
Therapeutic Strategies for RB1-Deficient Cancers: Intersecting Gene Regulation and Targeted Therapy



Abstract
Simple Summary
Abstract
1. Introduction
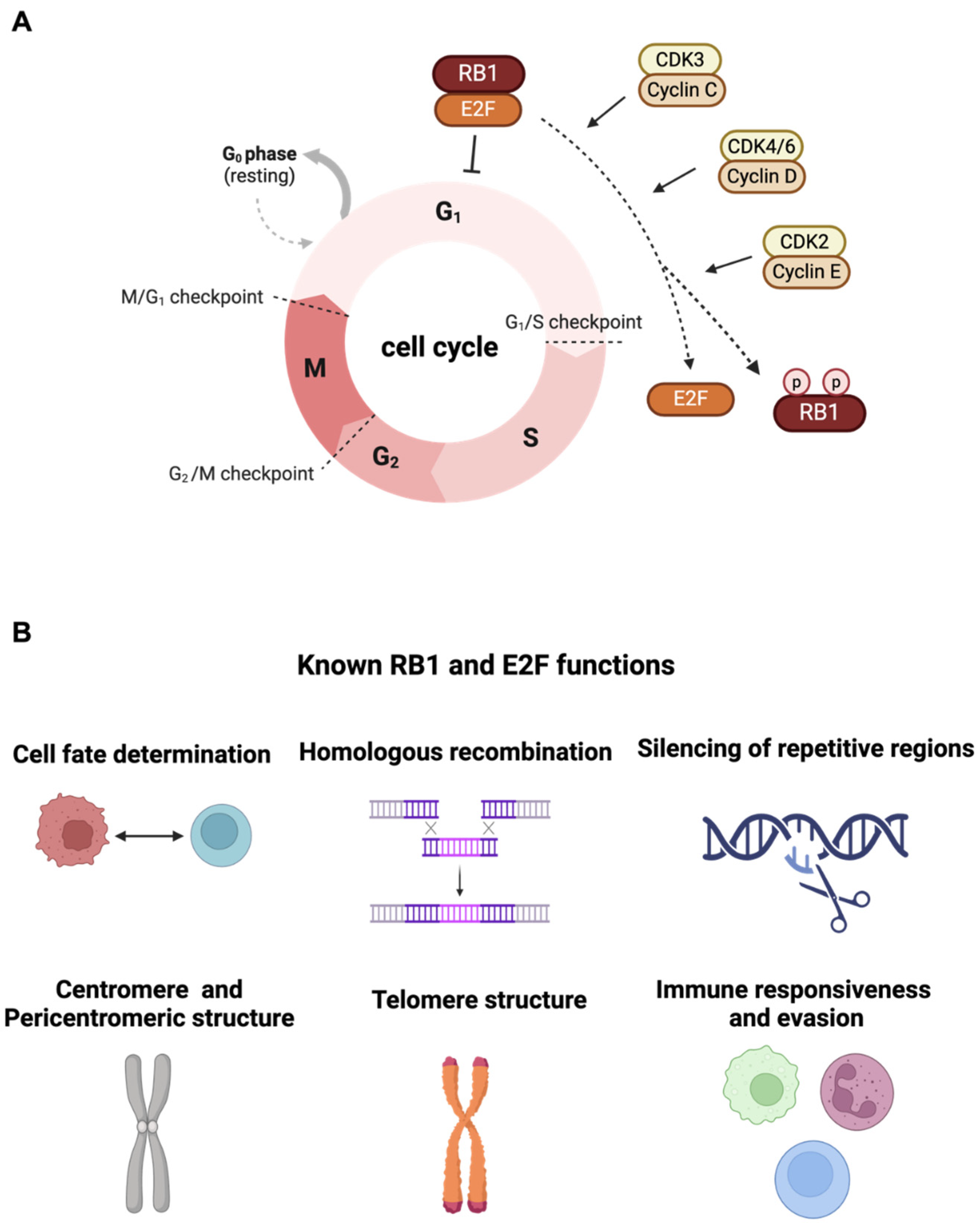
2. Canonical Function of RB1 Tumor Suppressor
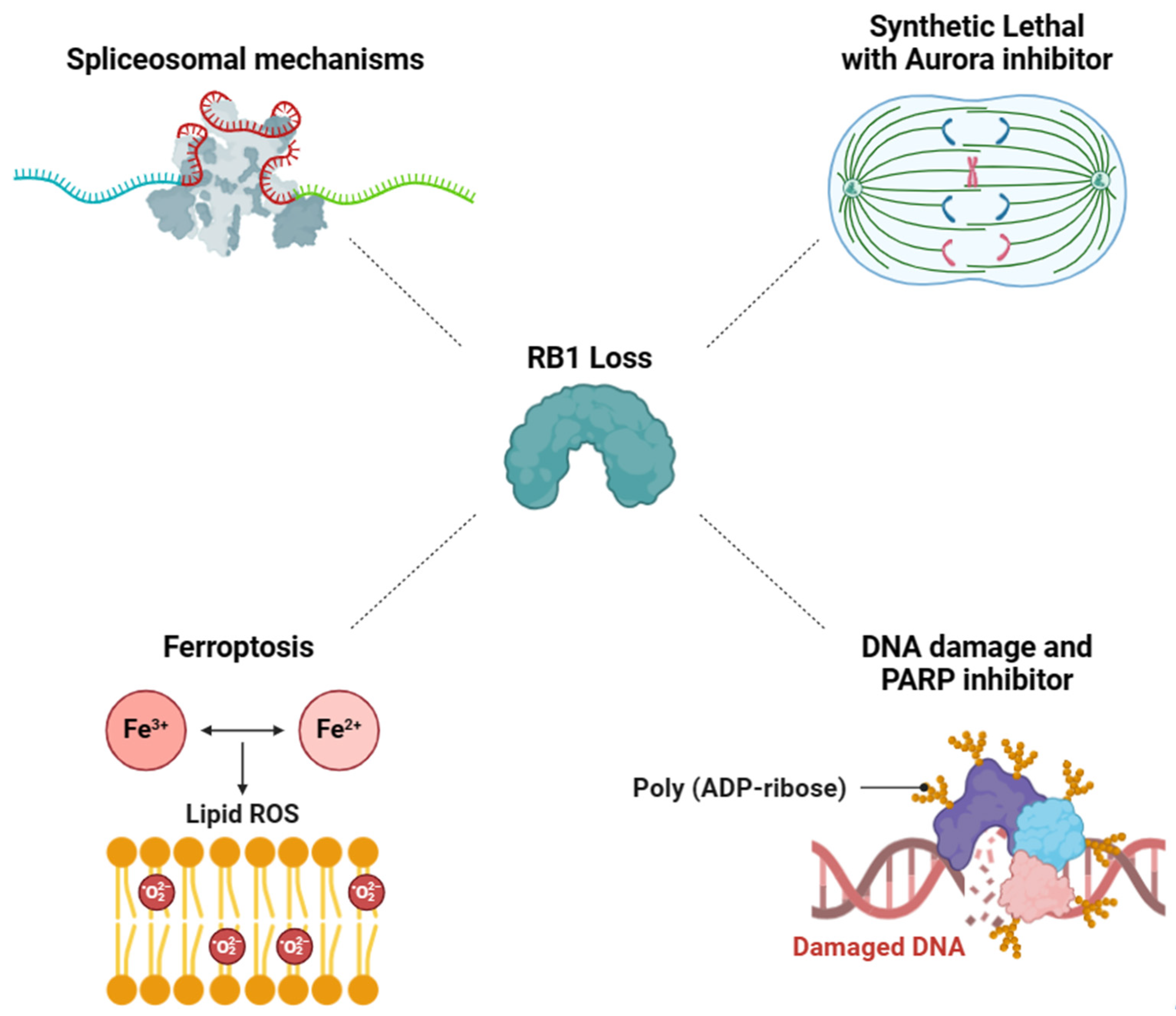
3. Exploiting Vulnerabilities Stemming from RB1 Deficiency-Associated Mechanisms for Targeted Therapy
3.1. RB1-Deficient Cancers Present a Vulnerability in Spliceosomal Mechanisms
3.2. Aurora Kinase Inhibitor-Induced Synthetic Lethality in RB1-Deficient Cancers
3.3. Synergistic Effect of DNA Damage and PARP Inhibitor on RB1-Deficient Cancers
3.4. Targeting RB1 Loss Cancer with Ferroptosis Inducer
3.5. Other Targets
3.5.1. Targeting RB1 Deficient Tumors through the Ubiquitin-Proteasome Pathway
3.5.2. Targeting Hyperactive E2F through Histone Demethylase LSD1 Inhibition in RB1-Deficient Tumors
3.5.3. Synergistic Chemo-Drug and Histone Methyltransferase DOT1L Inhibition for Treating Retinoblastoma
3.5.4. The Crosstalk of RB1 Loss and ESRRG
3.5.5. Targeting ER+/RB1-Knockout Breast Cancer with PRMT5 Inhibitor
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Zhao, Z. Characterization of Tumor-Suppressor Gene Inactivation Events in 33 Cancer Types. Cell Rep. 2019, 26, 496–506.e3. [Google Scholar] [CrossRef]
- Li, B.; Gordon, G.M.; Du, C.H.; Xu, J.; Du, W. Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer Cell 2010, 17, 469–480. [Google Scholar] [CrossRef]
- Kohno, S.; Linn, P.; Nagatani, N.; Watanabe, Y.; Kumar, S.; Soga, T.; Takahashi, C. Pharmacologically targetable vulnerability in prostate cancer carrying RB1-SUCLA2 deletion. Oncogene 2020, 39, 5690–5707. [Google Scholar] [CrossRef]
- Gordon, G.M.; Zhang, T.; Zhao, J.; Du, W. Deregulated G1-S control and energy stress contribute to the synthetic-lethal interactions between inactivation of RB and TSC1 or TSC2. J. Cell Sci. 2013, 126, 2004–2013. [Google Scholar] [CrossRef]
- Dynlacht, B.D.; Flores, O.; Lees, J.A.; Harlow, E. Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes Dev. 1994, 8, 1772–1786. [Google Scholar] [CrossRef]
- Aubry, A.; Pearson, J.D.; Huang, K.; Livne-Bar, I.; Ahmad, M.; Jagadeesan, M.; Khetan, V.; Ketela, T.; Brown, K.R.; Yu, T.; et al. Functional genomics identifies new synergistic therapies for retinoblastoma. Oncogene 2020, 39, 5338–5357. [Google Scholar] [CrossRef]
