jeong, H.Y.; Ham, I.-H.; Lee, S.H.; Ryu, D.; Son, S.-Y.; Han, S.-U.; Kim, T.-M.; Hur, H. Spatially Distinct Reprogramming of the Tumor Microenvironment Based on Tumor Invasion in Diffuse-Type Gastric Cancers. Clin. Cancer Res. 2021, 27, 6529–6542. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, Z.; Peng, G.; Xiao, Y.; Guo, J.; Wu, B.; Li, X.; Zhou, W.; Li, J.; Li, Z.; et al. Single-Cell RNA Sequencing Reveals a pro-Invasive Cancer-Associated Fibroblast Subgroup Associated with Poor Clinical Outcomes in Patients with Gastric Cancer. Theranostics 2022, 12, 620–638. [Google Scholar] [CrossRef]
- Hou, J.; Yan, D.; Liu, Y.; Huang, P.; Cui, H. The Roles of Integrin A5β1 in Human Cancer. OncoTargets Ther. 2020, 13, 13329–13344. [Google Scholar] [CrossRef]
- Dhaliwal, D.; Shepherd, T.G. Molecular and Cellular Mechanisms Controlling Integrin-Mediated Cell Adhesion and Tumor Progression in Ovarian Cancer Metastasis: A Review. Clin. Exp. Metastasis 2022, 39, 291–301. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, H.; Miyazaki, M. Heterocellular Adhesion in Cancer Invasion and Metastasis: Interactions between Cancer Cells and Cancer-Associated Fibroblasts. Cancers 2024, 16, 1636. https://doi.org/10.3390/cancers16091636
Yamaguchi H, Miyazaki M. Heterocellular Adhesion in Cancer Invasion and Metastasis: Interactions between Cancer Cells and Cancer-Associated Fibroblasts. Cancers. 2024; 16(9):1636. https://doi.org/10.3390/cancers16091636
Chicago/Turabian StyleYamaguchi, Hideki, and Makoto Miyazaki. 2024. "Heterocellular Adhesion in Cancer Invasion and Metastasis: Interactions between Cancer Cells and Cancer-Associated Fibroblasts" Cancers 16, no. 9: 1636. https://doi.org/10.3390/cancers16091636
APA StyleYamaguchi, H., & Miyazaki, M. (2024). Heterocellular Adhesion in Cancer Invasion and Metastasis: Interactions between Cancer Cells and Cancer-Associated Fibroblasts. Cancers, 16(9), 1636. https://doi.org/10.3390/cancers16091636