
Genetic Diagnosis of Retinoblastoma Using Aqueous Humour—Findings from an Extended Cohort

 , , , , ,
, , , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Sample Collection
- From primary (PE) or secondary (SE) enucleated eyes prior to opening the eye for tumour dissection;
- During IViC via the standard clinical protocol of AH aspiration prior to chemotherapy injection (Tx);
- By diagnostic anterior chamber tap from retained eyes that had received a minimum of one cycle of intravenous (IVC) or intra-arterial (IAC) chemotherapy (Dx1+).
2.2. Sample Processing and Analysis
2.3. Previous Publication of Data
2.4. Statistical Analysis
2.5. Clinical Diagnostic Testing
3. Results
3.1. Patient and Sample Characteristics
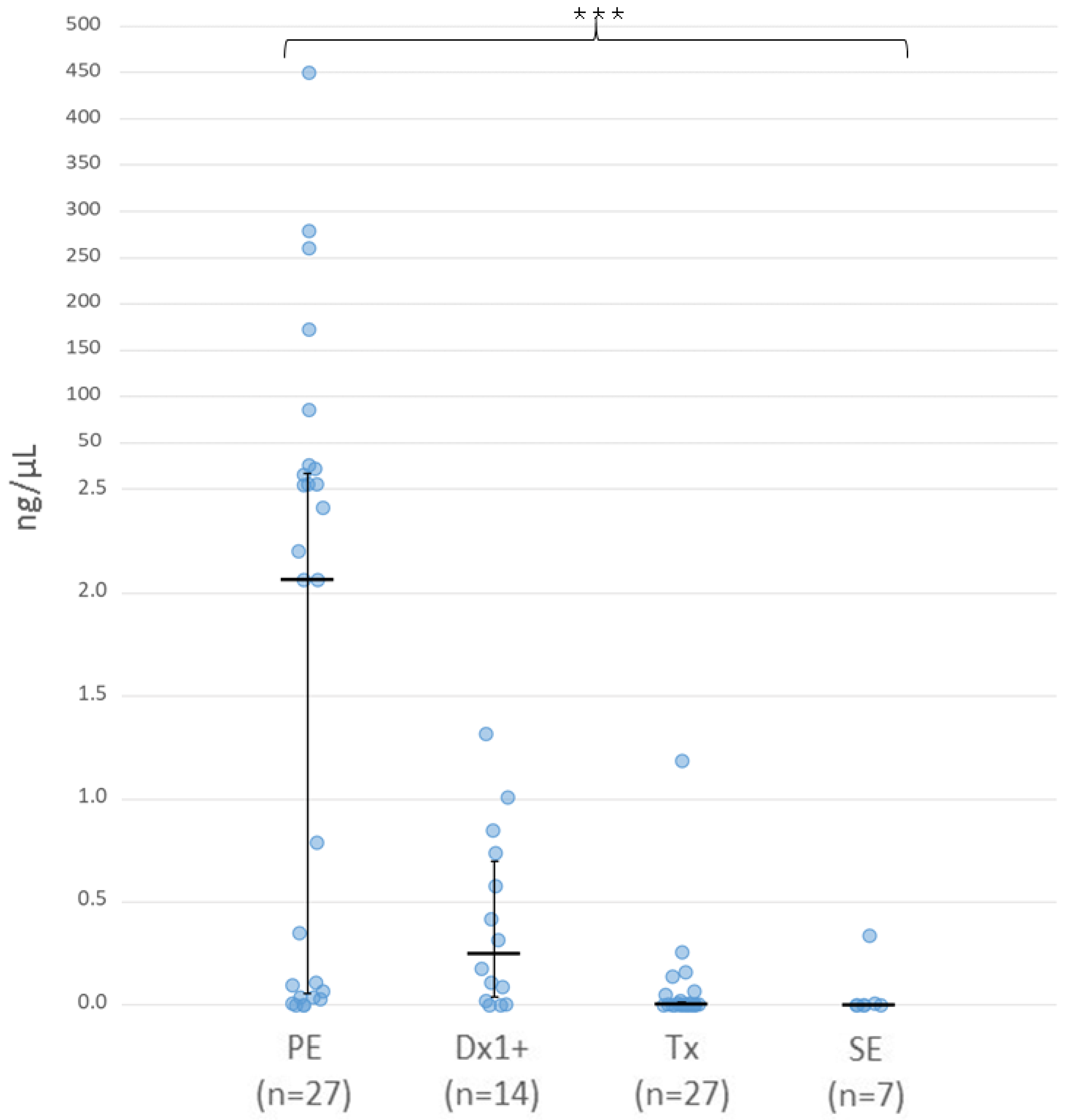
3.2. Cell-Free DNA Concentration in Aqueous Humour
3.3. RB1 Pathogenic Variant Detection Using Aqueous Humour
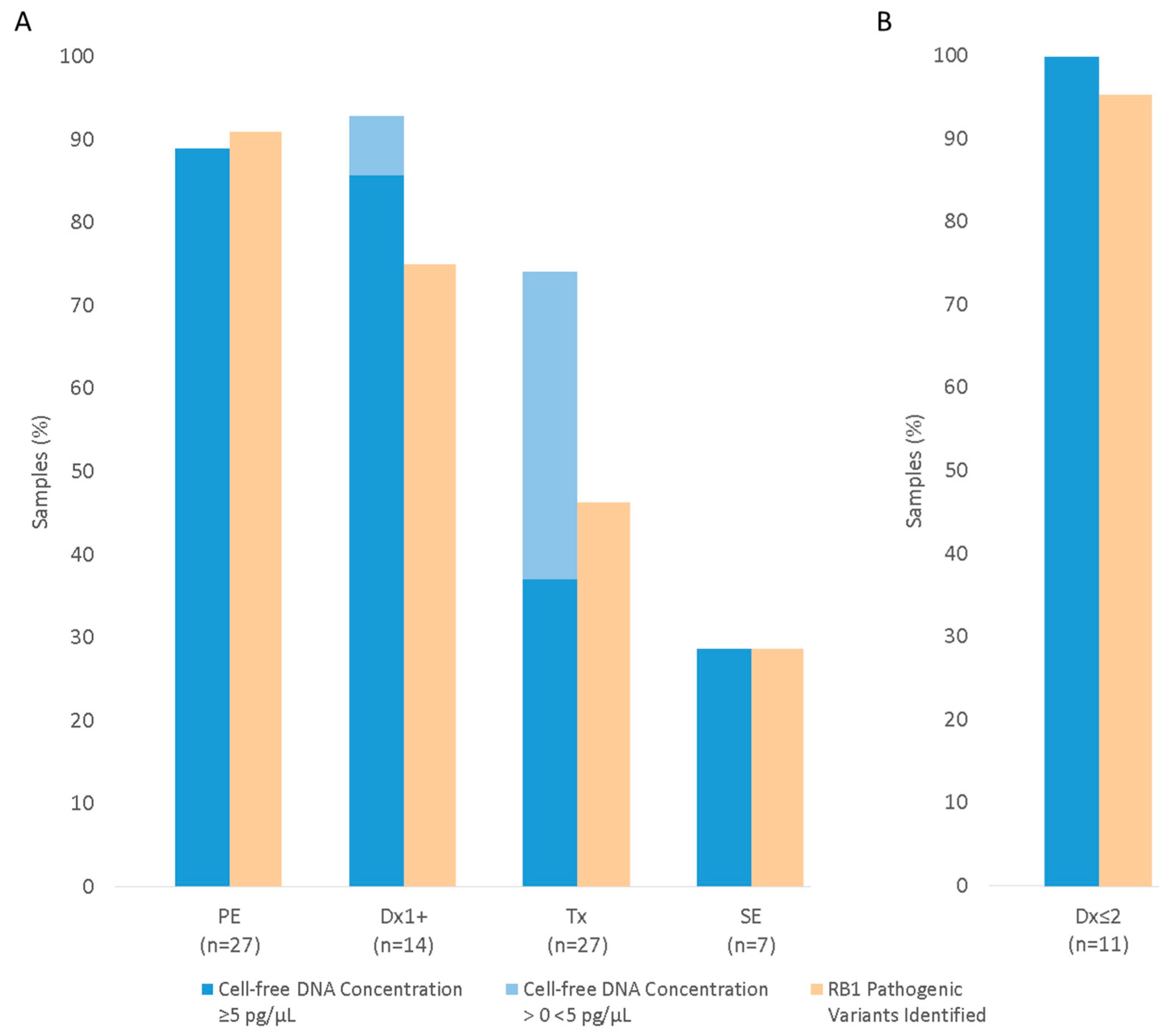
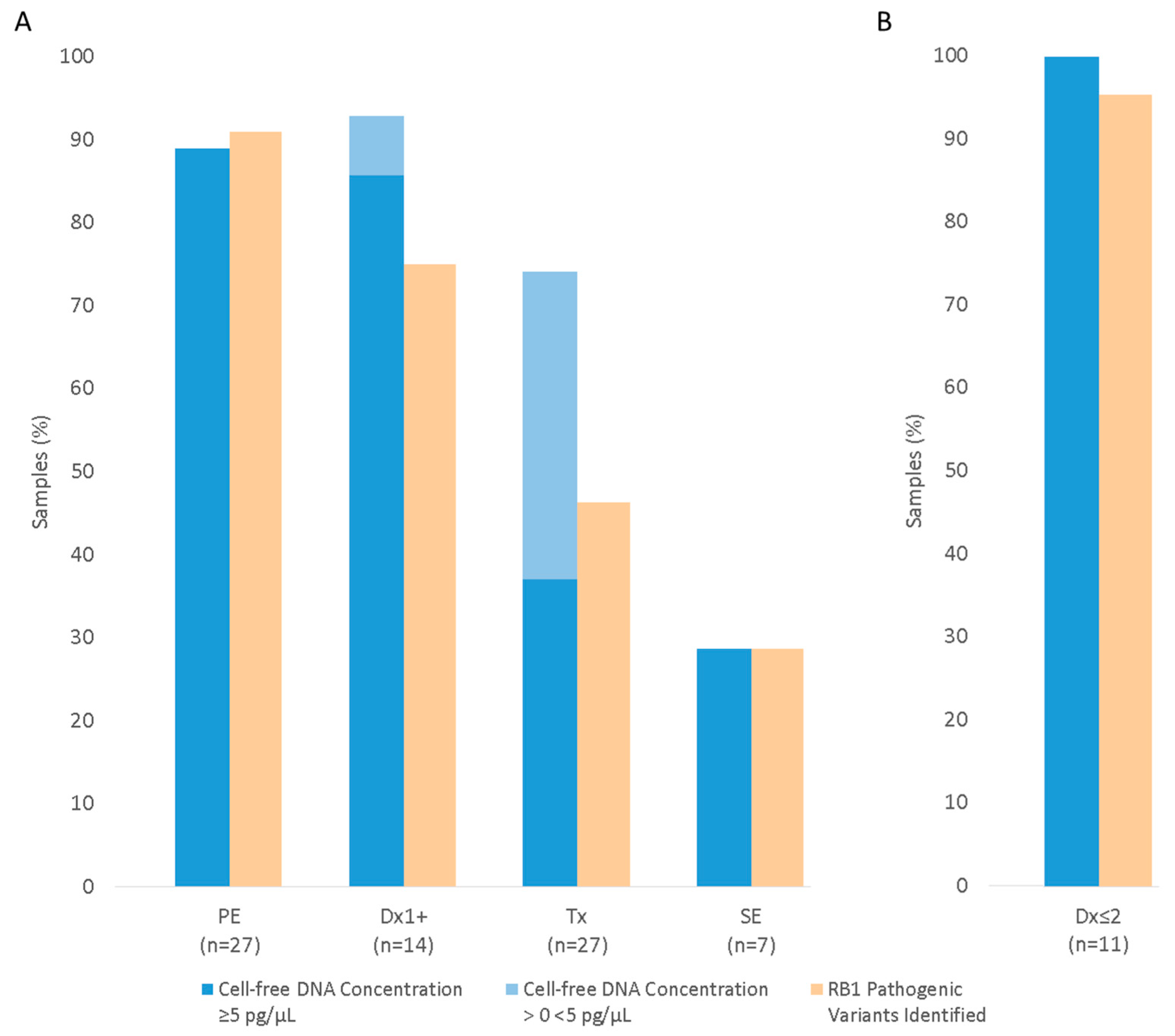
3.3.1. RB1 Pathogenic Variant Detection in AH from Enucleated Eyes
Primary Enucleation (PE)
Secondary Enucleation (SE)
3.3.2. RB1 Pathogenic Variant Detection in AH from Eyes Undergoing Conservative Treatment
Anterior Chamber Tap (Dx1+)
Eyes Undergoing IViC (Tx)
3.4. Determining Tumour-Derived Fraction of Cell-Free DNA
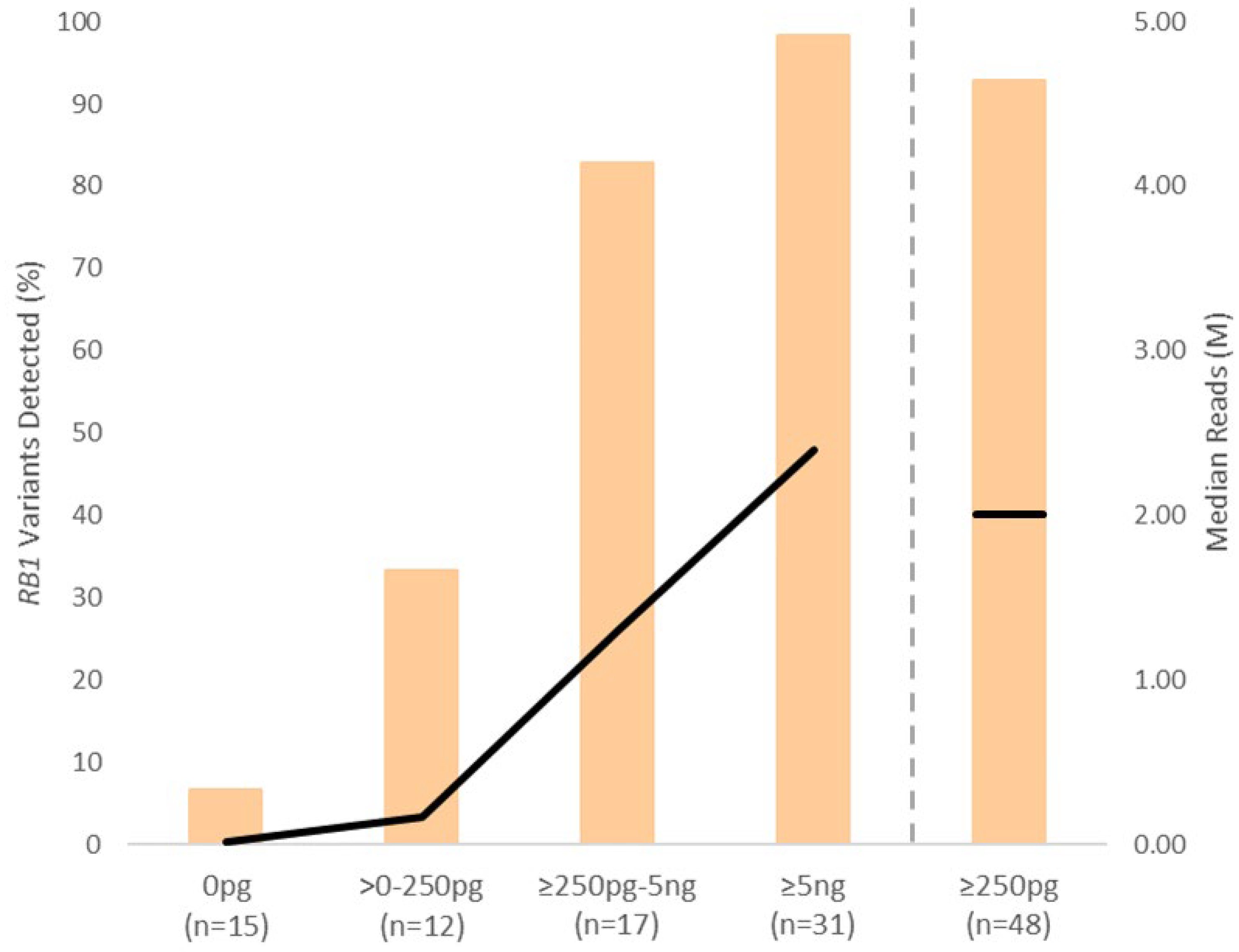
3.5. RB1 Pathogenic Variant Detection Correlates with cfDNA Concentration
3.6. Comparison of cfDNA Concentration and RB1 Pathogenic Variant Detection in Serial AH Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Appendix A
Appendix A.1. Sample Processing and DNA Extraction
