The Crucial Role of Hereditary Cancer Panel Testing in Unaffected Individuals with a Strong Family History of Cancer: A Retrospective Study of a Cohort of 103 Healthy Subjects

 ,
,  , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Genomic DNA Extraction
2.3. Next-Generation Sequencing (NGS)
2.4. Sanger Sequencing
2.5. Genetic Variant Classification
3. Results
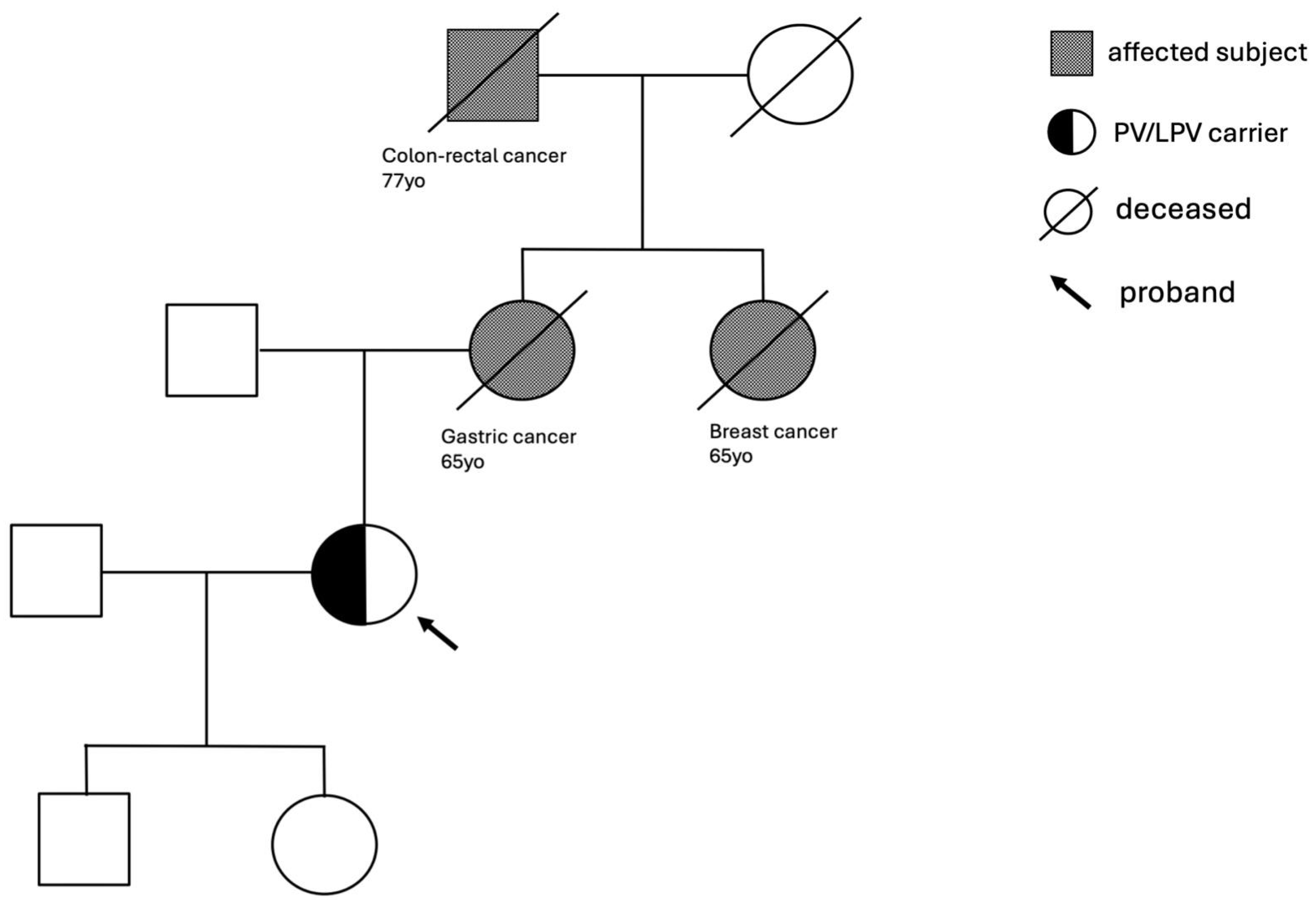
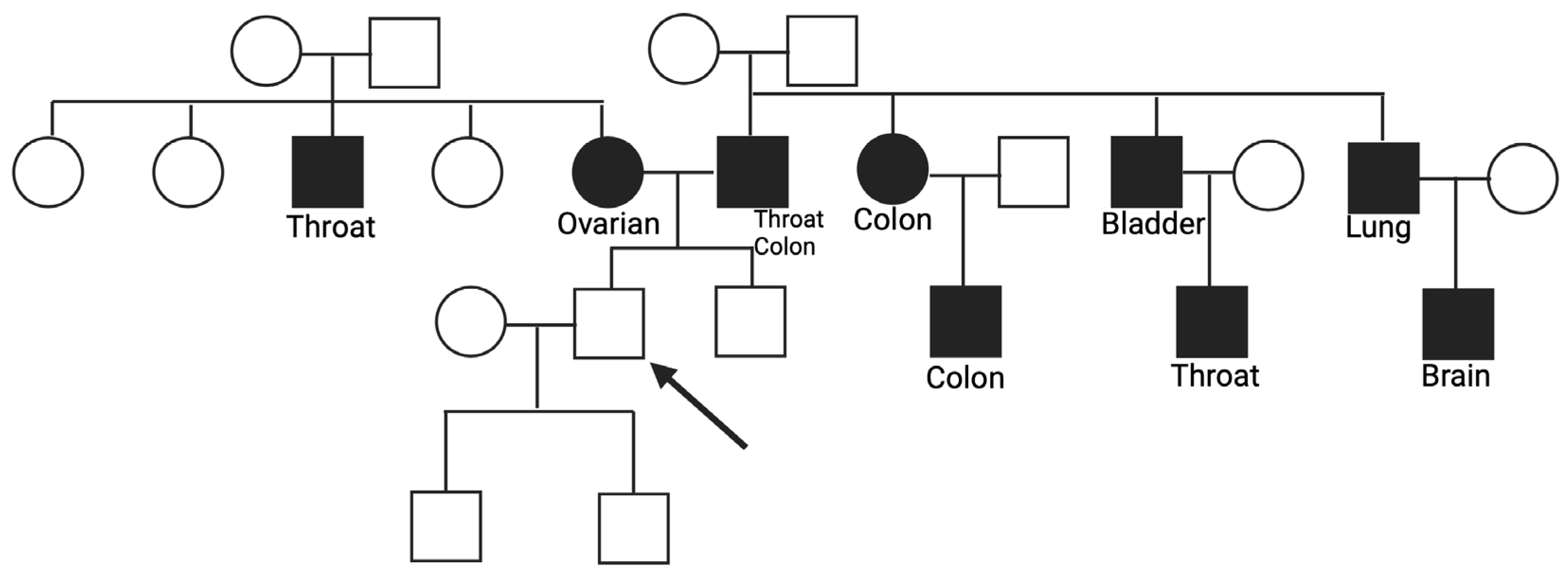
3.1. Genes Variants Linked to the Homologous Recombination (HR) and Related Family History
3.2. Genes Variants Linked to the Base Excision Repair (BER) and Related Family History
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antonucci, I.; Provenzano, M.; Sorino, L.; Rodrigues, M.; Palka, G.; Stuppia, L. A new case of “de novo” BRCA1 mutation in a patient with early-onset breast cancer. Clin. Case Rep. 2017, 5, 238–240. [Google Scholar] [CrossRef] [PubMed]
- NCCN. Detection, Prevention, and Risk Reduction. Available online: https://www.nccn.org/guidelines/category_2 (accessed on 27 February 2024).
- Knerr, S.; Guo, B.; Mittendorf, K.F.; Feigelson, H.S.; Gilmore, M.J.; Jarvik, G.P.; Kauffman, T.L.; Keast, E.; Lynch, F.L.; Muessig, K.R.; et al. Risk-reducing surgery in unaffected individuals receiving cancer genetic testing in an integrated health care system. Cancer 2022, 128, 3090–3098. [Google Scholar] [CrossRef] [PubMed]
- Kamps, R.; Brandão, R.D.; Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef] [PubMed]
- DQ Cancer Genetics Editorial Board. Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version. 6 March 2024. Available online: https://www.cancer.gov/publications/pdq/information-summaries/genetics/risk-assessment-hp-pdq (accessed on 27 February 2024).