- Harbour, J.W.; Dean, D.C. Chromatin remodeling and Rb activity. Curr. Opin. Cell Biol. 2000, 12, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Julian, L.M.; Palander, O.; Seifried, L.A.; Foster, J.E.; Dick, F.A. Characterization of an E2F1-specific binding domain in pRB and its implications for apoptotic regulation. Oncogene 2008, 27, 1572–1579. [Google Scholar] [CrossRef][Green Version]
- Dick, F.A.; Dyson, N. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol. Cell 2003, 12, 639–649. [Google Scholar] [CrossRef]
- Jori, F.P.; Melone, M.A.; Napolitano, M.A.; Cipollaro, M.; Cascino, A.; Giordano, A.; Galderisi, U. RB and RB2/p130 genes demonstrate both specific and overlapping functions during the early steps of in vitro neural differentiation of marrow stromal stem cells. Cell Death Differ. 2005, 12, 65–77. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bellan, C.; De Falco, G.; Tosi, G.M.; Lazzi, S.; Ferrari, F.; Morbini, G.; Bartolomei, S.; Toti, P.; Mangiavacchi, P.; Cevenini, G.; et al. Missing expression of pRb2/p130 in human retinoblastomas is associated with reduced apoptosis and lesser differentiation. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3602–3608. [Google Scholar]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 2010, 17, 376–387. [Google Scholar] [CrossRef]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef]
- Manning, A.L.; Yazinski, S.A.; Nicolay, B.; Bryll, A.; Zou, L.; Dyson, N.J. Suppression of genome instability in pRB-deficient cells by enhancement of chromosome cohesion. Mol. Cell 2014, 53, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Longworth, M.S.; Dyson, N.J. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010, 24, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Blasco, M.A. Role of Rb family in the epigenetic definition of chromatin. Cell Cycle 2005, 4, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Vasconcellos, I.; Schneider, R.; Anastasov, N.; Alonso-Rodriguez, S.; Sanli-Bonazzi, B.; Fernandez, J.L.; Atkinson, M.J. The Rb1 tumour suppressor gene modifies telomeric chromatin architecture by regulating TERRA expression. Sci. Rep. 2017, 7, 42056. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Garcia-Cao, M.; Fraga, M.F.; Schotta, G.; Peters, A.H.; Cotter, S.E.; Eguia, R.; Dean, D.C.; Esteller, M.; Jenuwein, T.; et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat. Cell Biol. 2005, 7, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cao, M.; Gonzalo, S.; Dean, D.; Blasco, M.A. A role for the Rb family of proteins in controlling telomere length. Nat. Genet. 