
Discovery of Novel Potential Prognostic Markers and Targeted Therapy to Overcome Chemotherapy Resistance in an Advanced-Stage Wilms Tumor


 ,
on behalf of the Thai Pediatric Cancer Atlas (TPCA) Consortium
,
on behalf of the Thai Pediatric Cancer Atlas (TPCA) Consortium
Abstract
Simple Summary
Abstract
1. Introduction
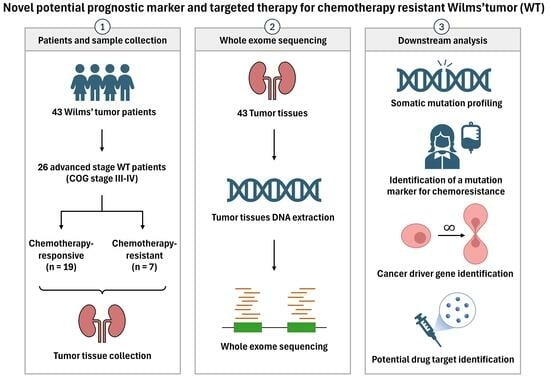
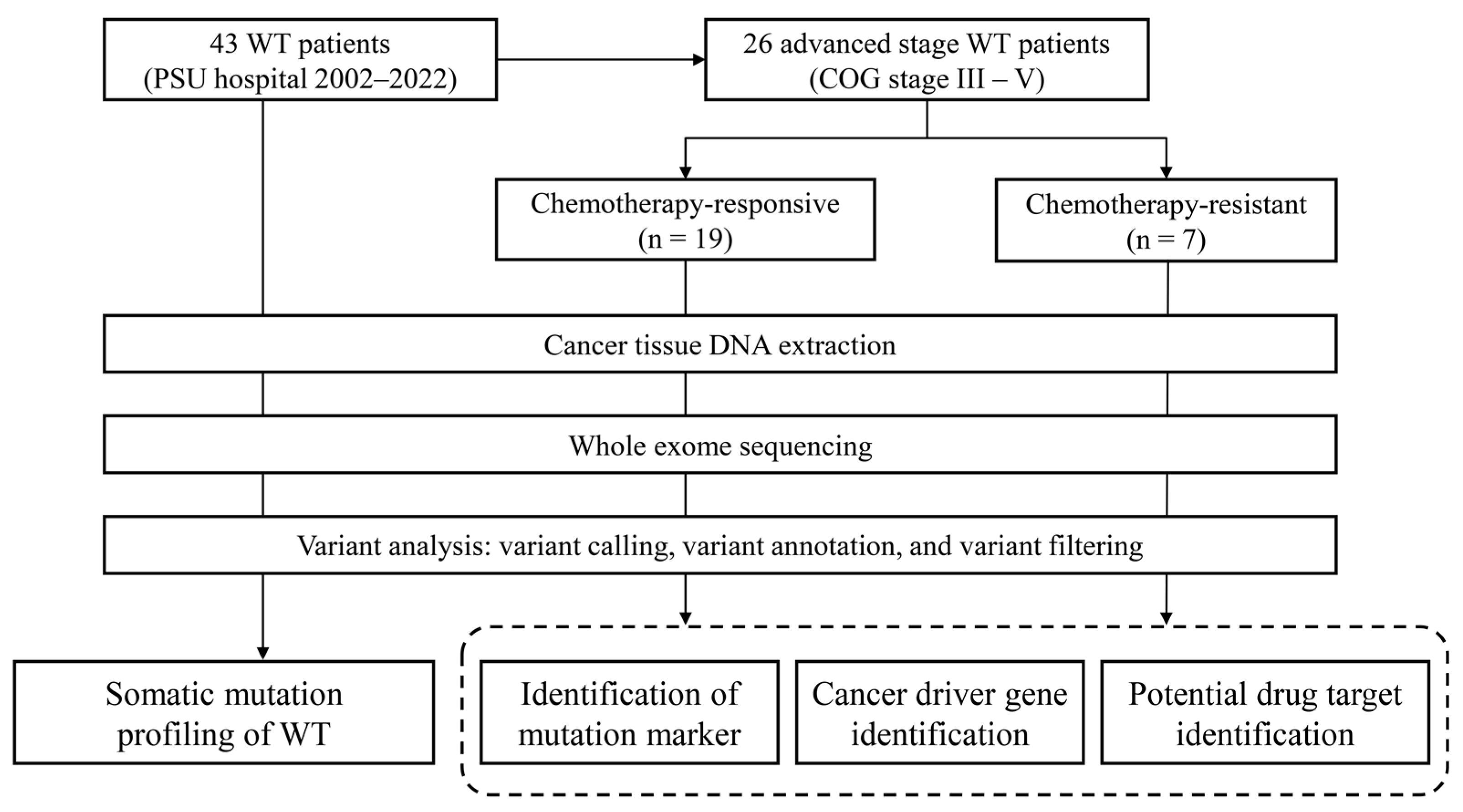
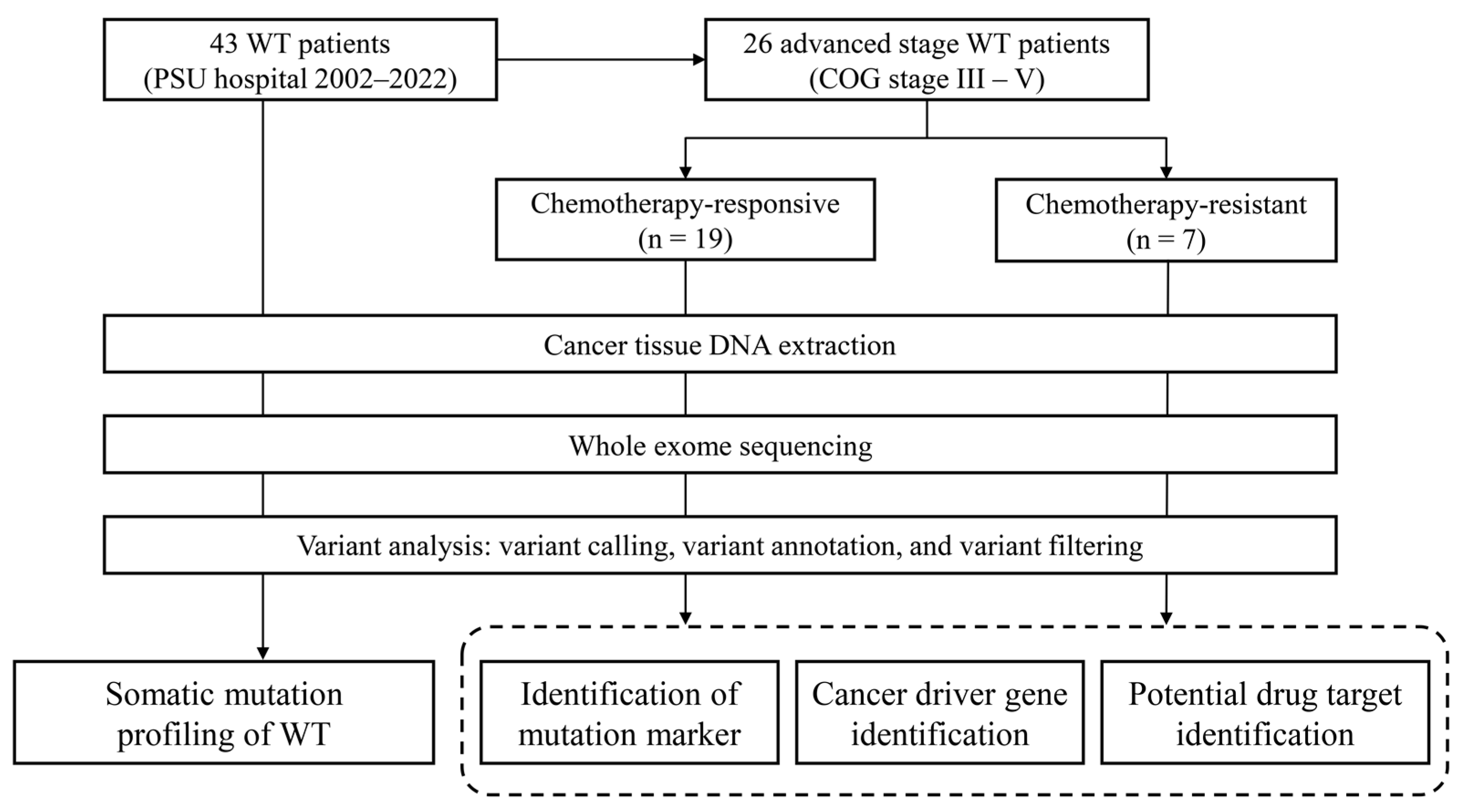
2. Materials and Methods
2.1. Biological Samples and Sequencing Library Preparation
2.2. Whole Exome Sequencing
2.3. Identification of Mutation Marker for Chemotherapy-Resistant WT
2.4. Identification of Cancer Driver Mutation and Their Potential Targeted Therapy
2.5. Statistical Analysis
3. Results
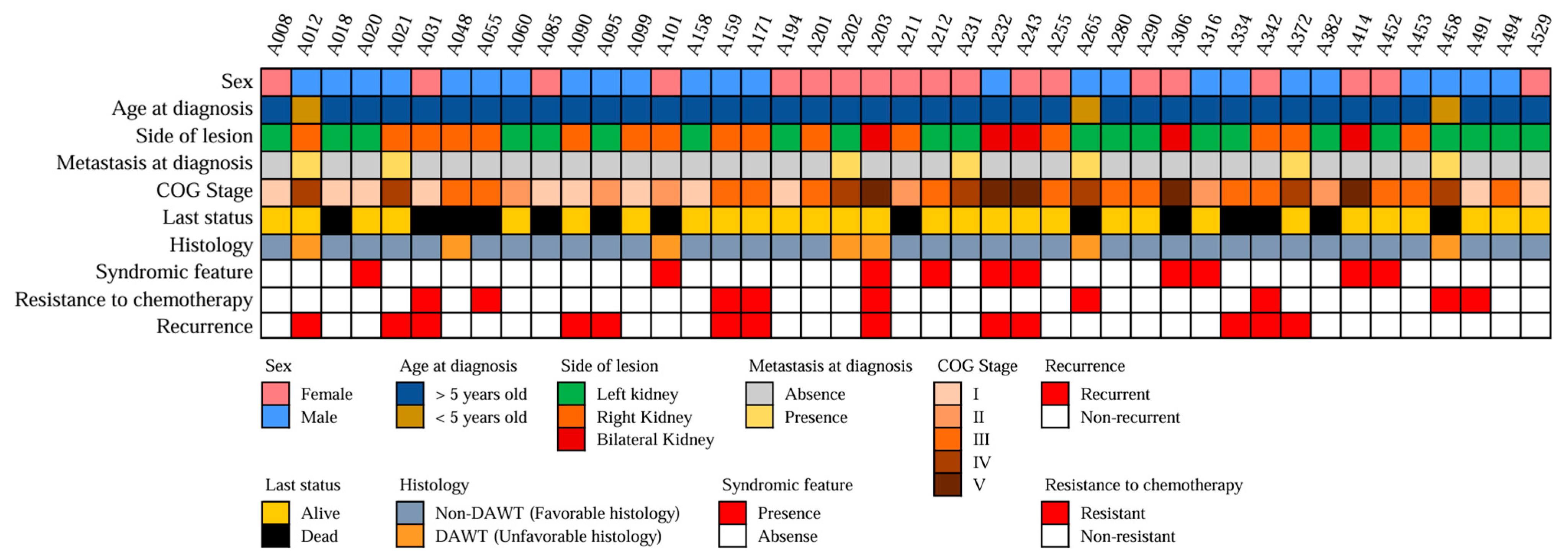
3.1. Clinical Characteristics of the Patients
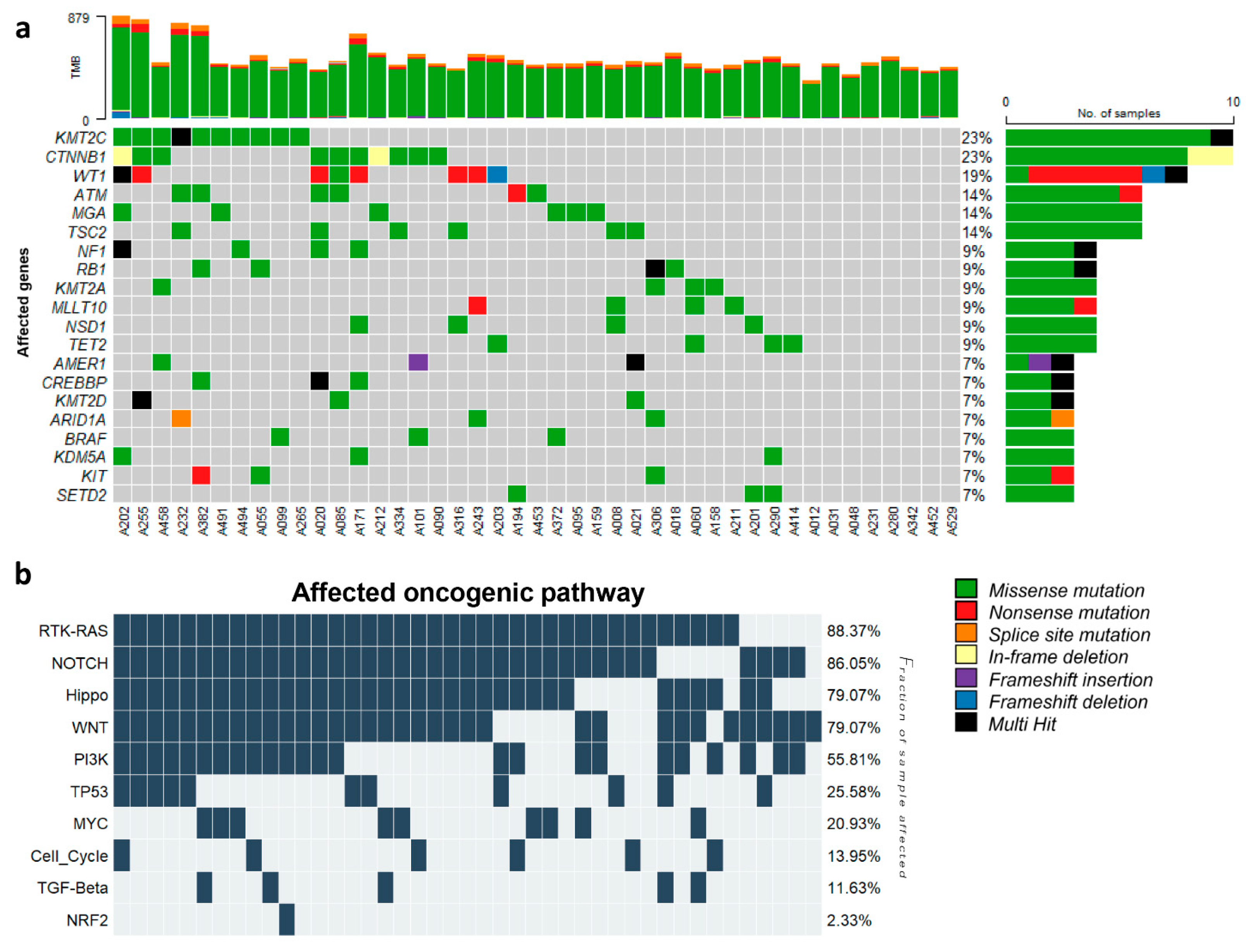
3.2. Somatic Mutation Profiling of WT and Clinical Relevance
3.3. Cancer Driver Gene and Potential Drug Target for Chemotherapy-Resistant WT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakata, K.; Colombet, M.; Stiller, C.A.; Pritchard-Jones, K.; Steliarova-Foucher, E.; IICC-3 Contributors. Incidence of childhood renal tumours: An international population-based study. Int. J. Cancer 2020, 147, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Doganis, D.; Karalexi, M.A.; NARECHEM-ST collaborating group; Panagopoulou, P.; Bouka, P.; Bouka, E.; Markozannes, G.; Ntzani, E.E.; Steliarova-Foucher, E.; Petridou, E.T. Incidence patterns of childhood non-Wilms renal tumors: Comparing data of the Nationwide Registry of Childhood Hematological Malignancies and Solid Tumors (NARECHEM-ST), Greece, and the Surveillance, Epidemiology, and End Results Program (SEER), USA. Cancer Epidemiol. 