
The Role of Estrogen and Estrogen Receptors in Head and Neck Tumors




Simple Summary
Abstract
1. Introduction
2. Head and Neck Tumors
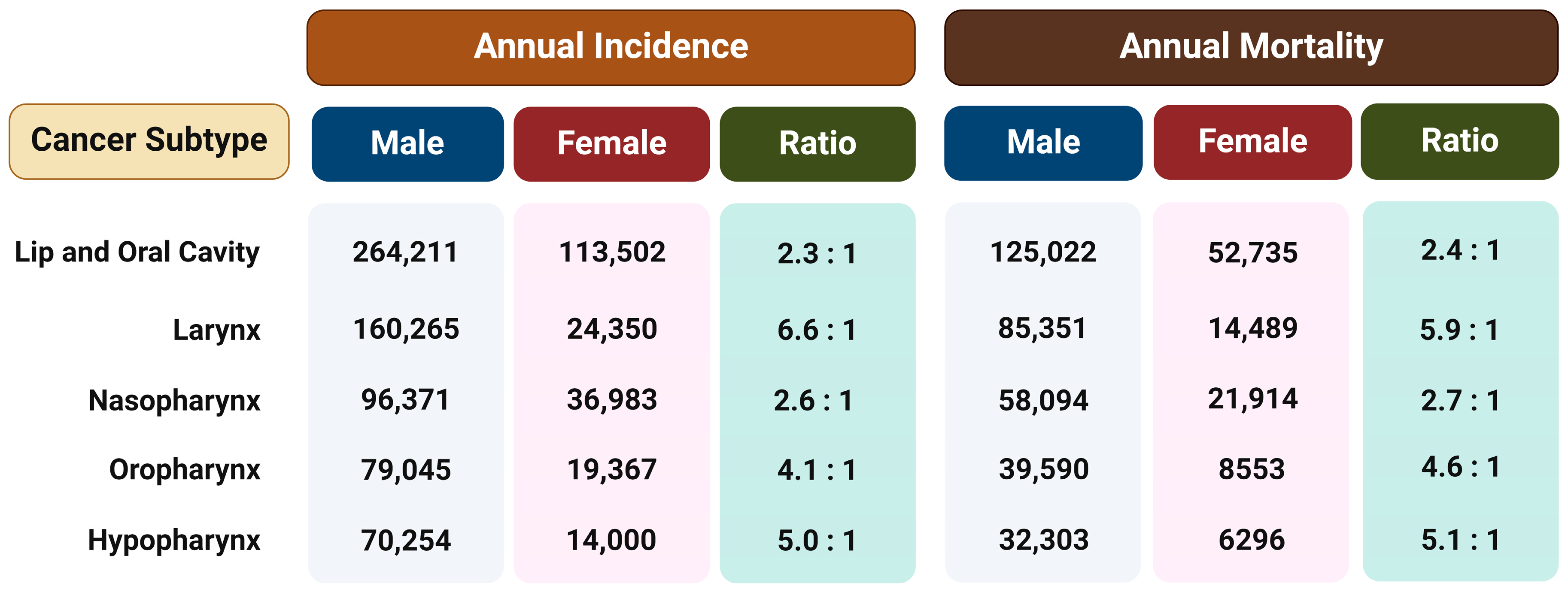
3. Sex-Related Disparities in HNSCC Incidence and Mortality
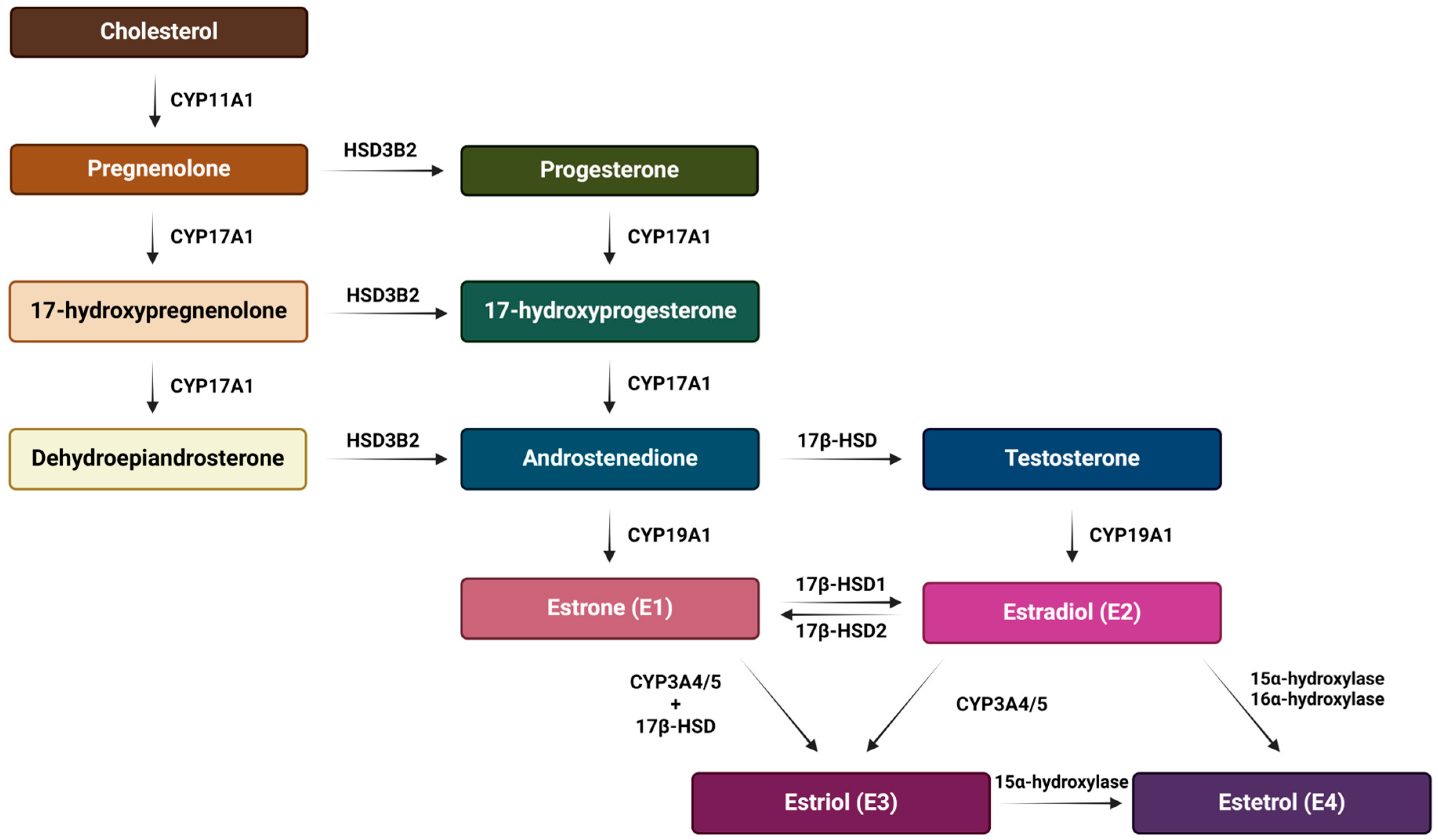
4. Estrogens
5. Estrogen Receptors
5.1. Nuclear Estrogen Receptors
5.2. Membrane Estrogen Receptors
5.2.1. ERα36
5.2.2. GPER1
5.2.3. NaV1.2 (SCN2A)
5.3. Mechanisms of ERs Action
5.3.1. Genomic Effects
5.3.2. Non-Genomic Effects
5.3.3. Other Mechanisms of Action
6. Role of Estrogen and Estrogen Receptors in Head and Neck Tumors
6.1. Role of Estrogen
6.2. Role of Nuclear Estrogen Receptors
6.3. Role of Membrane Estrogen Receptors
7. Antiestrogens and Phytoestrogens as a Therapy for Head and Neck Tumors
7.1. Impact of Antiestrogen Treatments on Head and Neck Tumors
7.2. Phytoestrogens in Head and Neck Tumor Prevention, Treatment and Pathogenesis
8. The Role of Estrogen Signaling in Modulation of Tumor Immune Microenvironment and Microbiome Composition and thus Associated Efficacy of Immunotherapy
9. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGuire, W.L.; Chamness, G.C.; Costlow, M.E.; Shepherd, R.E. Hormone Dependence in Breast Cancer. Metabolism 1974, 23, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Bonkhoff, H. Estrogen Receptor Signaling in Prostate Cancer: Implications for Carcinogenesis and Tumor Progression. Prostate 2018, 78, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Lubin, J.H.; Purdue, M.; Kelsey, K.; Zhang, Z.-F.; Winn, D.; Wei, Q.; Talamini, R.; Szeszenia-Dabrowska, N.; Sturgis, E.M.; Smith, E.; et al. Total Exposure and Exposure Rate Effects for Alcohol and Smoking and Risk of Head and Neck Cancer: A Pooled Analysis of Case-Control Studies. Am. J. Epidemiol. 2009, 170, 937–947. [Google Scholar] [CrossRef]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol Consumption and Site-Specific Cancer Risk: A Comprehensive Dose–Response Meta-Analysis. Br. J. Cancer 2015, 112, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, M.E.; Chiocca, S. Human Papillomavirus as a Driver of Head and Neck Cancers. Br. J. Cancer 2020, 122, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Goon, P.; Schürmann, M.; Oppel, F.; Shao, S.; Schleyer, S.; Pfeiffer, C.J.; Todt, I.; Brasch, F.; Scholtz, L.-U.; Göerner, M.; et al. Viral and Clinical Oncology of Head and Neck Cancers. Curr. Oncol. Rep. 2022, 24, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-I.; Lim, H.; Moon, A. Sex Differences in Cancer: Epidemiology, Genetics and Therapy. Biomol. Ther. 2018, 26, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhang, T.; Chou, J.; Patwa, H.S.; Sullivan, C.A.; Browne, J.D. Y-Chromosome-Linked Genes Are Associated With Sex-Related Head-Neck Squamous Cell Carcinoma Survival. Otolaryngol.–Head Neck Surg. 2023, 169, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Kumar, P.; Goel, R.; Kumar, A. Sex Hormones in Head and Neck Cancer: Current Knowledge and Perspectives. Clin. Cancer Investig. J. 2012, 1, 2. [Google Scholar] [CrossRef]
- Dotto, G.P.; Rustgi, A.K. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell 2016, 29, 622–637. [Google Scholar] [CrossRef]
- Bakhshi, P.; Ho, J.Q.; Zanganeh, S. Sex-Specific Outcomes in Cancer Therapy: The Central Role of Hormones. Front. Med. Technol. 2024, 6, 1320690. [Google Scholar] [CrossRef]
- Taningher, M.; Malacarne, D.; Izzotti, A.; Ugolini, D.; Parodi, S. Drug Metabolism Polymorphisms as Modulators of Cancer Susceptibility. Mutat. Res./Rev. Mutat. Res. 1999, 436, 227–261. [Google Scholar] [CrossRef] [PubMed]
- Čonkaš, J.; Sabol, M.; Ozretić, P. ‘Toxic Masculinity’: What Is Known about the Role of Androgen Receptors in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 3766. [Google Scholar] [CrossRef]
- Hashim, D.; Sartori, S.; Vecchia, C.L.; Serraino, D.; Maso, L.D.; Negri, E.; Smith, E.; Levi, F.; Boccia, S.; Cadoni, G.; et al. Hormone Factors Play a Favorable Role in Female Head and Neck Cancer Risk. Cancer Med. 2017, 6, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Vigneswaran, N.; Williams, M.D. Epidemiological Trends in Head and Neck Cancer and Aids in Diagnosis. Oral Maxillofac. Surg. Clin. N. Am. 2014, 26, 123–141. [Google Scholar] [CrossRef]
- Martino, R.; Ringash, J. Evaluation of Quality of Life and Organ Function in Head and Neck Squamous Cell Carcinoma. Hematol. Oncol. Clin. N. Am. 2008, 22, 1239–1256. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Incidencija Raka u Hrvatskoj 2019; Bilten 44; Hrvatski Zavod Za Javno Zdravstvo, Registar Za Rak Republike Hrvatske: Zagreb, Croatia, 2021; p. 37.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, V.; Lefebvre, J.-L.; Licitra, L.; Felip, E.; EHNS-ESMO-ESTRO Guidelines Working Group. Squamous Cell Carcinoma of the Head and Neck: EHNS-ESMO-ESTRO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2010, 21 (Suppl. S5), v184–v186. [Google Scholar] [CrossRef]
- Osazuwa-Peters, N.; Simpson, M.C.; Zhao, L.; Boakye, E.A.; Olomukoro, S.I.; Deshields, T.; Loux, T.M.; Varvares, M.A.; Schootman, M. Suicide Risk among Cancer Survivors: Head and Neck versus Other Cancers. Cancer 2018, 124, 4072–4079. [Google Scholar] [CrossRef]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical Update on Head and Neck Cancer: Molecular Biology and Ongoing Challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [PubMed]
- Nokovitch, L.; Maquet, C.; Crampon, F.; Taihi, I.; Roussel, L.-M.; Obongo, R.; Virard, F.; Fervers, B.; Deneuve, S. Oral Cavity Squamous Cell Carcinoma Risk Factors: State of the Art. J. Clin. Med. 2023, 12, 3264. [Google Scholar] [CrossRef]
