
Glioma Stem Cells—Features for New Therapy Design



Abstract
Simple Summary
Abstract
1. Introduction
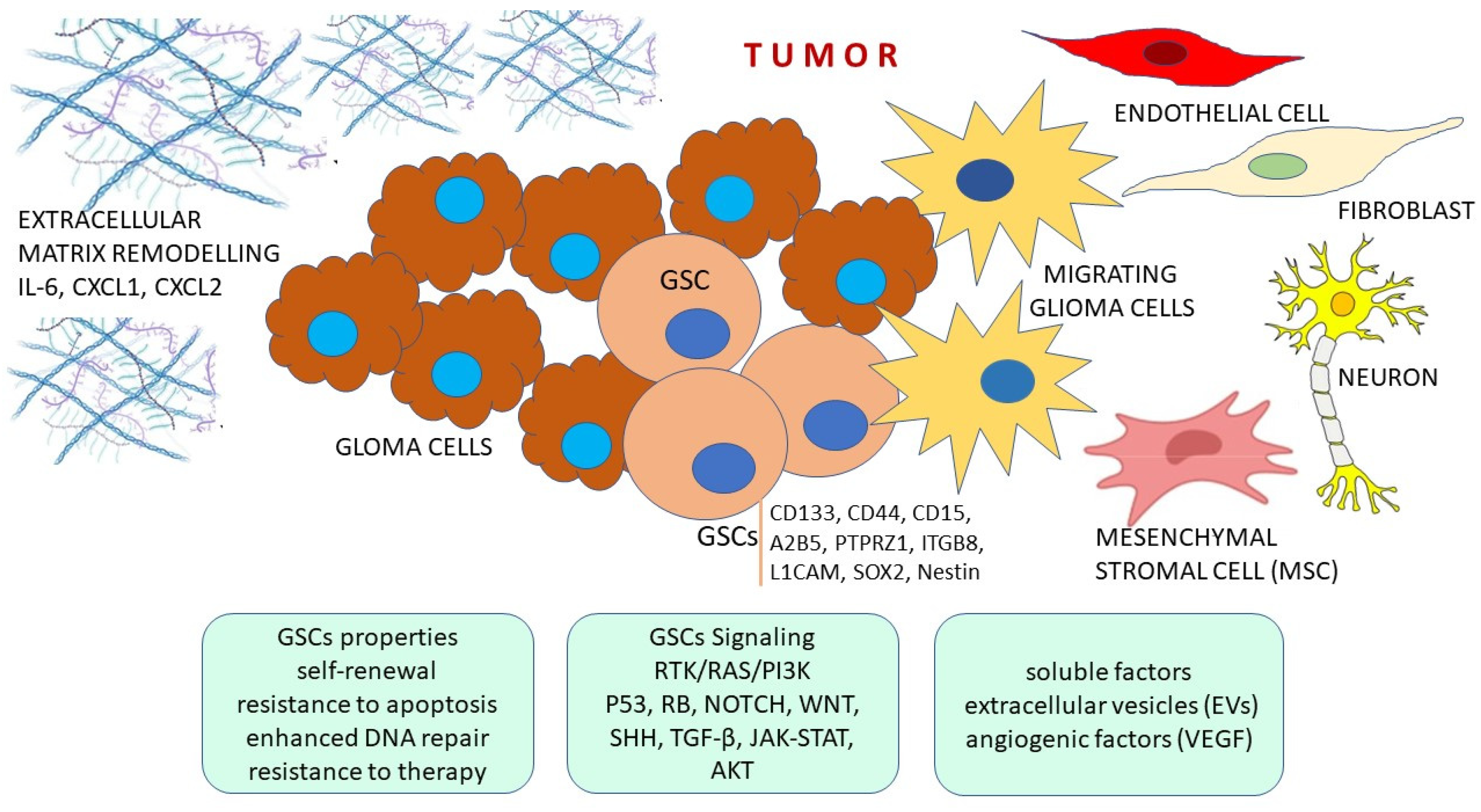
2. Features of Glioma Stem Cells
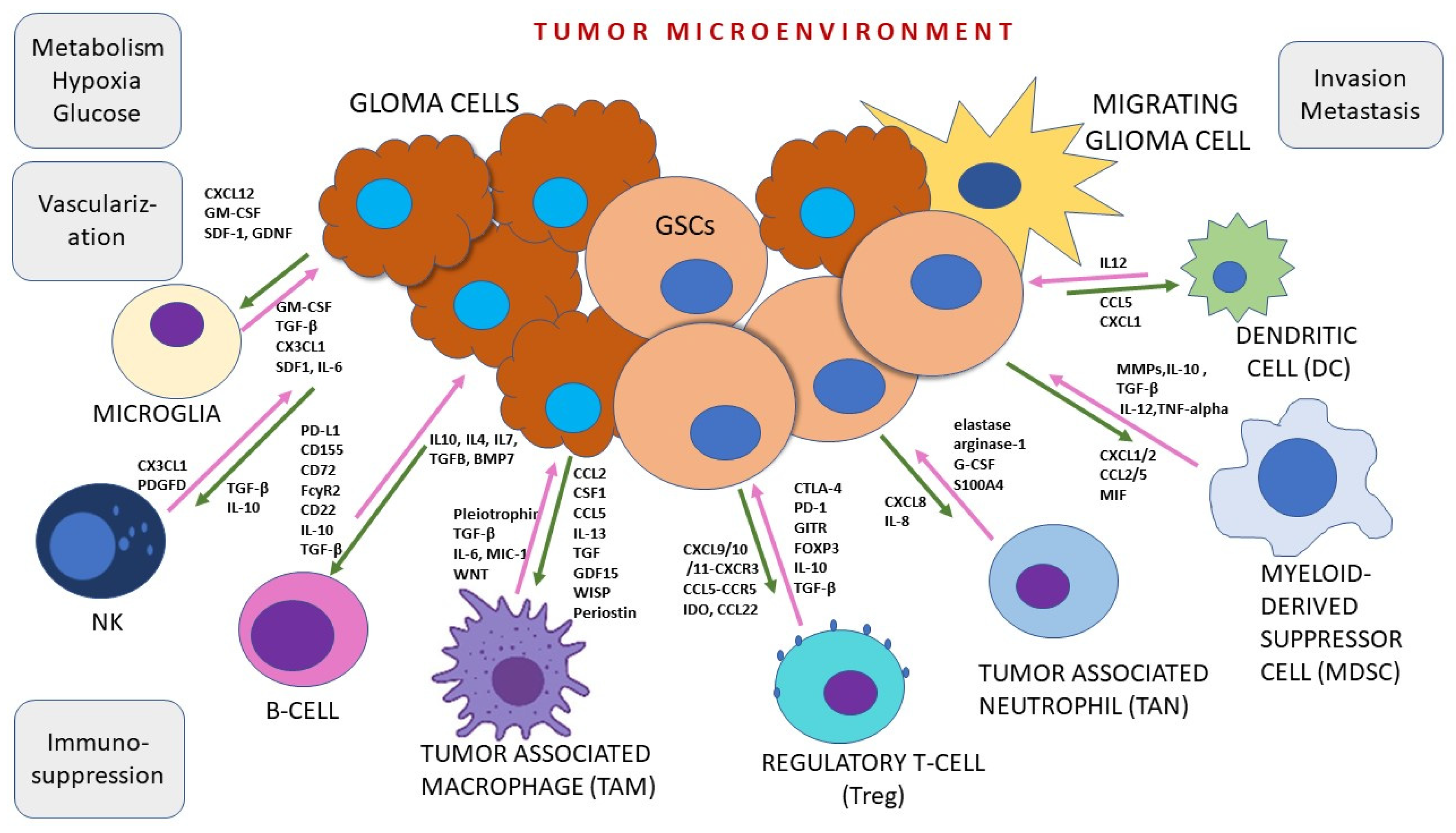
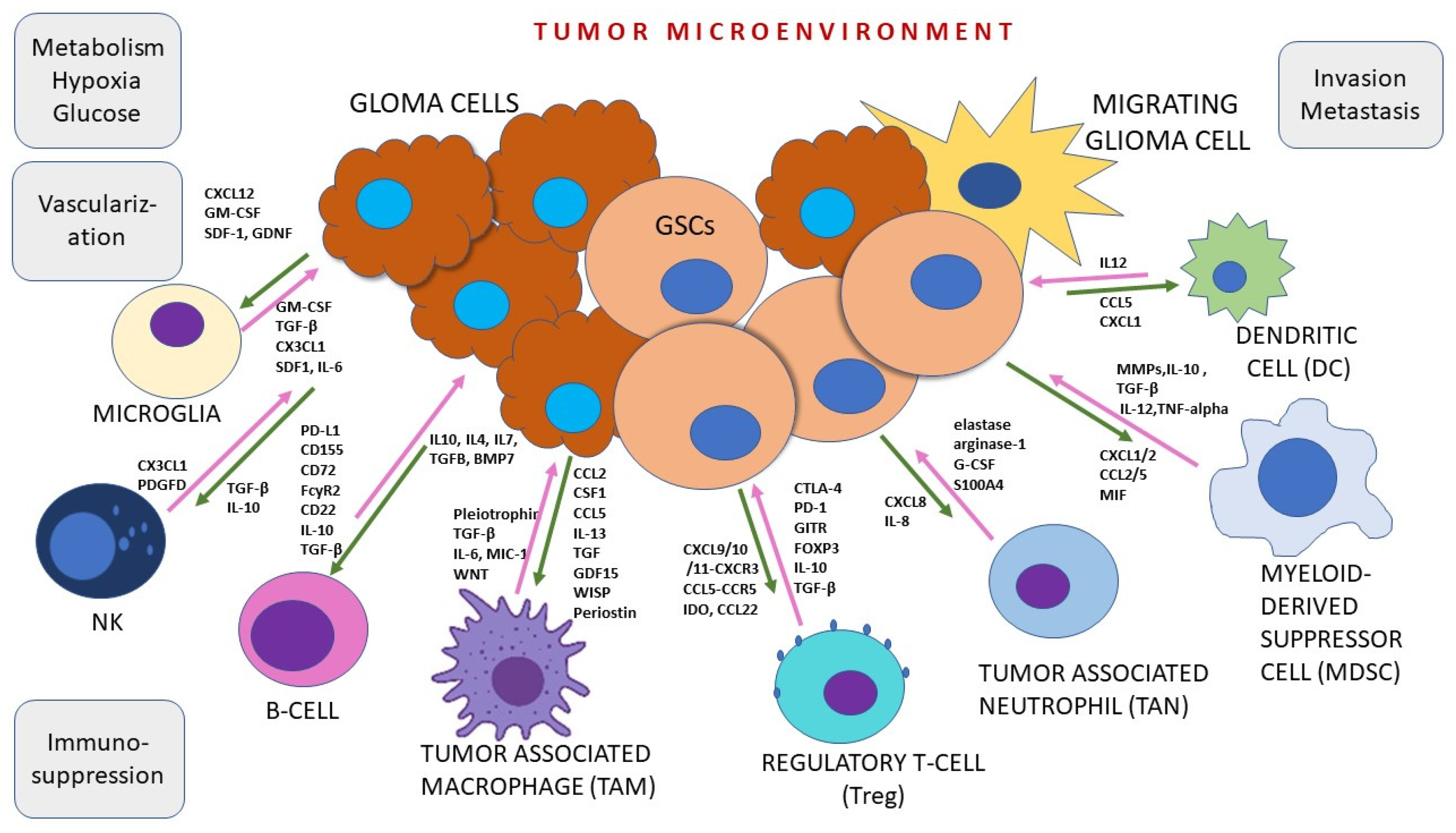
3. Tumor Microenvironment
4. Signaling Pathways in Glioma Cancer Stem Cells
5. Potential Therapeutic Approaches
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nasser, M.M.; Mehdipour, P. Exploration of involved key genes and signaling diversity in brain tumors. Cell Mol. Neurobiol. 2017, 38, 393–419. [Google Scholar] [CrossRef]
- Colopi, A.; Fuda, S.; Santi, S.; Onorato, A.; Cesarini, V.; Salvati, M.; Balistreri, C.R.; Dolci, S.; Guida, E. Impact of age and gender on glioblastoma onset, progression, and management. Mech. Ageing Dev. 2023, 211, 111801. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; von Deimling, A.; Cavenee, W.K. Diffuse astrocytic and oligodendroglial tumours—Introduction. In WHO Classification of Tumours of the Central Nervous System, 4th ed.; Louis, D.N., Ohgaki, H., Wiestler, O.D., Cavenee, W.K., Eds.; The International Agency for Research on Cancer: Lyon, France, 2016; pp. 15–17. [Google Scholar]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, B.W.; Priesterbach-Ackley, L.P.; Petersen, J.K.; Wesseling, P. Molecular Pathology of Tumors of the Central Nervous System. Ann. Oncol. 2019, 30, 1265–1278. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Waker, C.A.; Lober, R.M. Brain Tumors of Glial Origin. Adv. Exp. Med. Biol. 2019, 1190, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Melhem, J.M.; Detsky, J.; Lim-Fat, M.J.; Perry, J.R. Updates in IDH-Wildtype Glioblastoma. Neurotherapeutics 2022, 19, 1705–1723. [Google Scholar] [CrossRef]
- Barthel, F.P.; The GLASS Consortium; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, D.; Ulhøi, B.P.; Lukacova, S.; Alsner, J.; Stougaard, M.; Nyengaard, J.R. Impact of new molecular criteria on diagnosis and survival of adult glioma patients. IBRO Neurosci. Rep. 2022, 13, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Clausing, M.; William, D.; Preussler, M.; Biedermann, J.; Grützmann, K.; Richter, S.; Buchholz, F.; Temme, A.; Schröck, E.; Klink, B. Different Effects of RNAi-Mediated Downregulation or Chemical Inhibition of NAMPT in an Isogenic IDH Mutant and Wild-Type Glioma Cell Model. Int. J. Mol. Sci. 2022, 23, 5787. [Google Scholar] [CrossRef] [PubMed]