Appendix A.2. Targeted NGS
Appendix A.3. Bioinformatic Analysis
References
- Lohmann, D.R.; Gallie, B.L. Retinoblastoma. In GeneReviews(®); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J., Bird, T.D., Fong, C.-T., Mefford, H.C., Smith, R.J., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Price, E.A.; Price, K.; Kolkiewicz, K.; Hack, S.; Reddy, M.A.; Hungerford, J.L.; Kingston, J.E.; Onadim, Z. Spectrum of RB1 Mutations Identified in 403 Retinoblastoma Patients. J. Med. Genet. 2014, 51, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Dommering, C.J.; Mol, B.M.; Moll, A.C.; Burton, M.; Cloos, J.; Dorsman, J.C.; Meijers-Heijboer, H.; van der Hout, A.H. RB1 Mutation Spectrum in a Comprehensive Nationwide Cohort of Retinoblastoma Patients. J. Med. Genet. 2014, 51, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Rushlow, D.E.; Mol, B.M.; Kennett, J.Y.; Yee, S.; Pajovic, S.; Thériault, B.L.; Prigoda-Lee, N.L.; Spencer, C.; Dimaras, H.; Corson, T.W.; et al. Characterisation of Retinoblastomas without RB1 Mutations: Genomic, Gene Expression, and Clinical Studies. Lancet Oncol. 2013, 14, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Kamihara, J.; Bourdeaut, F.; Foulkes, W.D.; Molenaar, J.J.; Mossé, Y.P.; Nakagawara, A.; Parareda, A.; Scollon, S.R.; Schneider, K.W.; Skalet, A.H.; et al. Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin. Cancer Res. 2017, 23, e98–e106. [Google Scholar] [CrossRef]
- Feng, Y.-P.; Yang, T.-S.; Chung, C.-H.; Chien, W.-C.; Wong, C.-S. Early Childhood General Anesthesia Exposure Associated with Later Developmental Delay: A National Population-Based Cohort Study. PLoS ONE 2020, 15, e0238289. [Google Scholar] [CrossRef] [PubMed]