- Secretan, B.; Straif, K.; Baan, R.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens—Part E: Tobacco, Areca Nut, Alcohol, Coal Smoke, and Salted Fish. Lancet Oncol. 2009, 10, 1033–1034. [Google Scholar] [CrossRef]
- South, A.P.; den Breems, N.Y.; Richa, T.; Nwagu, U.; Zhan, T.; Poojan, S.; Martinez-Outschoorn, U.; Johnson, J.M.; Luginbuhl, A.J.; Curry, J.M. Mutation Signature Analysis Identifies Increased Mutation Caused by Tobacco Smoke Associated DNA Adducts in Larynx Squamous Cell Carcinoma Compared with Oral Cavity and Oropharynx. Sci. Rep. 2019, 9, 19256. [Google Scholar] [CrossRef] [PubMed]
- Philip, D.; Noronha, V.; Joshi, A.; Patil, V.; Ramaswamy, A.; Chougule, A.; Prabhash, K. Molecular Biology of Head and Neck Cancers. J. Head. Neck Physicians Surg. 2016, 4, 16. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of P53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Berthiller, J.; Straif, K.; Agudo, A.; Ahrens, W.; Bezerra Dos Santos, A.; Boccia, S.; Cadoni, G.; Canova, C.; Castellsague, X.; Chen, C.; et al. Low Frequency of Cigarette Smoking and the Risk of Head and Neck Cancer in the INHANCE Consortium Pooled Analysis. Int. J. Epidemiol. 2016, 45, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Neto, C.P.D.O.; Brito, H.O.; Costa, R.M.G.D.; Brito, L.M.O. Is There a Role for Sex Hormone Receptors in Head-and-Neck Cancer? Links with HPV Infection and Prognosis. Anticancer Res. 2021, 41, 3707–3716. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen Receptor Signaling Mechanisms. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 116, pp. 135–170. ISBN 978-0-12-815561-5. [Google Scholar]
- Vrtačnik, P.; Ostanek, B.; Mencej-Bedrač, S.; Marc, J. The Many Faces of Estrogen Signaling. Biochem. Med. 2014, 24, 329–342. [Google Scholar] [CrossRef]
- Pagana, K.D.; Pagana, T.J.; Pagana, T.N. Mosby’s Diagnostic and Laboratory Test Reference, 12th ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Treviño, L.S.; Gorelick, D.A. The Interface of Nuclear and Membrane Steroid Signaling. Endocrinology 2021, 162, 1–14. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen Receptor Interaction with Estrogen Response Elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The Nuclear Receptor Superfamily: A Structural Perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef]
- Gosden, J.R.; Middleton, P.G.; Rout, D. Localization of the Human Oestrogen Receptor Gene to Chromosome 6q24→q27 by in Situ Hybridization. Cytogenet. Genome Res. 1986, 43, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Flouriot, G.; Brand, H.; Denger, S.; Metivier, R.; Kos, M.; Reid, G.; Sonntag-Buck, V.; Gannon, F. Identification of a New Isoform of the Human Estrogen Receptor-alpha (hER-α) That Is Encoded by Distinct Transcripts and That Is Able to Repress hER-α Activation Function 1. EMBO J. 2000, 19, 4688–4700. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, Cloning, and Expression of Human Estrogen Receptor-α36, a Novel Variant of Human Estrogen Receptor-α66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027. [Google Scholar] [CrossRef]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.-Å. Human Estrogen Receptor β-Gene Structure, Chromosomal Localization, and Expression Pattern1. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S. Molecular Cloning and Characterization of Human Estrogen Receptor Betacx: A Potential Inhibitor Ofestrogen Action in Human. Nucleic Acids Res. 1998, 26, 3505–3512. [Google Scholar] [CrossRef]
- Marino, M.; Ascenzi, P. Membrane Association of Estrogen Receptor Alpha and Beta Influences 17beta-Estradiol-Mediated Cancer Cell Proliferation. Steroids 2008, 73, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Sula, A.; Hollingworth, D.; Ng, L.C.T.; Larmore, M.; DeCaen, P.G.; Wallace, B.A. A Tamoxifen Receptor within a Voltage-Gated Sodium Channel. Mol. Cell 2021, 81, 1160–1169. [Google Scholar] [CrossRef]
- Qiu, J.; Bosch, M.A.; Tobias, S.C.; Grandy, D.K.; Scanlan, T.S.; Rønnekleiv, O.K.; Kelly, M.J. Rapid Signaling of Estrogen in Hypothalamic Neurons Involves a Novel G-Protein-Coupled Estrogen Receptor That Activates Protein Kinase C. J. Neurosci. 2003, 23, 9529–9540. [Google Scholar] [CrossRef] [PubMed]
- Toran-Allerand, C.D.; Guan, X.; MacLusky, N.J.; Horvath, T.L.; Diano, S.; Singh, M.; Connolly, E.S.; Nethrapalli, I.S.; Tinnikov, A.A. ER-X: A Novel, Plasma Membrane-Associated, Putative Estrogen Receptor That Is Regulated during Development and after Ischemic Brain Injury. J. Neurosci. 2002, 22, 8391–8401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. A Variant of Estrogen Receptor-α, hER-α36: Transduction of Estrogen- and Antiestrogen-Dependent Membrane-Initiated Mitogenic Signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 9063–9068. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-L.; Yan, L.-Y.; Zhang, X.-T.; Yuan, J.; Li, M.; Qiao, J.; Wang, Z.-Y.; Sun, Q.-Y. ER-A36, a Variant of ER-α, Promotes Tamoxifen Agonist Action in Endometrial Cancer Cells via the MAPK/ERK and PI3K/Akt Pathways. PLoS ONE 2010, 5, e9013. [Google Scholar] [CrossRef]
- Tong, J.-S.; Zhang, Q.-H.; Wang, Z.-B.; Yang, C.-R.; Fu, X.-Q.; Hou, Y.; Wang, Z.-Y.; Sheng, J.; Sun, Q.-Y. ER-A36, a Novel Variant of ER-α, Mediates Estrogen-Stimulated Proliferation of Endometrial Carcinoma Cells via the PKCδ/ERK Pathway. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0015408 (accessed on 22 January 2024).