- Skoblar Vidmar, M.; Doma, A.; Smrdel, U.; Zevnik, K.; Studen, A. The Value of FET PET/CT in Recurrent Glioma with a Different IDH Mutation Status: The Relationship between Imaging and Molecular Biomarkers. Int. J. Mol. Sci. 2022, 23, 6787. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma [Internet]; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; Chapter 8. [Google Scholar] [CrossRef]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes. Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Abou-El-Ardat, K.; Seifert, M.; Becker, K.; Eisenreich, S.; Lehmann, M.; Hackmann, K.; Rump, A.; Meijer, G.; Carvalho, B.; Temme, A.; et al. Comprehensive molecular characterization of multifocal glioblastoma proves its monoclonal origin and reveals novel insights into clonal evolution and heterogeneity of glioblastomas. Neuro Oncol. 2017, 19, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Perry, A.; Wesseling, P. Histologic classification of gliomas. Handb. Clin. Neurol. 2016, 134, 71–95. [Google Scholar] [CrossRef]
- Jovčevska, I. Genetic secrets of long-term glioblastoma survivors. Bosn. J. Basic. Med. Sci. 2019, 19, 116–124. [Google Scholar] [CrossRef]
- Robertson, F.L.; Marqués-Torrejón, M.A.; Morrison, G.M.; Pollard, S.M. Experimental models and tools to tackle glioblastoma. Dis Model Mech. 2019, 12, dmm040386. [Google Scholar] [CrossRef]
- Noroxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef]
- Sant, M.; Minicozzi, P.; Lagorio, S.; Børge Johannesen, T.; Marcos-Gragera, R.; Francisci, S.; EUROCARE Working Group. Survival of European patients with central nervous system tumors. Int. J. Cancer. 2012, 131, 173–185. [Google Scholar] [CrossRef]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma: A systematic review. J. Clin. Neurosci. 2018, 54, 7–13. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Rick, J.; Chandra, A.; Aghi, M.K. Tumor treating fields: A new approach to glioblastoma therapy. J. Neuro. Oncol. 2018, 137, 447–453. [Google Scholar] [CrossRef]
- Paolillo, M.; Boselli, C.; Schinelli, S. Glioblastoma under Siege: An Overview of Current Therapeutic Strategies. Brain Sci. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Pellegatta, S. Perspectives for immunotherapy in glioblastoma treatment. Curr. Opin. Oncol. 2014, 26, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Artene, S.A.; Tuta, C.; Dragoi, A.; Alexandru, O.; Stefana Oana, P.; Tache, D.E.; Dănciulescu, M.M.; Boldeanu, M.V.; Siloşi, C.A.; Dricu, A. Current and emerging EGFR therapies for glioblastoma. J. Immunoassay Immunochem. 2018, 39, 1–11. [Google Scholar] [CrossRef]