- Reighard, C.; Junaid, S.; Jackson, W.M.; Arif, A.; Waddington, H.; Whitehouse, A.J.O.; Ing, C. Anesthetic Exposure During Childhood and Neurodevelopmental Outcomes: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2022, 5, e2217427. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.E.; Racher, H.; Zhang, C.; MacDonald, H.; Gallie, B.L. Genetics and Molecular Diagnostics in Retinoblastoma--An Update. Asia Pac. J. Ophthalmol. 2017, 6, 197–207. [Google Scholar] [CrossRef]
- Karcioglu, Z.A. Fine needle aspiration biopsy (fnab) for retinoblastoma. Retina 2002, 22, 707–710. [Google Scholar] [CrossRef]
- Munier, F.L.; Beck-Popovic, M.; Chantada, G.L.; Cobrinik, D.; Kivelä, T.T.; Lohmann, D.; Maeder, P.; Moll, A.C.; Carcaboso, A.M.; Moulin, A.; et al. Conservative Management of Retinoblastoma: Challenging Orthodoxy without Compromising the State of Metastatic Grace. “Alive, with Good Vision and No Comorbidity”. Prog. Retin. Eye Res. 2019, 73, 100764. [Google Scholar] [CrossRef]
- Gerrish, A.; Jenkinson, H.; Cole, T. The Impact of Cell-Free DNA Analysis on the Management of Retinoblastoma. Cancers 2021, 13, 1570. [Google Scholar] [CrossRef]
- Gerrish, A.; Stone, E.; Clokie, S.; Ainsworth, J.R.; Jenkinson, H.; McCalla, M.; Hitchcott, C.; Colmenero, I.; Allen, S.; Parulekar, M.; et al. Non-Invasive Diagnosis of Retinoblastoma Using Cell-Free DNA from Aqueous Humour. Br. J. Ophthalmol. 2019, 103, 721–724. [Google Scholar] [CrossRef]