- Schwartz, N.; Chaudhri, R.A.; Hadadi, A.; Schwartz, Z.; Boyan, B.D. 17Beta-Estradiol Promotes Aggressive Laryngeal Cancer Through Membrane-Associated Estrogen Receptor-Alpha 36. Horm. Cancer 2014, 5, 22–32. [Google Scholar] [CrossRef]
- Chaudhri, R.A.; Olivares-Navarrete, R.; Cuenca, N.; Hadadi, A.; Boyan, B.D.; Schwartz, Z. Membrane Estrogen Signaling Enhances Tumorigenesis and Metastatic Potential of Breast Cancer Cells via Estrogen Receptor-A36 (ERα36). J. Biol. Chem. 2012, 287, 7169–7181. [Google Scholar] [CrossRef]
- Rosano, C.; Ponassi, M.; Santolla, M.F.; Pisano, A.; Felli, L.; Vivacqua, A.; Maggiolini, M.; Lappano, R. Macromolecular Modelling and Docking Simulations for the Discovery of Selective GPER Ligands. AAPS J. 2016, 18, 41–46. [Google Scholar] [CrossRef]
- Arterburn, J.B.; Prossnitz, E.R. G Protein–Coupled Estrogen Receptor GPER: Molecular Pharmacology and Therapeutic Applications. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 295–320. [Google Scholar] [CrossRef]
- Masi, M.; Racchi, M.; Travelli, C.; Corsini, E.; Buoso, E. Molecular Characterization of Membrane Steroid Receptors in Hormone-Sensitive Cancers. Cells 2021, 10, 2999. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Thomas, P. Minireview: G Protein-Coupled Estrogen Receptor-1, GPER-1: Its Mechanism of Action and Role in Female Reproductive Cancer, Renal and Vascular Physiology. Endocrinology 2012, 153, 2953–2962. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef]
- Grande, F.; Occhiuzzi, M.A.; Lappano, R.; Cirillo, F.; Guzzi, R.; Garofalo, A.; Jacquot, Y.; Maggiolini, M.; Rizzuti, B. Computational Approaches for the Discovery of GPER Targeting Compounds. Front. Endocrinol. 2020, 11, 517. [Google Scholar] [CrossRef]
- Litt, M. Localization of a Human Brain Sodium Channel Gene (SCN2A) to Chromosome 2. Genomics 1989, 5, 204–208. [Google Scholar] [CrossRef] [PubMed]
- De Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.; Gillet, L.; Le Guennec, J.-Y.; Besson, P. Voltage-Gated Sodium Channels and Cancer: Is Excitability Their Primary Role? Front. Pharmacol. 2015, 6, 152. [Google Scholar] [CrossRef]
- Smitherman, K.A.; Sontheimer, H. Inhibition of Glial Na+ and K+ Currents by Tamoxifen. J. Membr. Biol. 2001, 181, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Porter, B.A.; Ortiz, M.A.; Bratslavsky, G.; Kotula, L. Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers 2019, 11, 1852. [Google Scholar] [CrossRef]
- Savatier, J.; Jalaguier, S.; Ferguson, M.L.; Cavaillès, V.; Royer, C.A. Estrogen Receptor Interactions and Dynamics Monitored in Live Cells by Fluorescence Cross-Correlation Spectroscopy. Biochemistry 2010, 49, 772–781. [Google Scholar] [CrossRef]
- Hall, J.M.; McDonnell, D.P.; Korach, K.S. Allosteric Regulation of Estrogen Receptor Structure, Function, and Coactivator Recruitment by Different Estrogen Response Elements. Mol. Endocrinol. 2002, 16, 469–486. [Google Scholar] [CrossRef]
- Hall, J.M.; McDonnell, D.P. The Estrogen Receptor β-Isoform (ERβ) of the Human Estrogen Receptor Modulates ERα Transcriptional Activity and Is a Key Regulator of the Cellular Response to Estrogens and Antiestrogens1. Endocrinology 1999, 140, 5566–5578. [Google Scholar] [CrossRef] [PubMed]
- Grober, O.M.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; De Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global Analysis of Estrogen Receptor Beta Binding to Breast Cancer Cell Genome Reveals an Extensive Interplay with Estrogen Receptor Alpha for Target Gene Regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Edvardsson, K.; Lewandowski, S.A.; Ström, A.; Gustafsson, J.-Å. A Genome-Wide Study of the Repressive Effects of Estrogen Receptor Beta on Estrogen Receptor Alpha Signaling in Breast Cancer Cells. Oncogene 2008, 27, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Lu, Y.; Zhang, H.; Zhang, Z.; Xu, W.; Wen, S.; Gao, W.; Wu, Y. Biological Roles and Clinical Significance of Estrogen and Androgen Receptors in Head and Neck Cancers. J. Cancer 2022, 13, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Soltysik, K.; Czekaj, P. Membrane Estrogen Receptors—Is It an Alternative Way of Estrogen Action? J. Physiol. Pharmacol. 2013, 64, 129–142. [Google Scholar] [PubMed]
- Pepermans, R.A.; Sharma, G.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor in Cancer and Stromal Cells: Functions and Novel Therapeutic Perspectives. Cells 2021, 10, 672. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Zhang, J.; Körner, H.; Jiang, Y.; Ying, S. The Emerging Role of Voltage-Gated Sodium Channels in Tumor Biology. Front. Oncol. 2019, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Micheli, A.; Ciampichini, R.; Oberaigner, W.; Ciccolallo, L.; de Vries, E.; Izarzugaza, I.; Zambon, P.; Gatta, G.; De Angelis, R.; EUROCARE Working Group. The Advantage of Women in Cancer Survival: An Analysis of EUROCARE-4 Data. Eur. J. Cancer 2009, 45, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, J.; Tuomainen, K.; Sallinen, T.; Faress, I.; Suleymanova, I.; Al-Samadi, A.; Salo, T.; Åström, P. Effect of Sex Steroid Hormones on Tongue Cancer Cells In Vitro. Anticancer Res. 2020, 40, 6029–6037. [Google Scholar] [CrossRef]
- Robbins, K.T.; Vu, T.P.; Diaz, A.; Varki, N.M. Growth Effects of Tamoxifen and Estradiol on Laryngeal Carcinoma Cell Lines. Arch. Otolaryngol.–Head Neck Surg. 1994, 120, 1261–1266. [Google Scholar] [CrossRef]
- Yoo, H.J.; Sepkovic, D.W.; Bradlow, H.L.; Yu, G.P.; Sirilian, H.V.; Schantz, S.P. Estrogen Metabolism as a Risk Factor for Head and Neck Cancer. Otolaryngol.–Head Neck Surg. 2001, 124, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Freedman, N.D.; Lacey, J.V.; Hollenbeck, A.R.; Leitzmann, M.F.; Schatzkin, A.; Abnet, C.C. The Association of Menstrual and Reproductive Factors with Upper Gastrointestinal Tract Cancers in the NIH-AARP Cohort. Cancer 2010, 116, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.-S.; Kim, B.Y. Relationship between Menopausal Hormone Therapy and Oral Cancer: A Cohort Study Based on the Health Insurance Database in South Korea. J. Clin. Med. 2022, 11, 5848. [Google Scholar] [CrossRef]
- Bristol, M.L.; James, C.D.; Wang, X.; Fontan, C.T.; Morgan, I.M. Estrogen Attenuates the Growth of Human Papillomavirus-Positive Epithelial Cells. mSphere 2020, 5, e00049-20. [Google Scholar] [CrossRef] [PubMed]
- Shatalova, E.G.; Klein-Szanto, A.J.P.; Devarajan, K.; Cukierman, E.; Clapper, M.L. Estrogen and Cytochrome P450 1B1 Contribute to Both Early- and Late-Stage Head and Neck Carcinogenesis. Cancer Prev. Res. 2011, 4, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Doll, C.; Arsenic, R.; Lage, H.; Jöhrens, K.; Hartwig, S.; Nelson, K.; Raguse, J.D. Expression of Estrogen Receptors in OSCC in Relation to Histopathological Grade. Anticancer Res. 2015, 35, 5867–5872. [Google Scholar] [PubMed]
- Ishida, H.; Wada, K.; Masuda, T.; Okura, M.; Kohama, K.; Sano, Y.; Nakajima, A.; Kogo, M.; Kamisaki, Y. Critical Role of Estrogen Receptor on Anoikis and Invasion of Squamous Cell Carcinoma. Cancer Sci. 2007, 98, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Marocchio, L.S.; Giudice, F.; Corrêa, L.; Pinto Junior, D.D.S.; De Sousa, S.O.M. Oestrogens and Androgen Receptors in Oral Squamous Cell Carcinoma. Acta Odontol. Scand. 2013, 71, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Akyu (Takei), R.; Tomihara, K.; Yamazaki, M.; Moniruzzaman, R.; Heshiki, W.; Sekido, K.; Tachinami, H.; Sakurai, K.; Yonesi, A.; Imaue, S.; et al. Protumor Role of Estrogen Receptor Expression in Oral Squamous Cell Carcinoma Cells. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2021, 132, 549–565. [Google Scholar] [CrossRef]
- Lukits, J.; Remenár, E.; Rásó, E.; Ladányi, A.; Kásler, M.; Tímár, J. Molecular Identification, Expression and Prognostic Role of Estrogen- and Progesterone Receptors in Head and Neck Cancer. Int. J. Oncol. 2007, 30, 155–160. [Google Scholar] [CrossRef][Green Version]