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 142. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Piper, K.; DePledge, L.; Karsy MCobbs, C. Glioma Stem Cells as Immunotherapeutic Targets: Advancements and Challenges. Front. Oncol. 2021, 11, 615704. [Google Scholar] [CrossRef] [PubMed]
- Gisina, A.; Kholodenko, I.; Kim, Y.; Abakumov, M.; Lupatov, A.; Yarygin, K. Glioma Stem Cells: Novel Data Obtained by Single-Cell Sequencing. Int. J. Mol. Sci. 2022, 23, 14224. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Gargini, R.; Segura-Collar, B.; Sánchez-Gómez, P. Cellular Plasticity and Tumor Microenvironment in Gliomas: The Struggle to Hit a Moving Target. Cancers 2020, 12, 1622. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Yang, H.; Mao, Y. The Oncogenesis of Glial Cells in Diffuse Gliomas and Clinical Opportunities. Neurosci. Bull. 2023, 39, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Tyler, T.C.; Treiber, J.M.; Taha, B.; Engin, H.B.; Carter, H.; Patel, K.S.; Dale, A.M.; Carter, B.S.; Chen, C.C. Glioblastomas located in proximity to the subventricular zone (SVZ) exhibited enrichment of gene expression profiles associated with the cancer stem cell state. J. Neurooncol. 2020, 148, 455–462. [Google Scholar] [CrossRef]
- De Silva, M.I.; Stringer, B.W.; Bardy, C. Neuronal and tumourigenic boundaries of glioblastoma plasticity. Trends Cancer. 2023, 9, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Landis, C.J.; Tran, A.N.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim. Biophys. Acta Rev. Cancer. 2018, 1869, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, R.; Allega, M.F.; Tardito, S. A map of the altered glioma metabolism. Trends Mol Med. 2021, 27, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.-C.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- van Noorden, C.J.F.; Hira, V.V.V.; van Dijck, A.J.; Novak, M.; Breznik, B.; Molenaar, R.J. Energy Metabolism in IDH1 Wild-Type and IDH1-Mutated Glioblastoma Stem Cells: A Novel Target for Therapy? Cells 2021, 10, 705. [Google Scholar] [CrossRef]