2022, 78, 102153. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, A.; Spreafico, F.; de Krijger, R.R.; Drost, J.; Brok, J.; Perotti, D.; van Tinteren, H.; Venkatramani, R.; Godzinski, J.; Rube, C.; et al. Prognostic Factors for Wilms Tumor Recurrence: A Review of the Literature. Cancers 2021, 13, 3142. [Google Scholar] [CrossRef]
- Dome, J.S.; Graf, N.; Geller, J.I.; Fernandez, C.V.; Mullen, E.A.; Spreafico, F.; Van den Heuvel-Eibrink, M.; Pritchard-Jones, K. Advances in Wilms Tumor Treatment and Biology: Progress through International Collaboration. J. Clin. Oncol. 2015, 33, 2999–3007. [Google Scholar] [CrossRef]
- Hontecillas-Prieto, L.; Garcia-Dominguez, D.J.; Vaca, D.P.; Garcia-Mejias, R.; Marcilla, D.; Ramirez-Villar, G.L.; Saez, C.; de Alava, E. Multidrug resistance transporter profile reveals MDR3 as a marker for stratification of blastemal Wilms tumour patients. Oncotarget 2017, 8, 11173–11186. [Google Scholar] [CrossRef] [PubMed]
- Sangkhathat, S.; Kanngurn, S.; Chaiyapan, W.; Gridist, P.; Maneechay, W. Wilms’ tumor 1 gene (WT1) is overexpressed and provides an oncogenic function in pediatric nephroblastomas harboring the wild-type WT1. Oncol. Lett. 2010, 1, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Brok, J.; Treger, T.D.; Gooskens, S.L.; van den Heuvel-Eibrink, M.M.; Pritchard-Jones, K. Biology and treatment of renal tumours in childhood. Eur. J. Cancer 2016, 68, 179–195. [Google Scholar] [CrossRef]
- Gadd, S.; Huff, V.; Walz, A.L.; Ooms, A.; Armstrong, A.E.; Gerhard, D.S.; Smith, M.A.; Auvil, J.M.G.; Meerzaman, D.; Chen, Q.R.; et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat. Genet. 2017, 49, 1487–1494. [Google Scholar] [CrossRef]