2002, 32, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Leong, H.S.; Lin, D.M.; Popkiss, S.; Pang, P.; Garsed, D.W.; Walkley, C.R.; Cullinane, C.; Ellul, J.; Haynes, N.M.; et al. Immune response to RB1-regulated senescence limits radiation-induced osteosarcoma formation. J. Clin. Investig. 2013, 123, 5351–5360. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Zoumpoulidou, G.; Luczynski, M.T.; Rieger, S.; Moquet, J.; Spanswick, V.J.; Hartley, J.A.; Rothkamm, K.; Huang, P.H.; Mittnacht, S. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. 2015, 10, 2006–2018. [Google Scholar] [CrossRef]
- Chen, P.L.; Riley, D.J.; Chen, Y.; Lee, W.H. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev. 1996, 10, 2794–2804. [Google Scholar] [CrossRef]
- Hagemeier, C.; Bannister, A.J.; Cook, A.; Kouzarides, T. The activation domain of transcription factor PU.1 binds the retinoblastoma (RB) protein and the transcription factor TFIID in vitro: RB shows sequence similarity to TFIID and TFIIB. Proc. Natl. Acad. Sci. USA 1993, 90, 1580–1584. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.; Miyamoto, H.; Nishimura, K.; Kang, H.; Ludlow, J.; Hsiao, P.; Wang, C.; Su, C.; Chang, C. Retinoblastoma, a tumor suppressor, is a coactivator for the androgen receptor in human prostate cancer DU145 cells. Biochem. Biophys. Res. Commun. 1998, 248, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Chang, C.Y.; Hu, N.; Wang, Y.C.; Lai, C.C.; Herrup, K.; Lee, W.H.; Bradley, A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 1992, 359, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Fazeli, A.; Schmitt, E.M.; Bronson, R.T.; Goodell, M.A.; Weinberg, R.A. Effects of an Rb mutation in the mouse. Nature 1992, 359, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Huo, Z.; Yu, Y.; Zhu, D.; Xu, A.; Huang, M.F.; Hu, R.; Wang, R.; Gingold, J.A.; Chen, Y.H.; et al. Hereditary retinoblastoma iPSC model reveals aberrant spliceosome function driving bone malignancies. Proc. Natl. Acad. Sci. USA 2022, 119, e2117857119. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Garcia, B.; Gunduz, V.; Vazquez-Rivera, V.; Cress, W.D.; Wright, G.; Bian, H.; Hinds, P.W.; Santiago-Cardona, P.G. A role for the retinoblastoma protein as a regulator of mouse osteoblast cell adhesion: Implications for osteogenesis and osteosarcoma formation. PLoS ONE 2010, 5, e13954. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, E.; Ketner, A.; Ransick, P.; Ardekani, D.; Bodenstine, T.; Chandar, N. Loss of Function of the Retinoblastoma Gene Affects Gap Junctional Intercellular Communication and Cell Fate in Osteoblasts. Biology 2024, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Kalathil, D.; John, S.; Nair, A.S. FOXM1 and Cancer: Faulty Cellular Signaling Derails Homeostasis. Front. Oncol. 2020, 10, 626836. [Google Scholar] [CrossRef]