- Badr, C.E.; Silver, D.J.; Siebzehnrubl, F.A.; Deleyrolle, L.P. Metabolic heterogeneity and adaptability in brain tumors. Cell Mol. Life Sci. 2020, 77, 5101–5119. [Google Scholar] [CrossRef]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer. 2020, 146, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Visweswaran, M.; Arfuso, F.; Warrier, S.; Dharmarajan, A. Aberrant lipid metabolism as an emerging therapeutic strategy to target cancer stem cells. Stem Cells 2020, 38, 6–14. [Google Scholar] [CrossRef]
- Mudassar, F.; Shen, H.; O’Neill, G.; Hau, E. Targeting tumor hypoxia and mitochondrial metabolism with antiparasitic drugs to improve radiation response in highgrade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 208. [Google Scholar] [CrossRef]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. Verteporfin inhibits oxidative phosphorylation and induces cell death specifically in glioma stem cells. FEBS J. 2020, 287, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Seliger, C.; Meyer, A.-L.; Leidgens, V.; Rauer, L.; Moeckel, S.; Jachnik, B.; Proske, J.; Dettmer, K.; Rothhammer-Hampl, T.; Kaulen, L.D.; et al. Metabolic Heterogeneity of Brain Tumor Cells of Proneural and Mesenchymal Origin. Int. J. Mol. Sci. 2022, 23, 11629. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.-W.; Chang, P.-C.; Chen, H.-Y.; Hueng, D.-Y.; Li, Y.-F.; Huang, S.-M. Exploring the Mechanism of Adjuvant Treatment of Glioblastoma Using Temozolomide and Metformin. Int. J. Mol. Sci. 2022, 23, 8171. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H.; Park, J.; Cho, H.J.; Shim, J.-K.; Moon, J.H.; Kim, E.H.; Chang, J.H.; Kim, S.Y.; Kang, S.-G. Combinatorial Therapeutic Effect of Inhibitors of Aldehyde Dehydrogenase and Mitochondrial Complex I, and the Chemotherapeutic Drug, Temozolomide against Glioblastoma Tumorspheres. Molecules 2021, 26, 282. [Google Scholar] [CrossRef] [PubMed]
- Hekmatshoar, Y.; Nakhle, J.; Galloni, M.; Vignais, M.L. The role of metabolism and tunneling nanotube-mediated intercellular mitochondria exchange in cancer drug resistance. Biochem. J. 2018, 475, 2305–2328. [Google Scholar] [CrossRef] [PubMed]
- Nakhle, J.; Khattar, K.; Özkan, T.; Boughlita, A.; Abba Moussa, D.; Darlix, A.; Lorcy, F.; Rigau, V.; Bauchet, L.; Gerbal-Chaloin, S.; et al. Mitochondria Transfer from Mesenchymal Stem Cells Confers Chemoresistance to Glioblastoma Stem Cells through Metabolic Rewiring. Cancer Res. Commun. 2023, 3, 1041–1056. [Google Scholar] [CrossRef]
- Wee, S.; Niklasson, M.; Marinescu, V.D.; Segerman, A.; Schmidt, L.; Hermansson, A.; Dirks, P.; Forsberg-Nilsson, K.; Westermark, B.; Uhrbom, L.; et al. Selective calcium sensitivity in immature glioma cancer stem cells. PLoS ONE 2014, 9, e115698. [Google Scholar] [CrossRef]