- Torrezan, G.T.; Ferreira, E.N.; Nakahata, A.M.; Barros, B.D.; Castro, M.T.; Correa, B.R.; Krepischi, A.C.; Olivieri, E.H.; Cunha, I.W.; Tabori, U.; et al. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat. Commun. 2014, 5, 4039. [Google Scholar] [CrossRef]
- Rakheja, D.; Chen, K.S.; Liu, Y.; Shukla, A.A.; Schmid, V.; Chang, T.C.; Khokhar, S.; Wickiser, J.E.; Karandikar, N.J.; Malter, J.S.; et al. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat. Commun. 2014, 2, 4802. [Google Scholar] [CrossRef]
- Hong, B.; Dong, R. Research advances in the targeted therapy and immunotherapy of Wilms tumor: A narrative review. Transl Cancer Res. 2021, 10, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Iwakuma, T. Drugs Targeting p53 Mutations with FDA Approval and in Clinical. Trials. Cancers (Basel) 2023, 15, 429. [Google Scholar] [CrossRef] [PubMed]
- Aungkawattanapong, N.; Techavichit, P.; Lauhasurayotin, S.; Chiengthong, K.; Sosothikul, D.; Monsereenusorn, C.; Supornsilchai, V.; Suphapeetiporn, K.; Teerapakpinyo, C.; Shuangshoti, S. A study of the TP53 Germline Mutation and Clinicopathologic Features in Thai Children with Adrenocortical Carcinoma. J. Health Sci. Med. Res. 2021, 39, 491–502. [Google Scholar] [CrossRef]
- Walz, A.L.; Ooms, A.; Gadd, S.; Gerhard, D.S.; Smith, M.A.; Guidry Auvil, J.M.; Meerzaman, D.; Chen, Q.R.; Hsu, C.H.; Yan, C.; et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 2015, 27, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Mahamdallie, S.; Yost, S.; Poyastro-Pearson, E.; Holt, E.; Zachariou, A.; Seal, S.; Elliott, A.; Clarke, M.; Warren-Perry, M.; Hanks, S.; et al. Identification of new Wilms tumour predisposition genes: An exome sequencing study. Lancet Child. Adolesc. Health 2019, 3, 322–331. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Heldenbrand, J.R.; Baheti, S.; Bockol, M.A.; Drucker, T.M.; Hart, S.N.; Hudson, M.E.; Iyer, R.K.; Kalmbach, M.T.; Kendig, K.I.; Klee, E.W.; et al. Recommendations for performance optimizations when using GATK3.8 and GATK4. BMC Bioinformatics 2019, 20, 557. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Group, T.T.P.O. National Protocol for the Treatment of Childhood Cancers, 1st ed.; M Print Corporation Co., Ltd.: Bangkok, Thailand, 2016. [Google Scholar]
- Petrov, I.; Alexeyenko, A. Individualized discovery of rare cancer drivers in global network context. eLife 2022, 11, e74010. [Google Scholar] [CrossRef] [PubMed]
- Ostroverkhova, D.; Przytycka, T.M.; Panchenko, A.R. Cancer driver mutations: Predictions and reality. Trends Mol. Med. 2023, 29, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Muinos, F.; Martinez-Jimenez, F.; Pich, O.; Gonzalez-Perez, A.; Lopez-Bigas, N. In silico saturation mutagenesis of cancer genes. Nature 2021, 596, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Klco, J.; Thomas, M., 3rd; Qi, W.; Walsh, M.; Ma, J.; Westover, T.; Abdelhamed, S.; Ezzell, L.; Rolle, C.; Xiong, E.; et al. Functional Characterization of Cooperating MGA Mutations in RUNX1::RUNX1T1 Acute Myeloid Leukemia. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, G.; Chen, B.; Wang, Y.; Guo, L.; Cao, L.; Ren, C.; Wen, L.; Liao, N. Association between histone lysine methyltransferase KMT2C mutation and clinicopathological factors in breast cancer. Biomed. Pharmacother. 2019, 116, 108997. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhong, A.; Zhang, S.; Chen, M.; Zhang, L.; Hang, X.; Zheng, J.; Wu, B.; Deng, X.; Pan, X.; et al. KMT2D Deficiency Promotes Myeloid Leukemias which Is Vulnerable to Ribosome Biogenesis Inhibition. Adv. Sci. 2023, 10, e2206098. [Google Scholar] [CrossRef]