- Fei, M.; Zhang, J.; Zhou, J.; Xu, Y.; Wang, J. Sex-Related Hormone Receptor in Laryngeal Squamous Cell Carcinoma: Correlation with Androgen Estrogen-ɑ and Prolactin Receptor Expression and Influence of Prognosis. Acta Oto-Laryngol. 2018, 138, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Liu, S.Y.W.; van Hasselt, C.A.; Vlantis, A.C.; Ng, E.K.W.; Zhang, H.; Dong, Y.; Ng, S.K.; Chu, R.; Chan, A.B.W.; et al. Estrogen Receptor α Induces Prosurvival Autophagy in Papillary Thyroid Cancer via Stimulating Reactive Oxygen Species and Extracellular Signal Regulated Kinases. J. Clin. Endocrinol. Metab. 2015, 100, E561–E571. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Dong, W.; Li, J.; Zhang, H.; Shan, Z.; Teng, W. Differential Expression Patterns and Clinical Significance of Estrogen Receptor-α and β in Papillary Thyroid Carcinoma. BMC Cancer 2014, 14, 383. [Google Scholar] [CrossRef]
- Grsic, K.; Opacic, I.L.; Sitic, S.; Milkovic Perisa, M.; Suton, P.; Sarcevic, B. The Prognostic Significance of Estrogen Receptor β in Head and Neck Squamous Cell Carcinoma. Oncol. Lett. 2016, 12, 3861–3865. [Google Scholar] [CrossRef]
- Koenigs, M.B.; Lefranc-Torres, A.; Bonilla-Velez, J.; Patel, K.B.; Hayes, D.N.; Glomski, K.; Busse, P.M.; Chan, A.W.; Clark, J.R.; Deschler, D.G.; et al. Association of Estrogen Receptor Alpha Expression With Survival in Oropharyngeal Cancer Following Chemoradiation Therapy. JNCI J. Natl. Cancer Inst. 2019, 111, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Ahn, S.-H.; Jeong, W.-J.; Jung, Y.H.; Bae, Y.J.; Paik, J.H.; Chung, J.-H.; Kim, H. Estrogen Receptor α as a Predictive Biomarker for Survival in Human Papillomavirus-Positive Oropharyngeal Squamous Cell Carcinoma. J. Transl. Med. 2020, 18, 240. [Google Scholar] [CrossRef]
- Doll, C.; Bestendonk, C.; Kreutzer, K.; Neumann, K.; Pohrt, A.; Trzpis, I.; Koerdt, S.; Dommerich, S.; Heiland, M.; Raguse, J.-D.; et al. Prognostic Significance of Estrogen Receptor Alpha in Oral Squamous Cell Carcinoma. Cancers 2021, 13, 5763. [Google Scholar] [CrossRef] [PubMed]
- Drake, V.; Bigelow, E.; Fakhry, C.; Windon, M.; Rooper, L.M.; Ha, P.; Miles, B.; Gourin, C.; Mandal, R.; Mydlarz, W.; et al. Biologic and Behavioral Associations of Estrogen Receptor Alpha Positivity in Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2021, 121, 105461. [Google Scholar] [CrossRef]
- Siegfried, J.M.; Stabile, L.P. Estrongenic Steroid Hormones in Lung Cancer. Semin. Oncol. 2014, 41, 5–16. [Google Scholar] [CrossRef]
- Chang, Y.-L.; Hsu, Y.-K.; Wu, T.-F.; Huang, C.-M.; Liou, L.-Y.; Chiu, Y.-W.; Hsiao, Y.-H.; Luo, F.-J.; Yuan, T.-C. Regulation of Estrogen Receptor α Function in Oral Squamous Cell Carcinoma Cells by FAK Signaling. Endocr.-Relat. Cancer 2014, 21, 555–565. [Google Scholar] [CrossRef]
- Brooks, Y.S.; Ostano, P.; Jo, S.-H.; Dai, J.; Getsios, S.; Dziunycz, P.; Hofbauer, G.F.L.; Cerveny, K.; Chiorino, G.; Lefort, K.; et al. Multifactorial ERβ and NOTCH1 Control of Squamous Differentiation and Cancer. J. Clin. Investig. 2014, 124, 2260–2276. [Google Scholar] [CrossRef] [PubMed]
- Anastasios, K.G.; Fuxe, J.; Varakis, J.; Repanti, M.; Goumas, P.; Papadaki, H. Estrogen Receptor-β Expression in Human Laryngeal Carcinoma: Correlation with the Expression of Epithelial-Mesenchymal Transition Specific Biomarkers. Oncol. Rep. 2009, 22, 1063–1068. [Google Scholar] [CrossRef][Green Version]
- Li, S.; Wang, B.; Tang, Q.; Liu, J.; Yang, X. Bisphenol A Triggers Proliferation and Migration of Laryngeal Squamous Cell Carcinoma via GPER Mediated Upregulation of IL-6. Cell Biochem. Funct. 2017, 35, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-C.; Wu, W.; Leng, X.-F.; Zhang, H.-W. Utility of G Protein-Coupled Oestrogen Receptor 1 as a Biomarker for Pan-Cancer Diagnosis, Prognosis and Immune Infiltration: A Comprehensive Bioinformatics Analysis. Aging 2023, 15, 12021–12067. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.-Y.; Weng, J.-R.; Hu, J.-L.; Wang, D.; Sargeant, A.M.; Chiu, C.-F. G15, a GPR30 Antagonist, Induces Apoptosis and Autophagy in Human Oral Squamous Carcinoma Cells. Chem.-Biol. Interact. 2013, 206, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Shen, Y.; Cai, J.; Lei, M.; Wang, Z. Expression of Voltage-Gated Sodium Channel α Subunit in Human Ovarian Cancer. Oncol. Rep. 2010, 23, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Pinatti, L.M.; Walline, H.M.; Carey, T.E. Human Papillomavirus Genome Integration and Head and Neck Cancer. J. Dent. Res. 2018, 97, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Irimie, A.; Braicu, C.; Cojocneanu, R.; Magdo, L.; Onaciu, A.; Ciocan, C.; Mehterov, N.; Dudea, D.; Buduru, S.; Berindan-Neagoe, I. Differential Effect of Smoking on Gene Expression in Head and Neck Cancer Patients. Int. J. Environ. Res. Public Health 2018, 15, 1558. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Schwartz, N.; Cohen, D.J.; Boyan, B.D.; Schwartz, Z. Estrogen Signaling and Estrogen Receptors as Prognostic Indicators in Laryngeal Cancer. Steroids 2019, 152, 108498. [Google Scholar] [CrossRef]
- Verma, A.; Schwartz, N.; Cohen, D.J.; Patel, V.; Nageris, B.; Bachar, G.; Boyan, B.D.; Schwartz, Z. Loss of Estrogen Receptors Is Associated with Increased Tumor Aggression in Laryngeal Squamous Cell Carcinoma. Sci. Rep. 2020, 10, 4227. [Google Scholar] [CrossRef]
- Lee, C.I.; Goodwin, A.; Wilcken, N. Fulvestrant for Hormone-sensitive Metastatic Breast Cancer. Cochrane Database Syst. Rev. 2017, 2017, CD011093. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, A.E. Similarities and Distinctions in the Mode of Action of Different Classes of Antioestrogens. Endocr.-Relat. Cancer 2000, 7, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Lee, J.-H.; Kim, Y.-K.; Myoung, H.; Yun, P.-Y. The Role of Tamoxifen in Combination with Cisplatin on Oral Squamous Cell Carcinoma Cell Lines. Cancer Lett. 2007, 245, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.K.; Bojar, H.; Eckel, J.; Van Lierop, A.; Balz, V.; Friebe-Hoffmann, U.; Hauser, U.; Bier, H. Effects of Tamoxifen on Human Squamous Cell Carcinoma Lines of the Head and Neck. Anti-Cancer Drugs 2002, 13, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Grenman, R.; Virolainen, E.; Shapira, A.; Carey, T. In Vitro Effects of Tamoxifen on UM-SCC Head and Neck Cancer Cell Lines: Correlation with the Estrogen and Progesterone Receptor Content. Int. J. Cancer 1987, 39, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Ku, T.K.S.; Crowe, D.L. Coactivator-Mediated Estrogen Response in Human Squamous Cell Carcinoma Lines. J. Endocrinol. 2007, 193, 147–155. [Google Scholar] [CrossRef][Green Version]
- Tavassoli, M. Tamoxifen Inhibits the Growth of Head and Neck Cancer Cells and Sensitizes These Cells to Cisplatin Induced-Apoptosis: Role of TGF-Beta1. Carcinogenesis 2002, 23, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Nakata, B.; Albright, K.D.; Barton, R.M.; Howell, S.B.; Los, G. Synergistic Interaction between Cisplatin and Tamoxifen Delays the Emergence of Cisplatin Resistance in Head and Neck Cancer Cell Lines. Cancer Chemother. Pharmacol. 1995, 35, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.; Helmstaedter, V.; Lage, H. The Influence of Tamoxifen on Growth Behavior and Cell–Cell Adhesion in OSCC in Vitro. Oral Oncol. 2007, 43, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ding, L.; Kang, L.; Wang, Z.-Y. Estrogen Receptor-Alpha 36 Mediates Mitogenic Antiestrogen Signaling in ER-Negative Breast Cancer Cells. PLoS ONE 2012, 7, e30174. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Yin, L. Estrogen Receptor Alpha-36 (ER-A36): A New Player in Human Breast Cancer. Available online: https://pubmed.ncbi.nlm.nih.gov/25917453/ (accessed on 22 January 2024).