- Xu, L.; Shen, L.; Polski, A.; Prabakar, R.K.; Shah, R.; Jubran, R.; Kim, J.W.; Biegel, J.; Kuhn, P.; Cobrinik, D.; et al. Simultaneous Identification of Clinically Relevant RB1 Mutations and Copy Number Alterations in Aqueous Humor of Retinoblastoma Eyes. Ophthalmic Genet. 2020, 41, 526–532. [Google Scholar] [CrossRef]
- Xu, L.; Kim, M.E.; Polski, A.; Prabakar, R.K.; Shen, L.; Peng, C.-C.; Reid, M.W.; Chévez-Barrios, P.; Kim, J.W.; Shah, R.; et al. Establishing the Clinical Utility of ctDNA Analysis for Diagnosis, Prognosis, and Treatment Monitoring of Retinoblastoma: The Aqueous Humor Liquid Biopsy. Cancers 2021, 13, 1282. [Google Scholar] [CrossRef]
- Le Gall, J.; Dehainault, C.; Benoist, C.; Matet, A.; Lumbroso-Le Rouic, L.; Aerts, I.; Jiménez, I.; Schleiermacher, G.; Houdayer, C.; Radvanyi, F.; et al. Highly Sensitive Detection Method of Retinoblastoma Genetic Predisposition and Biomarkers. J. Mol. Diagn. 2021, 23, 1714–1721. [Google Scholar] [CrossRef] [PubMed]
- Kletke, S.N.; Soliman, S.; Racher, H.; Mallipatna, A.; Shaikh, F.; Mireskandari, K.; Gallie, B.L. A Typical Anterior Retinoblastoma: Diagnosis by Aqueous Humor Cell-Free DNA Analysis. Ophthalmic Genet. 2022, 43, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.Y.; Xu, L.; Shen, L.; Kim, M.E.; Polski, A.; Prabakar, R.K.; Shah, R.; Jubran, R.; Kim, J.W.; Biegel, J.A.; et al. Inter-Eye Genomic Heterogeneity in Bilateral Retinoblastoma via Aqueous Humor Liquid Biopsy. NPJ Precis. Oncol. 2021, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Raval, V.; Racher, H.; Wrenn, J.; Singh, A.D. Aqueous Humor as a Surrogate Biomarker for Retinoblastoma Tumor Tissue. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2022, 26, 137.e1–137.e5. [Google Scholar] [CrossRef]