- Terrié, E.; Déliot, N.; Benzidane, Y.; Harnois, T.; Cousin, L.; Bois, P.; Oliver, L.; Arnault, P.; Vallette, F.; Constantin, B.; et al. Store-Operated Calcium Channels Control Proliferation and Self-Renewal of Cancer Stem Cells from Glioblastoma. Cancers 2021, 13, 3428. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Kim, Y.J.; Jung, H.J. Discovery of a New CaMKII-Targeted Synthetic Lethal Therapy against Glioblastoma Stem-like Cells. Cancers 2022, 14, 1315. [Google Scholar] [CrossRef]
- Han, J.M.; Jung, H.J. Synergistic Anticancer Effect of a Combination of Berbamine and Arcyriaflavin A against Glioblastoma Stem-like Cells. Molecules 2022, 27, 7968. [Google Scholar] [CrossRef]
- Johnson, A.L.; Laterra, J.; Lopez-Bertoni, H. Exploring glioblastoma stem cell heterogeneity: Immune microenvironment modulation and therapeutic opportunities. Front. Oncol. 2022, 12, 995498. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.M.; Faust Akl, C.; Wheeler, M.A.; Chiocca, E.A.; Reardon, D.A.; Quintana, F.J. Glial and myeloid heterogeneity in the brain tumour microenvironment. Nat. Rev. Cancer 2021, 21, 786–802. [Google Scholar] [CrossRef] [PubMed]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in glioblastoma research: Focus on the tumor microenvironment. Trends Cancer 2023, 9, 9–27, Erratum in Trends Cancer 2023, 9, 692. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Osswald, M.; Ratliff, M.; Dogan, H.; Xie, R.; Weil, S.; Hoffmann, D.C.; Kurz, F.T.; Kessler, T.; Heiland, S.; et al. Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma. Nat. Commun. 2021, 12, 1014. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Dai, Z.; Zhao, X.; Wang, G.; Lai, R. Anticarin β Inhibits Human Glioma Progression by Suppressing Cancer Stemness via STAT3. Front Oncol. 2021, 11, 715673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Yong, V.W. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012, 33, 312–319. [Google Scholar] [CrossRef]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neurooncol 2012, 14, 958–978. [Google Scholar] [CrossRef]
- Sielska, M.; Przanowski, P.; Wylot, B.; Gabrusiewicz, K.; Maleszewska, M.; Kijewska, M.; Zawadzka, M.; Kucharska, J.; Vinnakota, K.; Kettenmann, H.; et al. Distinct roles of CSF family cytokines in macrophage infiltration and activation in glioma progression and injury response. J. Pathol. 2013, 230, 310–321. [Google Scholar] [CrossRef]