- Available online: https://www.cancer.org/cancer/risk-prevention/genetics/genetic-testing-for-cancer-risk/understanding-genetic-testing-for-cancer.html (accessed on 23 April 2024).
- Di Rado, S.; Giansante, R.; Cicirelli, M.; Pilenzi, L.; Dell’Elice, A.; Anaclerio, F.; Rimoldi, M.; Grassadonia, A.; Grossi, S.; Canale, N.; et al. Detection of Germline Mutations in a Cohort of 250 Relatives of Mutation Carriers in Multigene Panel: Impact of Pathogenic Variants in Other Genes beyond BRCA1/2. Cancers 2023, 15, 5730. [Google Scholar] [CrossRef]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. 1998 Sep 4 [Updated 21 September 2023]. In GeneReviews® [Internet]; University of Washington, Seattle: Seattle, WA, USA, 1993–2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1247/ (accessed on 23 April 2024).
- Forbes, C.; Fayter, D.; de Kock, S.; Quek, R.G. A systematic review of international guidelines and recommendations for the genetic screening, diagnosis, genetic counseling, and treatment of BRCA-mutated breast cancer. Cancer Manag. Res. 2019, 11, 2321–2337. [Google Scholar] [CrossRef]
- Weitzel, J.N.; Lagos, V.I.; Cullinane, C.A.; Gambol, P.J.; Culver, J.O.; Blazer, K.R.; Palomares, M.R.; Lowstuter, K.J.; MacDonald, D.J. Limited family structure and BRCA gene mutation status in single cases of breast cancer. JAMA 2007, 297, 2587–2595. [Google Scholar] [CrossRef]
- Bramanti, S.M.; Trumello, C.; Lombardi, L.; Cavallo, A.; Stuppia, L.; Antonucci, I.; Babore, A. Uncertainty following an inconclusive result from the BRCA1/2 genetic test: A review about psychological outcomes. World J. Psychiatry 2021, 11, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Trottier, M.; Lunn, J.; Butler, R.; Curling, D.; Turnquest, T.; Francis, W.; Halliday, D.; Royer, R.; Zhang, S.; Li, S.; et al. Prevalence of founder mutations in the BRCA1 and BRCA2 genes among unaffected women from the Bahamas. Clin. Genet. 2016, 89, 328–331. [Google Scholar] [CrossRef]
- Bernstein-Molho, R.; Singer, A.; Laitman, Y.; Netzer, I.; Zalmanoviz, S.; Friedman, E. Multigene panel testing in unselected Israeli breast cancer cases: Mutational spectrum and use of BRCA1/2 mutation prediction algorithms. Breast. Cancer Res. Treat. 2019, 176, 165–170. [Google Scholar] [CrossRef]
- Ece Solmaz, A.; Yeniay, L.; Gökmen, E.; Zekioğlu, O.; Haydaroğlu, A.; Bilgen, I.; Özkınay, F.; Onay, H. Clinical Contribution of Next-Generation Sequencing Multigene Panel Testing for BRCA Negative High-Risk Patients With Breast Cancer. Clin. Breast. Cancer 2021, 21, e647–e653. [Google Scholar] [CrossRef]
- Anaclerio, F.; Pilenzi, L.; Dell’Elice, A.; Ferrante, R.; Grossi, S.; Ferlito, L.M.; Marinelli, C.; Gildetti, S.; Calabrese, G.; Stuppia, L.; et al. Clinical usefulness of NGS multi-gene panel testing in hereditary cancer analysis. Front. Genet. 2023, 14, 1060504. [Google Scholar] [CrossRef] [PubMed]
- Desrichard, A.; Bidet, Y.; Uhrhammer, N.; Bignon, Y.J. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast. Cancer Res. 2011, 13, R119. [Google Scholar] [CrossRef] [PubMed]