- Schweigert, A.; Fischer, C.; Mayr, D.; von Schweinitz, D.; Kappler, R.; Hubertus, J. Activation of the Wnt/beta-catenin pathway is common in wilms tumor, but rarely through beta-catenin mutation and APC promoter methylation. Pediatr. Surg. Int. 2016, 32, 1141–1146. [Google Scholar] [CrossRef]
- Fleming-de-Moraes, C.D.; Rocha, M.R.; Tessmann, J.W.; de Araujo, W.M.; Morgado-Diaz, J.A. Crosstalk between PI3K/Akt and Wnt/beta-catenin pathways promote colorectal cancer progression regardless of mutational status. Cancer Biol. Ther. 2022, 23, 1–13. [Google Scholar] [CrossRef]
- Langhammer, T.-S.; Roolf, C.; Krohn, S.; Kretzschmar, C.; Huebner, R.; Rolfs, A.; Freund, M.; Junghanss, C. PI3K/Akt Signaling Interacts With Wnt/β-Catenin Signaling but Does Not Induce an Accumulation of β-Catenin in the Nucleus of Acute Lymphoblastic Leukemia Cell Lines. Blood 2013, 122, 4886. [Google Scholar] [CrossRef]
- Bitaraf, M.; Mahmanzar, M.; Zafari, N.; Mohammadpour, H.; Vasei, M.; Moradi Matin, L.; Kajbafzadeh, A.M.; Majidi Zolbin, M. The potential key genes and pathways associated with Wilms tumor in quest of proper candidates for diagnostic and therapeutic purposes. Sci. Rep. 2022, 12, 17906. [Google Scholar] [CrossRef]
- Jimenez Martin, O.; Schlosser, A.; Furtwangler, R.; Wegert, J.; Gessler, M. MYCN and MAX alterations in Wilms tumor and identification of novel N-MYC interaction partners as biomarker candidates. Cancer Cell Int. 2021, 21, 555. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Ney, G.M.; McKay, L.; Koschmann, C.; Mody, R.; Li, Q. The Emerging Role of Ras Pathway Signaling in Pediatric Cancer. Cancer Res. 2020, 80, 5155–5163. [Google Scholar] [CrossRef]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef]