- Schmidt, M.J.; Prabakar, R.K.; Pike, S.; Yellapantula, V.; Peng, C.-C.; Kuhn, P.; Hicks, J.; Xu, L.; Berry, J.L. Simultaneous Copy Number Alteration and Single-Nucleotide Variation Analysis in Matched Aqueous Humor and Tumor Samples in Children with Retinoblastoma. Int. J. Mol. Sci. 2023, 24, 8606. [Google Scholar] [CrossRef] [PubMed]
- Cassoux, N.; Malaise, D.; Lumbroso-Le-Rouic, L.; Le Gall, J.; Golmard, L.; Cardoen, L.; Freneaux, P.; Bouchoucha, Y.; Aerts, I.; Doz, F.; et al. Diffuse Infiltrating Retinoblastoma with Anterior Chamber Involvement: Conservative Management and Identification of RB1 Alterations in Aqueous Humor. Ocul. Oncol. Pathol. 2023, 9, 96–100. [Google Scholar] [CrossRef]
- Sirivolu, S.; Xu, L.; Warren, M.; Prabakar, R.K.; Shah, R.; Kuhn, P.; Hicks, J.; Berry, J.L. Chromosome 6p Amplification Detected in Blood Cell-Free DNA in Advanced Intraocular Retinoblastoma. Ophthalmic Genet. 2022, 43, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Munier, F.L.; Soliman, S.; Moulin, A.P.; Gaillard, M.-C.; Balmer, A.; Beck-Popovic, M. Profiling Safety of Intravitreal Injections for Retinoblastoma Using an Anti-Reflux Procedure and Sterilisation of the Needle Track. Br. J. Ophthalmol. 2012, 96, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.H.; Abramson, D.H.; Ji, X.; Shields, C.L.; Teixeira, L.F.; Schefler, A.C.; Cassoux, N.; Hadjistilianou, D.; Berry, J.L.; Frenkel, S.; et al. Risk of Extraocular Extension in Eyes With Retinoblastoma Receiving Intravitreous Chemotherapy. JAMA Ophthalmol. 