- Mukhopadhyay, N.K.; Chand, V.; Pandey, A.; Kopanja, D.; Carr, J.R.; Chen, Y.J.; Liao, X.; Raychaudhuri, P. Plk1 Regulates the Repressor Function of FoxM1b by inhibiting its Interaction with the Retinoblastoma Protein. Sci. Rep. 2017, 7, 46017. [Google Scholar] [CrossRef] [PubMed]
- Chand, V.; Pandey, A.; Kopanja, D.; Guzman, G.; Raychaudhuri, P. Opposing Roles of the Forkhead Box Factors FoxM1 and FoxA2 in Liver Cancer. Mol. Cancer Res. 2019, 17, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.R.; Kiefer, M.M.; Park, H.J.; Li, J.; Wang, Z.; Fontanarosa, J.; DeWaal, D.; Kopanja, D.; Benevolenskaya, E.V.; Guzman, G.; et al. FoxM1 regulates mammary luminal cell fate. Cell Rep. 2012, 1, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Kopanja, D.; Chand, V.; O’Brien, E.; Mukhopadhyay, N.K.; Zappia, M.P.; Islam, A.; Frolov, M.V.; Merrill, B.J.; Raychaudhuri, P. Transcriptional Repression by FoxM1 Suppresses Tumor Differentiation and Promotes Metastasis of Breast Cancer. Cancer Res. 2022, 82, 2458–2471. [Google Scholar] [CrossRef] [PubMed]
- Chand, V.; Liao, X.; Guzman, G.; Benevolenskaya, E.; Raychaudhuri, P. Hepatocellular carcinoma evades RB1-induced senescence by activating the FOXM1-FOXO1 axis. Oncogene 2022, 41, 3778–3790. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef] [PubMed]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Ahlander, J.; Bosco, G. The RB/E2F pathway and regulation of RNA processing. Biochem. Biophys. Res. Commun. 2009, 384, 280–283. [Google Scholar] [CrossRef][Green Version]
- Adegbola, O.; Pasternack, G.R. A pp32-retinoblastoma protein complex modulates androgen receptor-mediated transcription and associates with components of the splicing machinery. Biochem. Biophys. Res. Commun. 2005, 334, 702–708. [Google Scholar] [CrossRef]
- Gingold, J.; Zhou, R.; Lemischka, I.R.; Lee, D.F. Modeling Cancer with Pluripotent Stem Cells. Trends Cancer 2016, 2, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Phase I First-in-Human Dose Escalation Study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 2021, 35, 3542–3550. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef]