- Zhi, X.; Tao, J.; Xie, K.; Zhu, Y.; Li, Z.; Tang, J.; Wang, W.; Xu, H.; Zhang, J.; Xu, Z. MUC4-induced nuclear translocation of beta-catenin: A novel mechanism for growth, metastasis and angiogenesis in pancreatic cancer. Cancer Lett. 2014, 346, 104–113. [Google Scholar] [CrossRef]
- Gao, L.; Liu, J.; Zhang, B.; Zhang, H.; Wang, D.; Zhang, T.; Xu, Z. Functional MUC4 suppress epithelial-mesenchymal transition in lung adenocarcinoma metastasis. Tumour Biol. 2014, 35, 1335–1341. [Google Scholar] [CrossRef]
- Giannakouros, P.; Comamala, M.; Matte, I.; Rancourt, C.; Piche, A. MUC16 mucin (CA125) regulates the formation of multicellular aggregates by altering beta-catenin signaling. Am. J. Cancer Res. 2015, 5, 219–230. [Google Scholar]
- Pai, P.; Rachagani, S.; Dhawan, P.; Sheinin, Y.M.; Macha, M.A.; Qazi, A.K.; Chugh, S. MUC4 is negatively regulated through the Wnt/beta-catenin pathway via the Notch effector Hath1 in colorectal cancer. Genes. Cancer 2016, 7, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Dann, R.B.; DeLoia, J.A.; Timms, K.M.; Zorn, K.K.; Potter, J.; Flake, D.D., 2nd; Lanchbury, J.S.; Krivak, T.C. BRCA1/2 mutations and expression: Response to platinum chemotherapy in patients with advanced stage epithelial ovarian cancer. Gynecol. Oncol. 2012, 125, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015, 75, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Anderson, B.; Burstein, H.J.; Chew, H.; Dang, C.; et al. Breast Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 691–722. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Patel, J.P.; Gonen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Villarroel, M.C.