- Giannopoulou, A.I.; Kanakoglou, D.S.; Papavassiliou, A.G.; Piperi, C. Insights into the multi-faceted role of Pioneer transcription factors in glioma formation and progression with targeting options. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188801. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Barthel, L.; Hadamitzky, M.; Dammann, P.; Schedlowski, M.; Sure, U.; Thakur, B.K.; Hetze, S. Glioma: Molecular signature and crossroads with tumor microenvironment. Cancer Metastasis Rev. 2022, 41, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef]
- Badie, B.; Schartner, J.; Klaver, J.; Vorpahl, J. In vitro modulation of microglia motility by glioma cells is mediated by hepatocyte growth factor/scatter factor. Neurosurgery 1999, 44, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, J.; Yao, X.; Wu, S.; Tian, W.; Gan, C.; Wan, X.; You, C.; Hu, F.; Zhang, S.; et al. Programmed Cell Death 10 Mediated CXCL2-CXCR2 Signaling in Regulating Tumor-Associated Microglia/Macrophages Recruitment in Glioblastoma. Front. Immunol. 2021, 12, 637053. [Google Scholar] [CrossRef] [PubMed]
- Tabu, K.; Taga, T. Cancer ego-system in glioma: An iron-replenishing niche network systemically self-organized by cancer stem cells. Inflamm. Regen. 2022, 42, 54. [Google Scholar] [CrossRef]
- Shivtiel, S.; Kollet, O.; Lapid, K.; Schajnovitz, A.; Goichberg, P.; Kalinkovich, A.; Shezen, E.; Tesio, M.; Netzer, N.; Petit, I.; et al. CD45 regulates retention, motility, and numbers of hematopoietic progenitors, and affects osteoclast remodeling of metaphyseal trabecules. J. Exp. Med. 2008, 205, 2381–2395. [Google Scholar] [CrossRef]
- Mosteiro, A.; Pedrosa, L.; Ferrés, A.; Diao, D.; Sierra, À.; González, J.J. The Vascular Microenvironment in Glioblastoma: A Comprehensive Review. Biomedicines 2022, 10, 1285. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Hitomi, M.; Venere, M.; Flavahan, W.A.; Yang, K.; Kim, Y.; Minhas, S.; Rich, J.N.; Hjelmeland, A.B. Glioma stem cell maintenance: The role of the microenvironment. Curr. Pharm. Des. 2011, 17, 2386–2401. [Google Scholar] [CrossRef]
- Silver, A.; Feier, D.; Ghosh, T.; Rahman, M.; Huang, J.; Sarkisian, M.R.; Deleyrolle, L.P. Heterogeneity of glioblastoma stem cells in the context of the immune microenvironment and geospatial organization. Front. Oncol. 2022, 12, 1022716. [Google Scholar] [CrossRef] [PubMed]