- Yang, W.; Jiang, X.X.; Zhao, X.Y.; Mao, P.A. Treatment of RB-deficient retinoblastoma with Aurora-A kinase inhibitor. Kaohsiung J. Med. Sci. 2022, 38, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Yang, E.J.; Zhang, B.; Wu, C.; Pardeshi, L.; Shi, C.; Mou, P.K.; Liu, Y.; Tan, K.; Shim, J.S. Synthetic lethality of RB1 and aurora A is driven by stathmin-mediated disruption of microtubule dynamics. Nat. Commun. 2020, 11, 5105. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Waqar, S.N.; Krug, L.M.; Dowlati, A.; Hann, C.L.; Chiappori, A.; Owonikoko, T.K.; Woo, K.M.; Cardnell, R.J.; Fujimoto, J.; et al. Randomized, Double-Blind, Phase II Study of Temozolomide in Combination with Either Veliparib or Placebo in Patients with Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2386–2394. [Google Scholar] [CrossRef]
- Zoumpoulidou, G.; Alvarez-Mendoza, C.; Mancusi, C.; Ahmed, R.M.; Denman, M.; Steele, C.D.; Tarabichi, M.; Roy, E.; Davies, L.R.; Manji, J.; et al. Therapeutic vulnerability to PARP1,2 inhibition in RB1-mutant osteosarcoma. Nat. Commun. 2021, 12, 7064. [Google Scholar] [CrossRef]
- Dong, Q.; Yu, T.; Chen, B.; Liu, M.; Sun, X.; Cao, H.; Liu, K.; Xu, H.; Wang, Y.; Zhuang, S.; et al. Mutant RB1 enhances therapeutic efficacy of PARPis in lung adenocarcinoma by triggering the cGAS/STING pathway. JCI Insight 2023, 8, e165268. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, G.; Armenia, J.; Mazzu, Y.Z.; Nandakumar, S.; Stopsack, K.H.; Atiq, M.O.; Komura, K.; Jehane, L.; Hirani, R.; Chadalavada, K.; et al. Significance of BRCA2 and RB1 Co-loss in Aggressive Prostate Cancer Progression. Clin. Cancer Res. 2020, 26, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Tsujino, T.; Takai, T.; Gui, F.; Tsutsumi, T.; Sztupinszki, Z.; Wang, Z.; Azuma, H.; Szallasi, Z.; Mouw, K.W.; et al. RB1 loss overrides PARP inhibitor sensitivity driven by RNASEH2B loss in prostate cancer. Sci. Adv. 2022, 8, eabl9794. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.E.; Chen, J.; Lu, Y.; Bawcom, A.R.; Wu, J.; Ou, J.; Asara, J.M.; Armstrong, A.J.; Wang, Q.; Li, L.; et al. RB1-deficient prostate tumor growth and metastasis are vulnerable to ferroptosis induction via the E2F/ACSL4 axis. J. Clin. Investig. 2023, 133, e166647. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 2014, 13, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Zhao, H.; Hoang, B.; Schwartz, E.L. Targeting the untargetable: RB1-deficient tumours are vulnerable to Skp2 ubiquitin ligase inhibition. Br. J. Cancer 2022, 127, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, H.; Bauzon, F.; Lu, Z.; Fu, H.; Cui, J.; Zhu, L. Deletions of Retinoblastoma 1 (Rb1) and Its Repressing Target S Phase Kinase-associated protein 2 (Skp2) Are Synthetic Lethal in Mouse Embryogenesis. J. Biol. Chem. 2016, 291, 10201–10209. [Google Scholar] [CrossRef]