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Jesus-Acosta, A.; Jones, S.; Maitra, A.; Hruban, R.H.; Eshleman, J.R.; Klein, A.; Laheru, D.; et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol. Cancer Ther. 2011, 10, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Cowin, P.A.; George, J.; Fereday, S.; Loehrer, E.; Van Loo, P.; Cullinane, C.; Etemadmoghadam, D.; Ftouni, S.; Galletta, L.; Anglesio, M.S.; et al. LRP1B deletion in high-grade serous ovarian cancers is associated with acquired chemotherapy resistance to liposomal doxorubicin. Cancer Res. 2012, 72, 4060–4073. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
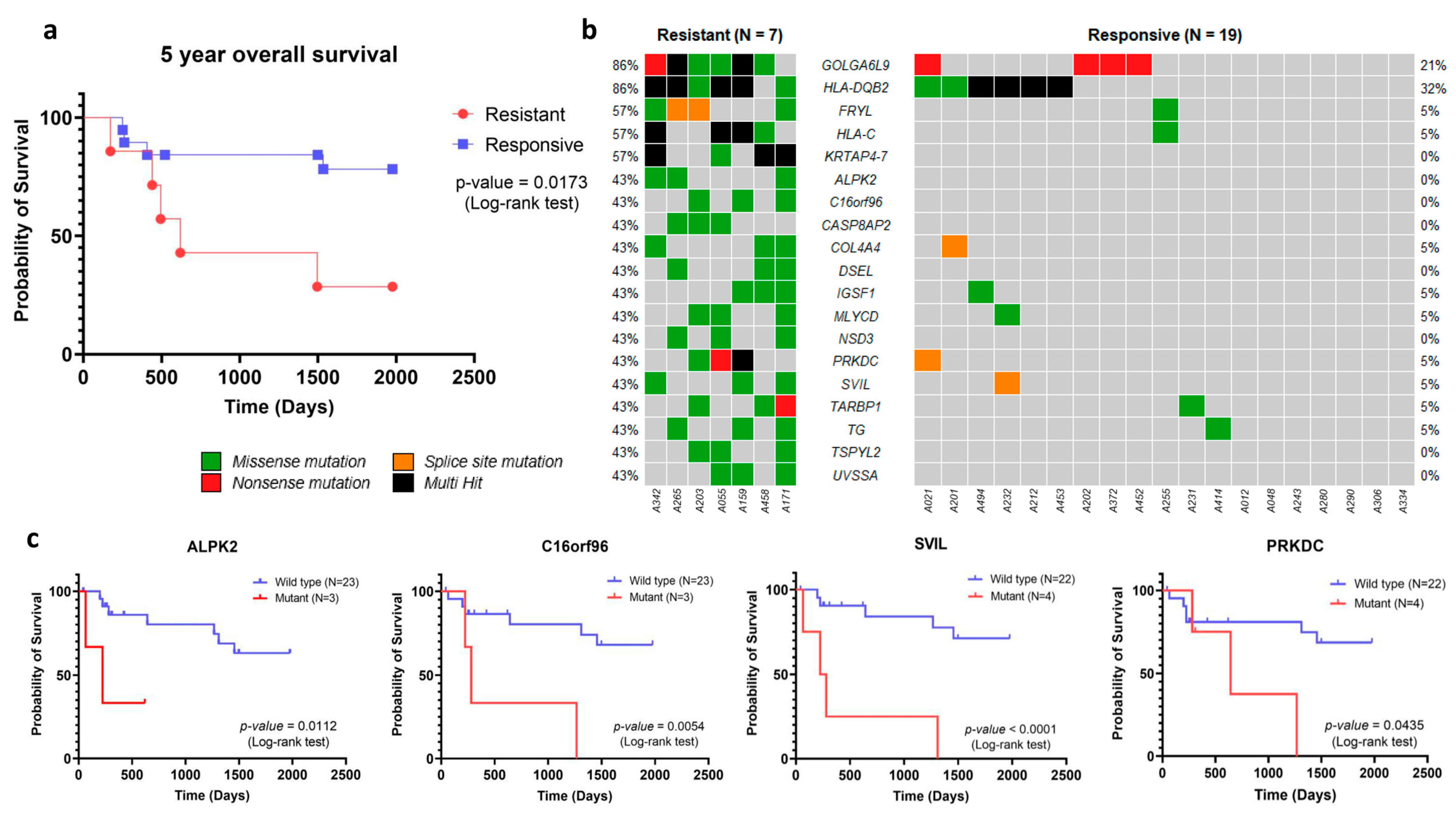
| Gene | Chemotherapy Response * | p-Value | |
|---|---|---|---|
| Resistant (N:7) | Responsive (N:19) | ||
| KRTAP4-7 | 4/7 (51.14%) | 0/19 (0.00%) | 0.002 |
| GOLGA6L9 | 6/7 (85.71%) | 4/19 (21.05%) | 0.005 |
| FRYL | 4/7 (51.14%) | 1/19 (0.05%) | 0.010 |
| HLA-C | 4/7 (51.14%) | 1/19 (0.05%) | 0.010 |
| ALPK2 | 3/7 (42.86%) | 0/19 (0.00%) | 0.013 |
| C16orf96 | 3/7 (42.86%) | 0/19 (0.00%) | 0.013 |
| CASP8AP2 | 3/7 (42.86%) | 0/19 (0.00%) | 0.013 |
| DSEL | 3/7 (42.86%) | 0/19 (0.00%) | 0.013 |
| NSD3 | 3/7 (42.86%) | 0/19 (0.00%) | 0.013 |
| TSPYL2 | 3/7 (42.86%) | 0/19 (0.00%) | 0.013 |
| UVSSA | 3/7 (42.86%) | 0/19 (0.00%) | 0.013 |
| HLA-DQB2 | 6/7 (85.71%) | 6/19 (31.58%) | 0.026 |
| COL4A4 | 3/7 (42.86%) | 1/19 (0.05%) | 0.047 |
| IGSF1 | 3/7 (42.86%) | 1/19 (0.05%) | 0.047 |
| MLYCD | 3/7 (42.86%) | 1/19 (0.05%) | 0.047 |
| PRKDC | 3/7 (42.86%) | 1/19 (0.05%) | 0.047 |
| SVIL | 3/7 (42.86%) | 1/19 (0.05%) | 0.047 |
| TARBP1 | 3/7 (42.86%) | 1/19 (0.05%) | 0.047 |