- Taiarol, L.; Formicola, B.; Fagioli, S.; Sierri, G.; D’Aloia, A.; Kravicz, M.; Renda, A.; Viale, F.; Dal Magro, R.; Ceriani, M.; et al. The 3.0 Cell Communication: New Insights in the Usefulness of Tunneling Nanotubes for Glioblastoma Treatment. Cancers 2021, 13, 4001. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, D.L.; Simone, L.; Svelto, M.; Pisani, F. Intercellular crosstalk mediated by tunneling nanotubes between central nervous system cells. What we need to advance. Front. Physiol. 2023, 14, 1214210. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito, S.; D’Amico, D.; Eugenin, E. Novel approaches for glioblastoma treatment: Focus on tumor heterogeneity, treatment resistance, and computational tools. Cancer Rep. 2019, 2, e1220. [Google Scholar] [CrossRef] [PubMed]
- Simone, L.; Capobianco, D.L.; Di Palma, F.; Binda, E.; Legnani, F.G.; Vescovi, A.L.; Svelto, M.; Pisani, F. GFAP serves as a structural element of tunneling nanotubes between glioblastoma cells and could play a role in the intercellular transfer of mitochondria. Front. Cell Dev. Biol. 2023, 11, 1221671. [Google Scholar] [CrossRef] [PubMed]
- Pinto, G.; Brou, C.; Zurzolo, C. Tunneling nanotubes: The fuel of tumor progression? Trends Cancer 2020, 6, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Salaud, C.; Alvarez-Arenas, A.; Geraldo, F.; Belmonte-Beitia, J.; Calvo, G.F.; Gratas, C.; Pecqueur, C.; Garnier, D.; Pérez-Garcià, V.; Vallette, F.M.; et al. Mitochondria transfer from tumor-activated stromal cells (TASC) to primary Glioblastoma cells. Biochem. Biophys. Res. Commun. 2020, 533, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Lokumcu, T.; Iskar, M.; Schneider, M.; Helm, D.; Klinke, G.; Schlicker, L.; Bethke, F.; Müller, G.; Richter, K.; Poschet, G.; et al. Proteomic, Metabolomic, and Fatty Acid Profiling of Small Extracellular Vesicles from Glioblastoma Stem-Like Cells and Their Role in Tumor Heterogeneity. ACS Nano. 2024, 18, 2500–2519. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, K.; Zong, H.; Shang, M.; Li, K.; He, X. Exosomal communication in glioma—A review. J. BUON. 2016, 21, 1368–1373. [Google Scholar]