2017, 135, 1426. [Google Scholar] [CrossRef] [PubMed]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Baple, E.L.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.E.; Eccles, D.M.; Abbs, S.; Scott, R.; Deans, Z.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease. 2020. Available online: https://www.Acgs.Uk.Com/Media/11631/Uk-Practice-Guidelines-for-Variant-Classification-v4-01-2020.Pdf (accessed on 31 October 2023).
- Zaiontz, C. Real Statistics Using Excel. Available online: https://www.Real-Statistics.Com (accessed on 30 April 2015).
- Linn Murphree, A. Intraocular Retinoblastoma: The Case for a New Group Classification. Ophthalmol. Clin. North. Am. 2005, 18, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Agilent Technologies, Inc. Agilent High Sensitivity D1000 ScreenTape System Quick Guide. 2015. Available online: https://www.Agilent.Com/Cs/Library/Usermanuals/Public/ScreenTape_HSD1000_QG.Pdf (accessed on 7 January 2019).
- Abramson, D.H.; Fabius, A.W.M.; Issa, R.; Francis, J.H.; Marr, B.P.; Dunkel, I.J.; Gobin, Y.P. Advanced Unilateral Retinoblastoma: The Impact of Ophthalmic Artery Chemosurgery on Enucleation Rate and Patient Survival at MSKCC. PLoS ONE 2015, 10, e0145436. [Google Scholar] [CrossRef] [PubMed]
- Im, D.H.; Pike, S.; Reid, M.W.; Peng, C.-C.; Sirivolu, S.; Grossniklaus, H.E.; Hubbard, G.B.; Skalet, A.H.; Bellsmith, K.N.; Shields, C.L.; et al. A Multicenter Analysis of Nucleic Acid Quantification Using Aqueous Humor Liquid Biopsy in Retinoblastoma: Implications for Clinical Testing. Ophthalmol. Sci. 2023, 3, 100289. [Google Scholar] [CrossRef] [PubMed]