- Wang, H.; Bauzon, F.; Ji, P.; Xu, X.; Sun, D.; Locker, J.; Sellers, R.S.; Nakayama, K.; Nakayama, K.I.; Cobrinik, D.; et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/− mice. Nat. Genet. 2010, 42, 83–88. [Google Scholar] [CrossRef]
- Ji, P.; Jiang, H.; Rekhtman, K.; Bloom, J.; Ichetovkin, M.; Pagano, M.; Zhu, L. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell 2004, 16, 47–58. [Google Scholar] [CrossRef]
- Aubry, A.; Yu, T.; Bremner, R. Preclinical studies reveal MLN4924 is a promising new retinoblastoma therapy. Cell Death Discov. 2020, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Liu, M.; Han, D.; Li, M.; Toure, A.A.; Wang, Z.; Besschetnova, A.; Patalano, S.; Macoska, J.A.; Gao, S.; et al. RB1 loss in castration-resistant prostate cancer confers vulnerability to LSD1 inhibition. Oncogene 2022, 41, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Theriault, B.L.; Dimaras, H.; Gallie, B.L.; Corson, T.W. The genomic landscape of retinoblastoma: A review. Clin. Exp. Ophthalmol. 2014, 42, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.; Malik, M.A.; Goswami, S.; Shukla, S.; Kaur, J. Epigenetic regulation of human retinoblastoma. Tumour Biol. 2016, 37, 14427–14441. [Google Scholar] [CrossRef] [PubMed]
- Benavente, C.A.; Dyer, M.A. Genetics and epigenetics of human retinoblastoma. Annu. Rev. Pathol. 2015, 10, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, J.K. Chromatin regulators in retinoblastoma: Biological roles and therapeutic applications. J. Cell Physiol. 2021, 236, 2318–2332. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Sun, Y.; Wu, Z.; Zheng, J.; Zhang, J.; Zeng, J.; Lee, C.; Kim, J.K. Targeting of histone methyltransferase DOT1L plays a dual role in chemosensitization of retinoblastoma cells and enhances the efficacy of chemotherapy. Cell Death Dis. 2021, 12, 1141. [Google Scholar] [CrossRef]
- Yi, Y.; Ge, S. Targeting the histone H3 lysine 79 methyltransferase DOT1L in MLL-rearranged leukemias. J. Hematol. Oncol. 2022, 15, 35. [Google Scholar] [CrossRef]
- Dimaras, H.; Corson, T.W.; Cobrinik, D.; White, A.; Zhao, J.; Munier, F.L.; Abramson, D.H.; Shields, C.L.; Chantada, G.L.; Njuguna, F.; et al. Retinoblastoma. Nat. Rev. Dis. Primers 2015, 1, 15021. [Google Scholar] [CrossRef]