| TG | 3/7 (42.86%) | 1/19 (0.05%) | 0.047 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choochuen, P.; Nokchan, N.; Khongcharoen, N.; Laochareonsuk, W.; Surachat, K.; Chotsampancharoen, T.; Sila, T.; Sangkhathat, S., on behalf of the Thai Pediatric Cancer Atlas (TPCA) Consortium. Discovery of Novel Potential Prognostic Markers and Targeted Therapy to Overcome Chemotherapy Resistance in an Advanced-Stage Wilms Tumor. Cancers 2024, 16, 1567. https://doi.org/10.3390/cancers16081567
Choochuen P, Nokchan N, Khongcharoen N, Laochareonsuk W, Surachat K, Chotsampancharoen T, Sila T, Sangkhathat S on behalf of the Thai Pediatric Cancer Atlas (TPCA) Consortium. Discovery of Novel Potential Prognostic Markers and Targeted Therapy to Overcome Chemotherapy Resistance in an Advanced-Stage Wilms Tumor. Cancers. 2024; 16(8):1567. https://doi.org/10.3390/cancers16081567
Chicago/Turabian StyleChoochuen, Pongsakorn, Natakorn Nokchan, Natthapon Khongcharoen, Wison Laochareonsuk, Komwit Surachat, Thirachit Chotsampancharoen, Thanit Sila, and Surasak Sangkhathat on behalf of the Thai Pediatric Cancer Atlas (TPCA) Consortium. 2024. "Discovery of Novel Potential Prognostic Markers and Targeted Therapy to Overcome Chemotherapy Resistance in an Advanced-Stage Wilms Tumor" Cancers 16, no. 8: 1567. https://doi.org/10.3390/cancers16081567
APA StyleChoochuen, P., Nokchan, N., Khongcharoen, N., Laochareonsuk, W., Surachat, K., Chotsampancharoen, T., Sila, T., & Sangkhathat, S., on behalf of the Thai Pediatric Cancer Atlas (TPCA) Consortium. (2024). Discovery of Novel Potential Prognostic Markers and Targeted Therapy to Overcome Chemotherapy Resistance in an Advanced-Stage Wilms Tumor. Cancers, 16(8), 1567. https://doi.org/10.3390/cancers16081567