- Su, X.; Xu, X.; Li, G.; Lin, B.; Cao, J.; Teng, L. ER-A36: A Novel Biomarker and Potential Therapeutic Target in Breast Cancer. Onco Targets Ther. 2014, 7, 1525–1533. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: An Oestrogen Receptor Antagonist with a Novel Mechanism of Action. Br. J. Cancer 2004, 90, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Fawell, S.E.; White, R.; Hoare, S.; Sydenham, M.; Page, M.; Parker, M.G. Inhibition of Estrogen Receptor-DNA Binding by the “Pure” Antiestrogen ICI 164,384 Appears to Be Mediated by Impaired Receptor Dimerization. Proc. Natl. Acad. Sci. USA 1990, 87, 6883–6887. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Wang, Z.-Y. Breast Cancer Cell Growth Inhibition by Phenethyl Isothiocyanate Is Associated with Down-Regulation of Oestrogen Receptor-Alpha36. J. Cell Mol. Med. 2010, 14, 1485–1493. [Google Scholar] [CrossRef]
- Grünow, J.; Rong, C.; Hischmann, J.; Zaoui, K.; Flechtenmacher, C.; Weber, K.-J.; Plinkert, P.; Hess, J. Regulation of Submaxillary Gland Androgen-Regulated Protein 3A via Estrogen Receptor 2 in Radioresistant Head and Neck Squamous Cell Carcinoma Cells. J. Exp. Clin. Cancer Res. 2017, 36, 25. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.; Helmstaedter, V.; Moreau, C.; Lage, H. Estradiol, Tamoxifen and ICI 182,780 Alter A3 and Β1 Integrin Expression and Laminin-1 Adhesion in Oral Squamous Cell Carcinoma Cell Cultures. Oral Oncol. 2008, 44, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.M. Centchroman, a Selective Estrogen Receptor Modulator, as a Contraceptive and for the Management of Hormone-related Clinical Disorders. Med. Res. Rev. 2001, 21, 302–347. [Google Scholar] [CrossRef]
- Lal, J. Clinical Pharmacokinetics and Interaction of Centchroman—A Mini Review. Contraception 2010, 81, 275–280. [Google Scholar] [CrossRef]
- Srivastava, V.K.; Gara, R.K.; Bhatt, M.L.B.; Sahu, D.P.; Mishra, D.P. Centchroman Inhibits Proliferation of Head and Neck Cancer Cells through the Modulation of PI3K/mTOR Pathway. Biochem. Biophys. Res. Commun. 2011, 404, 40–45. [Google Scholar] [CrossRef]
- Lecomte, S.; Demay, F.; Ferrière, F.; Pakdel, F. Phytochemicals Targeting Estrogen Receptors: Beneficial Rather Than Adverse Effects? Int. J. Mol. Sci. 2017, 18, 1381. [Google Scholar] [CrossRef]
- Kapinova, A.; Kubatka, P.; Golubnitschaja, O.; Kello, M.; Zubor, P.; Solar, P.; Pec, M. Dietary Phytochemicals in Breast Cancer Research: Anticancer Effects and Potential Utility for Effective Chemoprevention. Environ. Health Prev. Med. 2018, 23, 36. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, M. Bioavailability Challenges Associated with Development of Anti-Cancer Phenolics. Mini Rev. Med. Chem. 2010, 10, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, J.; Zou, H.; Zhang, J.; Ren, L. Estrogen Receptor-Mediated Health Benefits Ofphytochemicals: A Review. Food Funct. 2023, 14, 10681. [Google Scholar] [CrossRef]
- Coward, L.; Barnes, N.C.; Setchell, K.D.R.; Barnes, S. Genistein, Daidzein, and Their. Beta.-Glycoside Conjugates: Antitumor Isoflavones in Soybean Foods from American and Asian Diets. J. Agric. Food Chem. 1993, 41, 1961–1967. [Google Scholar] [CrossRef]
- Alhasan, S.A.; Pietrasczkiwicz, H.; Alonso, M.D.; Ensley, J.; Sarkar, F.H. Genistein-Induced Cell Cycle Arrest and Apoptosis in a Head and Neck Squamous Cell Carcinoma Cell Line. Nutr. Cancer 1999, 34, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Alhasan, S.A.; Ensley, J.F.; Sarkar, F.H. Genistein Induced Molecular Changes in a Squamous Cell Carcinoma of the Head and Neck Cell Line. Int. J. Oncol. 2000, 16, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Dev, A.; Sardoiwala, M.N.; Kushwaha, A.C.; Karmakar, S.; Choudhury, S.R. Genistein Nanoformulation Promotes Selective Apoptosis in Oral Squamous Cell Carcinoma through Repression of 3PK-EZH2 Signalling Pathway. Phytomedicine 2021, 80, 153386. [Google Scholar] [CrossRef]
- Alhasan, S.A.; Aranha, O.; Sarkar, F.H. Genistein Elicits Pleiotropic Molecular Effects on Head and Neck Cancer Cells. Clin. Cancer Res. 2001, 7, 4174–4181. [Google Scholar] [PubMed]
- Zhang, Q.; Cao, W.-S.; Wang, X.-Q.; Zhang, M.; Lu, X.-M.; Chen, J.-Q.; Chen, Y.; Ge, M.-M.; Zhong, C.-Y.; Han, H.-Y. Genistein Inhibits Nasopharyngeal Cancer Stem Cells through Sonic Hedgehog Signaling. Phytother. Res. 2019, 33, 2783–2791. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-L.; Liao, Y.-W.; Hsieh, C.-W.; Chen, P.-N.; Yu, C.-C. Soy Isoflavone Genistein Impedes Cancer Stemness and Mesenchymal Transition in Head and Neck Cancer through Activating miR-34a/RTCB Axis. Nutrients 2020, 12, 1924. [Google Scholar] [CrossRef]
- Han, H.; Zhong, C.; Zhang, X.; Liu, R.; Pan, M.; Tan, L.; Li, Y.; Wu, J.; Zhu, Y.; Huang, W. Genistein Induces Growth Inhibition and G2/M Arrest in Nasopharyngeal Carcinoma Cells. Nutr. Cancer 2010, 62, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Wu, J.; Dunn, T.; Yi, J.; Tong, X.; Zhang, D. Inhibition of Cyclooxygenase-2 Activity in Head and Neck Cancer Cells by Genistein. Cancer Lett. 2004, 211, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.M.; Attaai, A.H.; Zahran, A.M. Genistein Anticancer Efficacy during Induced Oral Squamous Cell Carcinoma: An Experimental Study. J. Egypt. Natl. Cancer Inst. 2022, 34, 37. [Google Scholar] [CrossRef] [PubMed]
- Myoung, H.; Hong, S.-P.; Yun, P.-Y.; Lee, J.-H.; Kim, M.-J. Anti-Cancer Effect of Genistein in Oral Squamous Cell Carcinoma with Respect to Angiogenesis and in Vitro Invasion. Cancer Sci. 2003, 94, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-H.; Zhang, Y.-X.; Tang, L.-H.; Yang, X.-J.; Cui, W.-M.; Han, C.-C.; Ji, W.-Y. MicroRNA-1469, a P53-Responsive microRNA Promotes Genistein Induced Apoptosis by Targeting Mcl1 in Human Laryngeal Cancer Cells. Biomed. Pharmacother. 2018, 106, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Pi, C.; Wang, G. Inhibition of PI3K/Akt/mTOR Pathway by Apigenin Induces Apoptosis and Autophagy in Hepatocellular Carcinoma Cells. Biomed. Pharmacother. 2018, 103, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Melstrom, L.G.; Salabat, M.R.; Ding, X.-Z.; Milam, B.M.; Strouch, M.; Pelling, J.C.; Bentrem, D.J. Apigenin Inhibits the GLUT-1 Glucose Transporter and the Phosphoinositide 3-Kinase/Akt Pathway in Human Pancreatic Cancer Cells. Pancreas 2008, 37, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-Y.; Wu, T.-T.; Zhou, S.-H.; Bao, Y.-Y.; Wang, Q.-Y.; Fan, J.; Huang, Y.-P. Apigenin Suppresses GLUT-1 and p-AKT Expression to Enhance the Chemosensitivity to Cisplatin of Laryngeal Carcinoma Hep-2 Cells: An in Vitro Study. Int. J. Clin. Exp. Pathol. 2014, 7, 3938–3947. [Google Scholar] [PubMed]
- Bao, Y.-Y.; Zhou, S.-H.; Lu, Z.-J.; Fan, J.; Huang, Y.-P. Inhibiting GLUT-1 Expression and PI3K/Akt Signaling Using Apigenin Improves the Radiosensitivity of Laryngeal Carcinoma In Vivo. Oncol. Rep. 2015, 34, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.-P.; Chou, T.-H.; Ding, H.-Y.; Chen, P.-R.; Chiang, F.-Y.; Kuo, P.-L.; Liang, C.-H. Apigenin Induces Apoptosis via Tumor Necrosis Factor Receptor- and Bcl-2-Mediated Pathway and Enhances Susceptibility of Head and Neck Squamous Cell Carcinoma to 5-Fluorouracil and Cisplatin. Biochim. Biophys. Acta 2012, 1820, 1081–1091. [Google Scholar] [CrossRef]
- Masuelli, L.; Marzocchella, L.; Quaranta, A.; Palumbo, C.; Pompa, G.; Izzi, V.; Canini, A.; Modesti, A.; Galvano, F.; Bei, R. Apigenin Induces Apoptosis and Impairs Head and Neck Carcinomas EGFR/ErbB2 Signaling. Front. Biosci. 2011, 16, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-S.; Kim, T.-H.; Park, J.-H.; Lim, H.; Cho, I.-A.; You, J.-S.; Lee, G.-J.; Seo, Y.-S.; Kim, D.K.; Kim, C.S.; et al. Formononetin Induces Apoptotic Cell Death through the Suppression of Mitogen-activated Protein Kinase and Nuclear factor-κB Phosphorylation in FaDu Human Head and Neck Squamous Cell Carcinoma Cells. Oncol. Rep. 2020, 43, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Xie, M.; Liang, J.; Li, H.; Li, Z.; Shi, S.; Yang, X.; Wang, Z.; Tang, A. Formononetin Targets the MAPK and PI3K/Akt Pathways to Induce Apoptosis in Human Nasopharyngeal Carcinoma Cells In Vitro and In Vivo. Int. J. Clin. Exp. Med. 2016, 9, 1180–1189. [Google Scholar]