- Cancellieri, F.; Peter, V.G.; Cuadrado, M.; Quinodoz, M.; Stathopoulos, C.; Munier, F.L.; Rivolta, C. An Integrated Approach to Study Genomics and Epigenomics of Retinoblastoma from Cell-Free DNA in the Aqueous Humor. In Proceedings of the International Society for Genetic Eye Diseases & Retinoblastoma (ISGEDR), São Paulo, Brazil, 6–8 July 2023. [Google Scholar]
- Xu, L.; Polski, A.; Prabakar, R.K.; Reid, M.W.; Chevez-Barrios, P.; Jubran, R.; Kim, J.W.; Kuhn, P.; Cobrinik, D.; Hicks, J.; et al. Chromosome 6p Amplification in Aqueous Humor Cell-Free DNA Is a Prognostic Biomarker for Retinoblastoma Ocular Survival. Mol. Cancer Res. 2020, 18, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Li, B.; Wu, S.; Chen, J.; Sain, D.; Xu, D.; Black, M.H.; Karam, R.; Gillespie, K.; Farwell Hagman, K.D.; et al. Detection of Structural Variation Using Target Captured Next-Generation Sequencing Data for Genetic Diagnostic Testing. Genet. Med. 2019, 21, 1603–1610. [Google Scholar] [CrossRef]
- Li, H.-T.; Xu, L.; Weisenberger, D.J.; Li, M.; Zhou, W.; Peng, C.-C.; Stachelek, K.; Cobrinik, D.; Liang, G.; Berry, J.L. Characterizing DNA Methylation Signatures of Retinoblastoma Using Aqueous Humor Liquid Biopsy. Nat. Commun. 2022, 13, 5523. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific Qubit® dsDNA HS Assay Kits. 2015. Available online: https://Tools.Thermofisher.Com/Content/Sfs/Manuals/Qubit_dsDNA_HS_Assay_UG.Pdf (accessed on 7 January 2019).