- Oros, K.K.; Ghadirian, P.; Greenwood, C.M.; Perret, C.; Shen, Z.; Paredes, Y.; Arcand, S.L.; Mes-Masson, A.M.; Narod, S.A.; Foulkes, W.D.; et al. Significant proportion of breast and/or ovarian cancer families of French Canadian descent harbor 1 of 5 BRCA1 and BRCA2 mutations. Int. J. Cancer 2004, 112, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Thiffault, I.; Saunders, C.; Jenkins, J.; Raje, N.; Canty, K.; Sharma, M.; Grote, L.; Welsh, H.I.; Farrow, E.; Twist, G.; et al. A patient with polymerase E1 deficiency (POLE1): Clinical features and overlap with DNA breakage/instability syndromes. BMC Med. Genet. 2015, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Jones, N.; Vogt, S.; Carli, M.; Vasen HF, A.; Sampson, J.R.; Aretz, S.; Hes, F.J. Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology 2009, 136, 471–476. [Google Scholar] [CrossRef]
- Imyanitov, E.N.; Kuligina, E.S.; Sokolenko, A.P.; Suspitsin, E.N.; Yanus, G.A.; Iyevleva, A.G.; Ivantsov, A.O.; Aleksakhina, S.N. Hereditary cancer syndromes. World J. Clin. Oncol. 2023, 14, 40–68. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, I.; Provenzano, M.; Sorino, L.; Balsamo, M.; Aceto, G.M.; Battista, P.; Euhus, D.; Cianchetti, E.; Ballerini, P.; Natoli, C.; et al. Comparison between CaGene 5.1 and 6.0 for BRCA1/2 mutation prediction: A retrospective study of 150 BRCA1/2 genetic tests in 517 families with breast/ovarian cancer. J. Hum. Genet. 2017, 62, 379–387. [Google Scholar] [CrossRef]
- Magrin, L.; Fanale, D.; Brando, C.; Fiorino, A.; Corsini, L.R.; Sciacchitano, R.; Filorizzo, C.; Dimino, A.; Russo, A.; Bazan, V. POLE, POLD1, and NTHL1: The last but not the least hereditary cancer-predisposing genes. Oncogene 2021, 40, 5893–5901. [Google Scholar] [CrossRef]
- AIOM (Associazione Italiana Oncologia Medica) Linee Guida Carcinoma Mammario in Stadio Precoce -Edizione. 2023. Available online: https://www.iss.it/documents/20126/8403839/Addendum%20LG_260_AIOM_Ca%20Mammella_ed2022 (accessed on 23 April 2024).
- Stolarova, L.; Kleiblova, P.; Zemankova, P.; Stastna, B.; Janatova, M.; Soukupova, J.; Achatz, M.I.; Ambrosone, C.; Apostolou, P.; Arun, B.K.; et al. ENIGMA CHEK2gether Project: A Comprehensive Study Identifies Functionally Impaired CHEK2 Germline Missense Variants Associated with Increased Breast Cancer Risk. Clin. Cancer Res. 2023, 29, 3037–3050. [Google Scholar] [CrossRef]
- Lombardi, L.; Bramanti, S.M.; Babore, A.; Stuppia, L.; Trumello, C.; Antonucci, I.; Cavallo, A. Psychological aspects, risk and protective factors related to BRCA genetic testing: A review of the literature. Support. Care Cancer 2019, 27, 3647–3656. [Google Scholar] [CrossRef]
- Babore, A.; Bramanti, S.M.; Lombardi, L.; Stuppia, L.; Trumello, C.; Antonucci, I.; Cavallo, A. The role of depression and emotion regulation on parenting stress in a sample of mothers with cancer. Support. Care Cancer 2019, 27, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Cicalini, I.; Cufaro, M.C.; Consalvo, A.; Upadhyaya, P.; Sala, G.; Antonucci, I.; Del Boccio, P.; Stuppia, L.; De Laurenzi, V. Breast cancer in the era of integrating “Omics” approaches. Oncogenesis 2022, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Ahsan, M.D.; Webster, E.; Levi, S.R.; Brewer, J.T.; Lin, J.; Blank, S.V.; Krinsky, H.; Nchako, C.; Wolfe, I.; et al. Web-based tool for cancer family history collection: A prospective randomized controlled trial. Gynecol. Oncol. 2023, 173, 22–30. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Multi-Gene Panel | ||