- Ying, K.; Liu, Y.; Zhang, C.; Shangguan, M. Medical Findings of Nasopharyngeal Carcinoma Patients and Anti-Tumor Benefits of Formononetin. Eur. J. Pharmacol. 2019, 861, 172619. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.H.; Wang, Y.; Xin, M. Low Concentration of Formononetin Stimulates the Proliferation of Nasopharyngeal Carcinoma Cell Line CNE2 by Upregulating Bcl-2 and p-ERK1/2 Expression. Pharm. Biol. 2016, 54, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Gu, M.; Takahata, T.; Frederick, B.; Agarwal, C.; Siriwardana, S.; Agarwal, R.; Sclafani, R.A. Resveratrol Selectively Induces DNA Damage, Independent of Smad4 Expression, in Its Efficacy against Human Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2011, 17, 5402–5411. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-S.; Tsai, C.-W.; Yang, J.-S.; Hsu, Y.-M.; Shih, L.-C.; Chiu, H.-Y.; Bau, D.-T.; Tsai, F.-J. Resveratrol Inhibited the Metastatic Behaviors of Cisplatin-Resistant Human Oral Cancer Cells via Phosphorylation of ERK/p-38 and Suppression of MMP-2/9. J. Food Biochem. 2021, 45, e13666. [Google Scholar] [CrossRef] [PubMed]
- Heiduschka, G.; Bigenzahn, J.; Brunner, M.; Thurnher, D. Resveratrol Synergistically Enhances the Effect of Etoposide in HNSCC Cell Lines. Acta Otolaryngol. 2014, 134, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Shrotriya, S.; Agarwal, R.; Sclafani, R.A. A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma. Adv. Exp. Med. Biol. 2015, 815, 333–348. [Google Scholar] [CrossRef]
- Cho, I.-A.; You, S.-J.; Kang, K.-R.; Kim, S.-G.; Oh, J.-S.; You, J.-S.; Lee, G.-J.; Seo, Y.-S.; Kim, D.K.; Kim, C.S.; et al. Biochanin-A Induces Apoptosis and Suppresses Migration in FaDu Human Pharynx Squamous Carcinoma Cells. Oncol. Rep. 2017, 38, 2985–2992. [Google Scholar] [CrossRef]
- Dorsey, M.; Benghuzzi, H.; Tucci, M.; Cason, Z. Growth and Cell Viability of Estradiol and IP-6 Treated Hep-2 Laryngeal Carcinoma Cells. Biomed. Sci. Instrum. 2005, 41, 205–210. [Google Scholar] [PubMed]
- Helton, W.B.; Choi, E.-Y.; Gairola, C.G.; Valentino, J.; Hollie, S. R409—Apigenin and Kaempferol Effects on Oral Cancer FADU Cells. Otolaryngol.–Head Neck Surg. 2008, 139, P180. [Google Scholar] [CrossRef]
- Liu, F.; Pan, Q.; Wang, L.; Yi, S.; Liu, P.; Huang, W. Anticancer Targets and Mechanisms of Calycosin to Treat Nasopharyngeal Carcinoma. Biofactors 2020, 46, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Swanson, H.I.; Choi, E.-Y.; Helton, W.B.; Gairola, C.G.; Valentino, J. Impact of Apigenin and Kaempferol on Human Head and Neck Squamous Cell Carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 214–220. [Google Scholar] [CrossRef]
- Levin, E.R. Integration of the Extranuclear and Nuclear Actions of Estrogen. Mol. Endocrinol. 2005, 19, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Mughees, M.; Sengupta, A.; Khowal, S.; Wajid, S. Mechanism of Tumour Microenvironment in the Progression and Development of Oral Cancer. Mol. Biol. Rep. 2021, 48, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi Rad, H.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the Tumor Microenvironment for Effective Immunotherapy. Med. Res. Rev. 2021, 41, 1474–1498. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, K.; Naruse, T.; Yanamoto, S. Tumor Microenvironmental Modification by the Current Target Therapy for Head and Neck Squamous Cell Carcinoma. J. Exp. Clin. Cancer Res. 2023, 42, 114. [Google Scholar] [CrossRef] [PubMed]
- Goel, B.; Tiwari, A.K.; Pandey, R.K.; Singh, A.P.; Kumar, S.; Sinha, A.; Jain, S.K.; Khattri, A. Therapeutic Approaches for the Treatment of Head and Neck Squamous Cell Carcinoma–An Update on Clinical Trials. Transl. Oncol. 2022, 21, 101426. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, L.; Denaro, N.; Vivenza, D.; Varamo, C.; Strola, G.; Fortunato, M.; Chamorey, E.; Comino, A.; Monteverde, M.; Lo Nigro, C.; et al. Elevated Basal Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC) and High Epidermal Growth Factor Receptor (EGFR) Expression Predict Favourable Outcome in Patients with Locally Advanced Head and Neck Cancer Treated with Cetuximab and Radiotherapy. Cancer Immunol. Immunother. 2017, 66, 573–579. [Google Scholar] [CrossRef]
- Fessas, P.; Lee, H.; Ikemizu, S.; Janowitz, T. A Molecular and Preclinical Comparison of the PD-1–Targeted T-Cell Checkpoint Inhibitors Nivolumab and Pembrolizumab. Semin. Oncol. 2017, 44, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab Alone or with Chemotherapy versus Cetuximab with Chemotherapy for Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-048): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Burcher, K.M.; Lantz, J.W.; Gavrila, E.; Abreu, A.; Burcher, J.T.; Faucheux, A.T.; Xie, A.; Jackson, C.; Song, A.H.; Hughes, R.T.; et al. Relationship between Tumor Mutational Burden, PD-L1, Patient Characteristics, and Response to Immune Checkpoint Inhibitors in Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 5733. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex Differences in Immune Responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, A.; Rothenberger, N.J.; Stabile, L.P. The Impact of Estrogen in the Tumor Microenvironment. In Tumor Microenvironment; Birbrair, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1277, pp. 33–52. ISBN 978-3-030-50223-2. [Google Scholar]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.-C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin. Cancer Res. 2017, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-R.; Slavin, S.; Da, J.; Hsu, I.; Luo, J.; Xiao, G.-Q.; Ding, J.; Chou, F.-J.; Yeh, S. Estrogen Receptor α in Cancer Associated Fibroblasts Suppresses Prostate Cancer Invasion via Reducing CCL5, IL6 and Macrophage Infiltration in the Tumor Microenvironment. Mol. Cancer 2016, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell–Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Kozasa, K.; Mabuchi, S.; Matsumoto, Y.; Kuroda, H.; Yokoi, E.; Komura, N.; Kawano, M.; Takahashi, R.; Sasano, T.; Shimura, K.; et al. Estrogen Stimulates Female Cancer Progression by Inducing Myeloid-Derived Suppressive Cells: Investigations on Pregnant and Non-Pregnant Experimental Models. Oncotarget 2019, 10, 1887–1902. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Lee, S.; Margolin, K. Cytokines in Cancer Immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef]
- Ciucci, A.; Zannoni, G.F.; Buttarelli, M.; Lisi, L.; Travaglia, D.; Martinelli, E.; Scambia, G.; Gallo, D. Multiple Direct and Indirect Mechanisms Drive Estrogen-Induced Tumor Growth in High Grade Serous Ovarian Cancers. Oncotarget 2016, 7, 8155–8171. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ellison, S.J.; Alarid, E.T.; Shapiro, D.J. Interplay between the Levels of Estrogen and Estrogen Receptor Controls the Level of the Granzyme Inhibitor, Proteinase Inhibitor 9 and Susceptibility to Immune Surveillance by Natural Killer Cells. Oncogene 2007, 26, 4106–4114. [Google Scholar] [CrossRef] [PubMed]
- Tai, P.; Wang, J.; Jin, H.; Song, X.; Yan, J.; Kang, Y.; Zhao, L.; An, X.; Du, X.; Chen, X.; et al. Induction of Regulatory T Cells by Physiological Level Estrogen. J. Cell. Physiol. 2008, 214, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Márquez-Garbán, D.C.; Deng, G.; Comin-Anduix, B.; Garcia, A.J.; Xing, Y.; Chen, H.-W.; Cheung-Lau, G.; Hamilton, N.; Jung, M.E.; Pietras, R.J. Antiestrogens in Combination with Immune Checkpoint Inhibitors in Breast Cancer Immunotherapy. J. Steroid Biochem. Mol. Biol. 2019, 193, 105415. [Google Scholar] [CrossRef]
- Hamilton, D.H.; Griner, L.M.; Keller, J.M.; Hu, X.; Southall, N.; Marugan, J.; David, J.M.; Ferrer, M.; Palena, C. Targeting Estrogen Receptor Signaling with Fulvestrant Enhances Immune and Chemotherapy-Mediated Cytotoxicity of Human Lung Cancer. Clin. Cancer Res. 2016, 22, 6204–6216. [Google Scholar] [CrossRef] [PubMed]
- Capone, I.; Marchetti, P.; Ascierto, P.A.; Malorni, W.; Gabriele, L. Sexual Dimorphism of Immune Responses: A New Perspective in Cancer Immunotherapy. Front. Immunol. 2018, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Wallis, C.J.D.; Butaney, M.; Satkunasivam, R.; Freedland, S.J.; Patel, S.P.; Hamid, O.; Pal, S.K.; Klaassen, Z. Association of Patient Sex With Efficacy of Immune Checkpoint Inhibitors and Overall Survival in Advanced Cancers: A Systematic Review and Meta-Analysis. JAMA Oncol. 2019, 5, 529. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.; Martins, D.; Mendes, F. Immunotherapy in Head and Neck Cancer When, How, and Why? Biomedicines 2022, 10, 2151. [Google Scholar] [CrossRef]