- Raymond, C.K.; Raymond, F.C.; Hill, K. UltraPrep Is a Scalable, Cost-Effective, Bead-Based Method for Purifying Cell-Free DNA. PLoS ONE 2020, 15, e0231854. [Google Scholar] [CrossRef] [PubMed]
- Tyner, C.; Barber, G.P.; Casper, J.; Clawson, H.; Diekhans, M.; Eisenhart, C.; Fischer, C.M.; Gibson, D.; Gonzalez, J.N.; Guruvadoo, L.; et al. The UCSC Genome Browser Database: 2017 Update. Nucleic Acids Res. 2017, 45, D626–D634. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast Duplicate Marking and Structural Variant Read Extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; WGS500 Consortium; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating Mapping-, Assembly- and Haplotype-Based Approaches for Calling Variants in Clinical Sequencing Applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Sample Type | Samples (n) | Patients/Eyes (n) | Laterality U/B (n) | IIRC Grade C/D/E (n) | Chemotherapy Cycles 1 Mean (Range) |
|---|---|---|---|---|---|
| PE | 27 | 27/27 | 21/6 | 0/9/18 | 0 |
| Dx1+ | 14 | 14/14 | 13/1 | 5/7/2 | 1.6 (1–3) |
| Tx | 27 | 26/27 | 17/9 | 2/19/6 | 5.7 (2–12) |
| SE | 7 | 7/7 | 2/5 | 0/4/3 | 5.5 (2–8) |
| All | 75 | 68/71 | 49/19 | 6/38/27 | 2.8 (0–12) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerrish, A.; Mashayamombe-Wolfgarten, C.; Stone, E.; Román-Montañana, C.; Abbott, J.; Jenkinson, H.; Millen, G.; Gurney, S.; McCalla, M.; Staveley, S.-J.; et al. Genetic Diagnosis of Retinoblastoma Using Aqueous Humour—Findings from an Extended Cohort. Cancers 2024, 16, 1565. https://doi.org/10.3390/cancers16081565
Gerrish A, Mashayamombe-Wolfgarten C, Stone E, Román-Montañana C, Abbott J, Jenkinson H, Millen G, Gurney S, McCalla M, Staveley S-J, et al. Genetic Diagnosis of Retinoblastoma Using Aqueous Humour—Findings from an Extended Cohort. Cancers. 2024; 16(8):1565. https://doi.org/10.3390/cancers16081565
Chicago/Turabian StyleGerrish, Amy, Chipo Mashayamombe-Wolfgarten, Edward Stone, Claudia Román-Montañana, Joseph Abbott, Helen Jenkinson, Gerard Millen, Sam Gurney, Maureen McCalla, Sarah-Jane Staveley, and et al. 2024. "Genetic Diagnosis of Retinoblastoma Using Aqueous Humour—Findings from an Extended Cohort" Cancers 16, no. 8: 1565. https://doi.org/10.3390/cancers16081565
APA StyleGerrish, A., Mashayamombe-Wolfgarten, C., Stone, E., Román-Montañana, C., Abbott, J., Jenkinson, H., Millen, G., Gurney, S., McCalla, M., Staveley, S.-J., Kainth, A., Kirk, M., Bowen, C., Cavanagh, S., Bunstone, S., Carney, M., Mohite, A., Clokie, S., Reddy, M. A., ... Cole, T. (2024). Genetic Diagnosis of Retinoblastoma Using Aqueous Humour—Findings from an Extended Cohort. Cancers, 16(8), 1565. https://doi.org/10.3390/cancers16081565