- Chan, H.S.; Gallie, B.L.; Munier, F.L.; Beck Popovic, M. Chemotherapy for retinoblastoma. Ophthalmol. Clin. N. Am. 2005, 18, 55–63. [Google Scholar] [CrossRef]
- Corson, T.W.; Gallie, B.L. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer 2007, 46, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C., Jr.; Squire, J.A.; et al. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Field, M.G.; Kuznetsoff, J.N.; Zhang, M.G.; Dollar, J.J.; Durante, M.A.; Sayegh, Y.; Decatur, C.L.; Kurtenbach, S.; Pelaez, D.; Harbour, J.W. RB1 loss triggers dependence on ESRRG in retinoblastoma. Sci. Adv. 2022, 8, eabm8466. [Google Scholar] [CrossRef] [PubMed]
- Ao, A.; Wang, H.; Kamarajugadda, S.; Lu, J. Involvement of estrogen-related receptors in transcriptional response to hypoxia and growth of solid tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 7821–7826. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Gantner, M.L.; Smith, L.E.H. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Prog. Retin. Eye Res. 2018, 64, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Wangsa-Wirawan, N.D.; Linsenmeier, R.A. Retinal oxygen: Fundamental and clinical aspects. Arch. Ophthalmol. 2003, 121, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Tripathy, A.; Yu, W.; Eberhart, C.G.; Asnaghi, L. Hypoxia inhibits growth, proliferation, and increases response to chemotherapy in retinoblastoma cells. Exp. Eye Res. 2017, 162, 48–61. [Google Scholar] [CrossRef]
- Sudhakar, J.; Venkatesan, N.; Lakshmanan, S.; Khetan, V.; Krishnakumar, S.; Biswas, J. Hypoxic tumor microenvironment in advanced retinoblastoma. Pediatr. Blood Cancer 2013, 60, 1598–1601. [Google Scholar] [CrossRef]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2020, 382, 514–524. [Google Scholar] [CrossRef]
- Wander, S.A.; Cohen, O.; Gong, X.; Johnson, G.N.; Buendia-Buendia, J.E.; Lloyd, M.R.; Kim, D.; Luo, F.; Mao, P.; Helvie, K.; et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov. 2020, 10, 1174–1193. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Chang, T.C.; Wang, Y.; Guo, L.; Gao, Y.; Bikorimana, E.; Lemoff, A.; Fang, Y.V.; Zhang, H.; Zhang, Y.; et al. PRMT5 is an actionable therapeutic target in CDK4/6 inhibitor-resistant ER+/RB-deficient breast cancer. Nat. Commun. 2024, 15, 2287. [Google Scholar] [CrossRef] [PubMed]