- Gaeta, A.; Tagliabue, M.; D’Ecclesiis, O.; Ghiani, L.; Maugeri, P.; De Berardinis, R.; Veneri, C.; Gaiaschi, C.; Cacace, M.; D’Andrea, L.; et al. Are Sex and Gender Considered in Head and Neck Cancer Clinical Studies? NPJ Precis. Onc. 2023, 7, 84. [Google Scholar] [CrossRef]
- Ulrich, B.C.; Guibert, N. Immunotherapy Efficacy and Gender: Discovery in Precision Medicine. Transl. Lung Cancer Res. 2018, 7, S211–S213. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, J.Y.; Lim, S.H.; Park, K.; Sun, J.-M.; Ko, Y.H.; Baek, C.-H.; Son, Y.; Jeong, H.S.; Ahn, Y.C.; et al. Association between PD-L1 and HPV Status and the Prognostic Value of PD-L1 in Oropharyngeal Squamous Cell Carcinoma. Cancer Res. Treat. 2016, 48, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-M.; Sung, W.-W.; Hsieh, M.-J.; Tsai, S.-C.; Lai, H.-W.; Yang, S.-M.; Shen, K.-H.; Chen, M.-K.; Lee, H.; Yeh, K.-T.; et al. High PD-L1 Expression Correlates with Metastasis and Poor Prognosis in Oral Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0142656. [Google Scholar] [CrossRef]
- Lenouvel, D.; González-Moles, M.Á.; Ruiz-Ávila, I.; Gonzalez-Ruiz, L.; Gonzalez-Ruiz, I.; Ramos-García, P. Prognostic and Clinicopathological Significance of PD-L1 Overexpression in Oral Squamous Cell Carcinoma: A Systematic Review and Comprehensive Meta-Analysis. Oral Oncol. 2020, 106, 104722. [Google Scholar] [CrossRef] [PubMed]
- Reynoso-García, J.; Miranda-Santiago, A.E.; Meléndez-Vázquez, N.M.; Acosta-Pagán, K.; Sánchez-Rosado, M.; Díaz-Rivera, J.; Rosado-Quiñones, A.M.; Acevedo-Márquez, L.; Cruz-Roldán, L.; Tosado-Rodríguez, E.L.; et al. A Complete Guide to Human Microbiomes: Body Niches, Transmission, Development, Dysbiosis, and Restoration. Front. Syst. Biol. 2022, 2, 951403. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, Y.; Zhang, L. Role of the Microbiome in Oral Cancer Occurrence, Progression and Therapy. Microb. Pathog. 2022, 169, 105638. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Yeh, Y.-M.; Yu, H.-Y.; Chin, C.-Y.; Hsu, C.-W.; Liu, H.; Huang, P.-J.; Hu, S.-N.; Liao, C.-T.; Chang, K.-P.; et al. Oral Microbiota Community Dynamics Associated With Oral Squamous Cell Carcinoma Staging. Front. Microbiol. 2018, 9, 862. [Google Scholar] [CrossRef] [PubMed]
- Beltran, J.F.; Viafara-Garcia, S.; Labrador, A.P.; Basterrechea, J. The Role of Periodontopathogens and Oral Microbiome in the Progression of Oral Cancer. A Review. Open Dent. J. 2021, 15, 367–376. [Google Scholar] [CrossRef]
- Wang, J.; Gao, B. Mechanisms and Potential Clinical Implications of Oral Microbiome in Oral Squamous Cell Carcinoma. Curr. Oncol. 2023, 31, 168–182. [Google Scholar] [CrossRef]
- Chapadgaonkar, S.S.; Bajpai, S.S.; Godbole, M.S. Gut Microbiome Influences Incidence and Outcomes of Breast Cancer by Regulating Levels and Activity of Steroid Hormones in Women. Cancer Rep. 2023, 6, e1847. [Google Scholar] [CrossRef] [PubMed]
- Farhat, E.K.; Sher, E.K.; Džidić-Krivić, A.; Banjari, I.; Sher, F. Functional Biotransformation of Phytoestrogens by Gut Microbiota with Impact on Cancer Treatment. J. Nutr. Biochem. 2023, 118, 109368. [Google Scholar] [CrossRef] [PubMed]
- Cornejo Ulloa, P.; Krom, B.P.; Van Der Veen, M.H. Sex Steroid Hormones as a Balancing Factor in Oral Host Microbiome Interactions. Front. Cell. Infect. Microbiol. 2021, 11, 714229. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut Microbiome Influences Efficacy of PD-1–Based Immunotherapy against Epithelial Tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- d’Afflitto, M.; Upadhyaya, A.; Green, A.; Peiris, M. Association Between Sex Hormone Levels and Gut Microbiota Composition and Diversity—A Systematic Review. J. Clin. Gastroenterol. 2022, 56, 384–392. [Google Scholar] [CrossRef]
- Wang, L.; Tang, L.; Zhai, D.; Song, M.; Li, W.; Xu, S.; Jiang, S.; Meng, H.; Liang, J.; Wang, Y.; et al. The Role of the Sex Hormone-Gut Microbiome Axis in Tumor Immunotherapy. Gut Microbes 2023, 15, 2185035. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serum (pg/mL) | Urine (µg/24 h) | ||
|---|---|---|---|
| Child (<10 years old) | <15 | 0−6 | |
| Adult male | 10−50 | 0−6 | |
| Adult female | Follicular phase | 20−350 | 0−13 |
| Midcycle peak | 150−750 | 4−14 | |
| Luteal phase | 30−450 | 4−10 | |
| Post-menopause | ≤20 | 0−4 | |
| Type of Estrogen Receptor | Receptor | Receptor Function | Significance in HNTs | Reference |
|---|---|---|---|---|
| Nuclear | ERα, ERβ | Transcription factor | Increased ERβ expression in HNSCC compared to normal tissue, no difference in expression between males and females, neither in tumors nor in normal tissue | [80] |
| Contradictory findings: Predominant ERβ expression in most OSCC studies; Predominant ERα expression in oral cavity and laryngeal/hypopharyngeal cancers | [81,82,83,84,85] | |||
| Frequent co-expression (~40% of cases) of ER and progesterone receptors independent of primary tumor site | [85] | |||
| Higher level of both ESR1 and ERα in laryngeal cancer | [86] | |||
| Mutually opposite effect in same type of HNT: ERα induces, ERβ inhibits growth of papillary thyroid cancer | [87,88] | |||
| ERβ expression in OPSCC associated with higher survival rates compared to ERβ-negative OPSCC | [89] | |||
| Higher ERα expression associated with improved survival rates in OPSCC patients receiving primary chemoradiation; ERα as a biomarker for better overall survival in patients with HPV+ OPSCC | [90,91] | |||
| Significant influence of ERα expression on decrease in overall and relapse-free survival only in male OSCC cohort, in comparison to ERα-negative patients | [92] | |||
| HPV positivity and smaller HNSCC tumor size (≤T2) were independently associated with ERα positivity | [93] | |||
| Elevated expression of FAK is associated with increased ERα phosphorylation, transcription, and cell growth of OSCC cells | [95] | |||
| Increased ERβ expression or ERβ agonist treatment inhibited SCC cells proliferation and promoted NOTCH1 expression in vitro and in mouse xenotransplants | [96] | |||
| ERβ expressed in majority of laryngeal carcinomas (83%); expression is positively correlated with maintenance of E-cadherin and ß-catenin at cell junctions of tumor cells plasma membranes, and negatively correlated with increased TNM stage, nuclear translocation of β-catenin, and loss of E-cadherin | [97] | |||
| Membrane | ERα36 | Membrane-initiated estrogen signaling | Increases PKC activity in laryngeal cancer cells, increasing proliferation and survival, and enhancing expression of metastatic and angiogenic factors in response to E2; ERα36 expression positively correlated with VEGF in laryngeal tumor samples and with metastasis to regional lymph nodes | [50] |
| Inverse correlation between ERα66 and ERα36 expression and clinical cancer stage: ERα66 was decreased with ascending tumor aggressiveness, while ERα36 expression was increased | [104,105] | |||
| GPER1 | G protein-coupled receptor | Upregulates IL-6, promoting proliferation and migration of LSCC cells in response to estrogen mimetic bisphenol A | [98] | |
| Elevated expression in HNSCC compared to normal tissue; Lower expression was associated with poor prognosis | [99] | |||
| GPER1 antagonist G15 shows antitumor effect in OSCC cell lines (SCC-4, SCC-9, HSC-3): induces dose-dependent cytotoxicity, G2/M cycle arrest, and apoptosis; downregulates expression of AKT, cell cycle-related proteins, and mitogen-activated protein kinases; induces formation of autophagosomes | [100] | |||
| NaV1.2 | Sodium ion channel | HPV viral integration into SCN2A genomic region observed in oral and oropharyngeal cancers: fusion of HPV L2 gene into SCN2A intron 16 results in gene disruption and homozygous loss of SCN2A | [102] | |
| Upregulated SCN2A expression in smoking HNSCC patients compared to never-smokers | [103] |
| Antiestrogen | Effect on HNTs | Dose | Cell Lines | Reference |
|---|---|---|---|---|
| Tamoxifen | Inhibits growth of laryngeal cancer cells in vitro and in vivo | 3–8 µM | UM-SCC-5, UM-SCC-11B | [75] |
| Inhibits growth of ER-negative HNT cells | 5–10 µM | UM-SCC-11B, UM-SCC-14C, UM-SCC-22B | [109] | |
| Insensitivity to TAM treatment observed in three ER-negative HNT cells, while it had an inhibitory effect on three ER-positive HNT cells | 5 µM | UM-SCC-5, UM-SCC-9, UM-SCC-12; UM-SCC-1, UM-SCC-3, UM-SCC-14B | [110] | |