|---|---|---|
| APC | ATM | BARD1 |
| BRCA1 | BRCA2 | BRIP1 |
| CDK4 | CDK12 | CDKN2A |
| CDH1 | CHEK2 | EPCAM |
| MLH1 | MSH2 | MSH6 |
| MUTYH | NBN | NF1 |
| PALB2 | POLE | POLE |
| POLD1 | PTEN | RAD51C |
| RAD51D | SMAD4 | TP53 |
| NAME | EXON | SEQUENCE |
|---|---|---|
| BRCA2_EX11F | Exon 11 | attgagatcacagctgcccc |
| BRCA2_EX11R | Exon 11 | tgaagtctgactcacagaagttt |
| CHEK2_EX13F | Exon 13 | atgtggatgtgagtcagccag |
| CHEK2_EX13R | Exon 13 | atcagctccttaagcccagacta |
| BRCA1_EX10F | Exon 10 | ttggtcagctttctgtaatcg |
| BRCA1_EX10R | Exon 10 | ccataccacgacatttgaca |
| POLE_EX8F | Exon 8 | gtcgctgctcacatgaattt |
| POLE_EX8R | Exon 8 | atttgggggaaaagcagcaa |
| MUTYH_13F | Exon 13 | agggcagtggcatgagtaac |
| MUTYH_13R | Exon 13 | gggtcaaggggttcaaatag |
| CASE ID | GENE | OMIM | REFSEQ | CODING | PROTEIN |
|---|---|---|---|---|---|
| Subject 1 | BRCA2 | 164757 | NM_000059.3 | c.4914dupA | V1639fs |
| Subject 2 | CHEK2 | 604373 | NM_007194.4 | c.1427C>T | T476M |
| Subject 3 | BRCA1 | 113705 | NM_007294.4 | c.1953dup | K652fs |
| Subject 4 | POLE | 174762 | NM_006231.3 | c.778C>T | R260* |
| Subject 5 | MUTYH | 608456 | NM_001128425.2 | c.1187G>A | G396D |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilenzi, L.; Anaclerio, F.; Dell’Elice, A.; Minelli, M.; Giansante, R.; Cicirelli, M.; Tinari, N.; Grassadonia, A.; Pantalone, A.; Grossi, S.; et al. The Crucial Role of Hereditary Cancer Panel Testing in Unaffected Individuals with a Strong Family History of Cancer: A Retrospective Study of a Cohort of 103 Healthy Subjects. Cancers 2024, 16, 2327. https://doi.org/10.3390/cancers16132327
Pilenzi L, Anaclerio F, Dell’Elice A, Minelli M, Giansante R, Cicirelli M, Tinari N, Grassadonia A, Pantalone A, Grossi S, et al. The Crucial Role of Hereditary Cancer Panel Testing in Unaffected Individuals with a Strong Family History of Cancer: A Retrospective Study of a Cohort of 103 Healthy Subjects. Cancers. 2024; 16(13):2327. https://doi.org/10.3390/cancers16132327
Chicago/Turabian StylePilenzi, Lucrezia, Federico Anaclerio, Anastasia Dell’Elice, Maria Minelli, Roberta Giansante, Michela Cicirelli, Nicola Tinari, Antonino Grassadonia, Andrea Pantalone, Simona Grossi, and et al. 2024. "The Crucial Role of Hereditary Cancer Panel Testing in Unaffected Individuals with a Strong Family History of Cancer: A Retrospective Study of a Cohort of 103 Healthy Subjects" Cancers 16, no. 13: 2327. https://doi.org/10.3390/cancers16132327
APA StylePilenzi, L., Anaclerio, F., Dell’Elice, A., Minelli, M., Giansante, R., Cicirelli, M., Tinari, N., Grassadonia, A., Pantalone, A., Grossi, S., Canale, N., Bruno, A., Calabrese, G., Ballerini, P., Stuppia, L., & Antonucci, I. (2024). The Crucial Role of Hereditary Cancer Panel Testing in Unaffected Individuals with a Strong Family History of Cancer: A Retrospective Study of a Cohort of 103 Healthy Subjects. Cancers, 16(13), 2327. https://doi.org/10.3390/cancers16132327