- Fennell, D.A.; Porter, C.; Lester, J.; Danson, S.; Blackhall, F.; Nicolson, M.; Nixon, L.; Gardner, G.; White, A.; Griffiths, G.; et al. Olaparib maintenance versus placebo monotherapy in patients with advanced non-small cell lung cancer (PIN): A multicentre, randomised, controlled, phase 2 trial. EClinicalMedicine 2022, 52, 101595. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drugs | Pharmacological Function | Cancer Types | Genome Status | References |
|---|---|---|---|---|
| Pladienolide B | Spliceosomal inhibitors | Osteosarcoma | RB1 mutation | [36] |
| Sudemycin D6 | Osteosarcoma | RB1 mutation | [36] | |
| Barasertib-HQPA (AZD2811) | Aurora kinase B inhibitors | Small Cell Lung Cancer | RB1 deficient | [52] |
| LY3295668 Erbumine | Aurora kinase A inhibitors | Small Cell Lung Cancer | RB1 deficient | [56] |
| ENMD-2076 | Non Small Cell Lung Cancer | RB1 deficient | [58] | |
| Lynparza (Olaparib) | PARP inhibitors | Lung Adenocarcinoma | RB1 mutation | [61,94] |
| Osteosarcoma | RB1 mutation | [60] | ||
| Rucaparib | Lung Adenocarcinoma | RB1 mutation | [61] | |
| Zejula (Niraparib) | Lung Adenocarcinoma | RB1 mutation | [61] | |
| Osteosarcoma | RB1 mutation | [60] | ||
| JKE-1674 | Ferroptosis inducer | Prostate Cancer | RB1 deficient | [65] |
| Pevonedistat (MLN4924) | SKP2 inhibitors | Retinoblastoma | RB1 deficient, MYC amplification | [71] |
| GSK2879552 | LSD1 inhibitors | Prostate Cancer | RB1 deficient | [72] |
| GSK5182 | ESRRG inhibitors | Retinoblastoma | RB1 deficient | [83] |
| Pemrametostat (GSK3326595) | PRMT5 inhibitors | Breast Cancer | ER+, RB1 deficient | [93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.-F.; Wang, Y.-X.; Chou, Y.-T.; Lee, D.-F. Therapeutic Strategies for RB1-Deficient Cancers: Intersecting Gene Regulation and Targeted Therapy. Cancers 2024, 16, 1558. https://doi.org/10.3390/cancers16081558
Huang M-F, Wang Y-X, Chou Y-T, Lee D-F. Therapeutic Strategies for RB1-Deficient Cancers: Intersecting Gene Regulation and Targeted Therapy. Cancers. 2024; 16(8):1558. https://doi.org/10.3390/cancers16081558
Chicago/Turabian StyleHuang, Mo-Fan, Yuan-Xin Wang, Yu-Ting Chou, and Dung-Fang Lee. 2024. "Therapeutic Strategies for RB1-Deficient Cancers: Intersecting Gene Regulation and Targeted Therapy" Cancers 16, no. 8: 1558. https://doi.org/10.3390/cancers16081558
APA StyleHuang, M.-F., Wang, Y.-X., Chou, Y.-T., & Lee, D.-F. (2024). Therapeutic Strategies for RB1-Deficient Cancers: Intersecting Gene Regulation and Targeted Therapy. Cancers, 16(8), 1558. https://doi.org/10.3390/cancers16081558