| Proliferation inhibition of SCC cells by inhibiting G1/S phase progression; this inhibition correlated with the upregulation of p27 and downregulation of cyclin E and CDK6 | 100 nM | SCC-4, SCC-9, SCC-25 | [111] | |
| Inhibits invasion of SCC cells and induces anoikis as a direct result of adhesion inhibition and disruption of survival signals, due to the reduction in phosphorylation of FAK, ERK, and MAPK | 3–30 µM | SCCTF, SCCKN, SAS, Ca9-22 | [82] | |
| TAM in combination with cisplatin enhanced cytotoxic and apoptotic effect on OSCC cells, possibly through inhibition of PKC activity and upregulation of the TGFB1 | 5 µM TAM, 5 µg/mL cisplatin | A-253, HSC-3, KB | [108] | |
| Induced G1 cell cycle arrest independently of p53 status and increased level of hypophosphorylated active RB in OSCC; TAM combined with cisplatin induced apoptosis more effectively and resulted in increased secretion of TGFB1 | 1 µM TAM, 5 µg/mL cis-platin | HN-6, HN-5 | [112] | |
| Delayed development of cisplatin resistance in HNT cells | 3.5 µM TAM, 6.5 µM cisplatin | UM-SCC-10B | [113] | |
| Induced growth inhibition and increased the OSCC cells aggregation ability | 5 µM | UM-SCC-14A, UM-SCC-14B, UM-SCC-14C | [114] | |
| Significantly sensitized HNSCC cells to fractionated irradiation (IR) | 1 µM | FaDu | [121] | |
| Reduced β1 integrin transcription and α3 integrin cell surface expression and inhibited the growth of OSCC cells | 1, 5 µM | UM-SCC-14A, UM-SCC-14B, UM-SCC-14C | [122] | |
| Fulvestrant | Significantly sensitized HNSCC cells to fractionated irradiation (IR) | 10 nM | FaDu | [121] |
| Reduced laminin-1 adhesion and inhibited growth of OSCC cells | 1, 5 µM | UM-SCC-14A, UM-SCC-14B, UM-SCC-14C | [122] | |
| Restored estrogen-mediated decrease of apoptosis in pre-malignant oral leukoplakia cells | 1 µM | MSK-Leuk1 | [80] | |
| Centchroman | Antiproliferative effect in HNSCC cells; induces apoptosis and inhibits AKT/mTOR and STAT3 signaling; inhibits colony formation of HNSCC cells and alters proteins associated with DNA damage and cell cycle regulation | 2.5, 5, 10 µM | FaDu, CAL-27, SCC-9, SCC-25 | [125] |
| Phytoestrogen | Effect on HNTs | Dose | Cell Line or Organism | Reference |
|---|---|---|---|---|
| Genistein | Induced time-dependent and irreversible proliferation inhibition, S/G2-M phase cell cycle arrest and apoptosis | 5–50 µM | HN4 | [131] |
| Down-regulation of CDK1 and cyclin B1, up-regulation of CDK inhibitor p21WAF1 and apoptosis regulator BAX | 25 µM | HN4 | [132] | |
| Treatment with genistein nanoparticles selectively induced apoptosis by increasing ROS production, leading to translocation of BAX proteins to mitochondria and caspase 3 activation | 40 µM | JHU-011 | [133] | |
| Down-regulation of MMP-2 and MMP-9 secretion and NF-κB DNA binding activity, inhibition of HNT invasion potential, decreased level of phosphorylated AKT, induced telomere shortening | 5–50 µM | HN4 | [134] | |
| Inhibited tumorsphere formation and induced apoptosis of nasopharyngeal cancer stem cells, through the suppression of SHH signaling | 100 μM | CNE-2, HONE-1 | [135] | |
| Decreased HNT TICs proliferation; downregulation of EMT; potentiated cell death caused by doxorubicin, cisplatin, 5-FU chemotherapeutics; increased ROS production induced by miR-34a, which resulted in reduced migration, invasion, self-renewal, and increased apoptosis rate | 20 μM | HNT-TICs | [136] | |
| Induced dose-dependent G2/M phase arrest, where SERMs (FULV, propyl pyrazole triol, diarylprepionitrile) did not affect genistein-induced growth inhibition | 30–120 µM | CNE-2 | [137] | |
| Growth inhibition via G2/M phase arrest, decreased proliferating cell nuclear antigen expression, no difference in number of apoptotic cells | 50–200 µM | SCC-25 | [138] | |
| Delayed the clinicopathological change of DMBA-induced carcinogenesis of OSCC in vivo | 20 mg/kg | Syrian hamster | [139] | |
| Down-regulation in VEGF mRNA expression, reduced tumor invasion through artificial basement membrane and gelatinolytic activity in vitro; Lower CD31 immunoreactivity in vivo, no difference in tumor growth and metastatic behaviors | 27.3 μg/mL; 0.5 mg/kg | HSC-3; HSC-3 cell xenograft mouse model | [140] | |
| Elevated miR-1469 expression through p53 activation MCL1 inhibition | 100 μM | Tu 212 | [141] | |
| Apigenin | Dose-dependent inhibitory effect on GLUT-1 mRNA and protein expression, resulting in the PI3K/AKT pathway downregulation in cisplatin-treated HNT cells | 40–160 µM | HEp-2 | [144] |
| Xenograft growth inhibition and enhanced xenograft radiosensitivity as the result of suppressing GLUT-1 expression via the PI3K/AKT pathway in vivo | Intraperitoneal injection with 50 or 100 µg | HEp-2 cell xenograft mouse model | [145] | |
| Inhibits growth, induces G2/M phase cell cycle arrest, induces apoptosis through upregulation of TNF-R and TRAIL-R pathways | 10, 20 µM | SCC-25 | [146] | |
| Dose-dependent survival inhibition and apoptosis induction; reduction in ligand-induced phosphorylation of EGFR and ErbB2 | 6–100 µM; 50 µM | CAL-27, SCC-15, FaDu | [147] | |
| Formononetin | Increased cell death by activation of caspase cascade; dose-dependent suppression of the mitogen-activated protein kinases ERK1/2 and p38, and NF-κB phosphorylation in vitro; delayed tumor growth in vivo after oral administration | 5–50 µM; 10 mg/kg | FaDu; FaDu cell xenograft mouse model | [148] |
| Inhibited proliferation and induced apoptosis, decreased AKT phosphorylation, enhanced phosphorylation of JNK/SAPK and p38 MAPK, upregulated pro-apoptotic factors BAXand caspase 3, and downregulated the anti-apoptotic BCL-2; slowed down tumor growth rate in vivo | 5–40 µM; intraperitoneal injection with 10 or 20 mg/kg | CNE-1; CNE-1 cell xenograft mouse model | [149] | |
| Inhibited proliferation and induced apoptosis, decreased wound healing process and migratory capability | 10, 20, 40 μM | CNE-2 | [150] | |
| Inhibitory effect on apoptosis by upregulating BCL-2 and p-ERK1/2 | 0.1–1 µM | CNE-2 | [151] | |
| Resveratrol | Suppressed viability and induced DNA damage, induced S-phase arrest and apoptosis, together with activation of BRCA1 and γ-H2AX foci | 5–50 µM | FaDu, CAL-27 | [152] |
| Suppressed migration and invasion potential of cisplatin-resistant human OSCC cells in a dose-dependent manner; decreased expression of p-ERK, p-p38, MMP-2, and MMP-9, resulting in decreased overall metastatic potential | 25, 50, 75 µM; 50 μM | cisplatin-resistant CAL-27 | [153] | |
| Synergistic effect with etoposide on the induction of apoptosis and necrosis | 20–240 μM, etoposide:resveratrol = 1:4 | CAL-27, SCC-25, FaDu | [154] | |
| Biochanin A | Induced dose- and time-dependent cell death; increased activation of extrinsic (FASL and caspase-8) and intrinsic apoptotic factors (BAD and caspase-9), decreased expression of intrinsic anti-apoptotic factors (BCL-2 and BCL-XL); inhibited wound healing potential through MMP-2 and MMP-9 suppression, via downregulation of p38, MAPK, NF-κB, and AKT signaling pathways | 25, 50 µM | FaDu | [156] |
| Inositol-6 phosphate | Decrease in cell number without initiating apoptosis | 1 mM | HEp-2 | [157] |
| Kaempferol | Dose-dependent decrease in cell viability | 0.1–100 µM | FaDu | [160] |
| Calycosin | Dose-dependent reduction of cell survival rate and increased apoptosis in vitro; upregulated expression of TP53 and CASP8, and reduced MAPK14 expression in vitro and in vivo; dose-dependent reduction in tumor mass in vivo | 20, 40, 80 µM; 20, 30, 60 mg/kg | CNE-1; CNE-1 cell xenograft mouse model | [159] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kranjčević, J.-K.; Čonkaš, J.; Ozretić, P. The Role of Estrogen and Estrogen Receptors in Head and Neck Tumors. Cancers 2024, 16, 1575. https://doi.org/10.3390/cancers16081575
Kranjčević J-K, Čonkaš J, Ozretić P. The Role of Estrogen and Estrogen Receptors in Head and Neck Tumors. Cancers. 2024; 16(8):1575. https://doi.org/10.3390/cancers16081575
Chicago/Turabian StyleKranjčević, Jacqueline-Katrin, Josipa Čonkaš, and Petar Ozretić. 2024. "The Role of Estrogen and Estrogen Receptors in Head and Neck Tumors" Cancers 16, no. 8: 1575. https://doi.org/10.3390/cancers16081575
APA StyleKranjčević, J.-K., Čonkaš, J., & Ozretić, P. (2024). The Role of Estrogen and Estrogen Receptors in Head and Neck Tumors. Cancers, 16(8), 1575. https://doi.org/10.3390/cancers16081575