- Davidson, C.L.; Vengoji, R.; Jain, M.; Batra, S.K.; Shonka, N. Biological, diagnostic and therapeutic implications of exosomes in glioma. Cancer Lett. 2024, 582, 216592. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sui, R.; Piao, H. Tumor-derived small extracellular vesicles: Potential roles and mechanism in glioma. J. Nanobiotechnology 2022, 20, 383. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Qiu, W.; Wang, C.; Qi, Y.; Li, B.; Wang, S.; Zhao, R.; Cheng, B.; Han, X.; Du, H.; et al. Neuronal Activity Promotes Glioma Progression by Inducing Proneural-to-Mesenchymal Transition in Glioma Stem Cells. Cancer Res. 2024, 84, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Peñaranda-Fajardo, N.M.; Meijer, C.; Liang, Y.; Dijkstra, B.M.; Aguirre-Gamboa, R.; den Dunnen, W.F.A.; Kruyt, F.A.E. ER stress and UPR activation in glioblastoma: Identification of a noncanonical PERK mechanism regulating GBM stem cells through SOX2 modulation. Cell Death Dis. 2019, 10, 690. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.A.; Maghe, C.; Gavard, J. Lysosomes in glioblastoma: Pump up the volume. Cell Cycle. 2020, 19, 2094–2104. [Google Scholar] [CrossRef]
- Maghe, C.; Trillet, K.; André-Grégoire, G.; Kerhervé, M.; Merlet, L.; Jacobs, K.A.; Schauer, K.; Bidère, N.; Gavard, J. The paracaspase MALT1 controls cholesterol homeostasis in glioblastoma stem-like cells through lysosome proteome shaping. Cell Rep. 2024, 43, 113631. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wei, M.; Li, Y.; Ma, Q.; Zhang, H. Research Progress on the Regulation Mechanism of Key Signal Pathways Affecting the Prognosis of Glioma. Front. Mol. Neurosci. 2022, 15, 910543. [Google Scholar] [CrossRef] [PubMed]
- Nasrolahi, A.; Azizidoost, S.; Radoszkiewicz, K.; Najafi, S.; Ghaedrahmati, F.; Anbiyaee, O.; Khoshnam, S.E.; Farzaneh, M.; Uddin, S. Signaling pathways governing glioma cancer stem cells behavior. Cell Signal. 2023, 101, 110493. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Diao, S.; Wang, Q.; Zhu, C.; Sun, X.; Yin, B.; Zhang, X.; Meng, X.; Wang, B. IL-17A promotes cell migration and invasion of glioblastoma cells via activation of PI 3K/AKT signalling pathway. J. Cell Mol. Med. 2019, 23, 357–369. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440. [Google Scholar] [CrossRef]
- Tuncel, G.; Kalkan, R. Receptor tyrosine kinase-Ras-PI 3 kinase-Akt signaling network in glioblastoma multiforme. Med. Oncol 2018, 35, 122. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, S.; Lin, Y.; Cao, X.; Liu, J. TGF-β promotes glioma cell growth via activating Nodal expression through Smad and ERK1/2 pathways. Biochem. Biophys. Res. Commun. 2014, 443, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- García-Gómez, P.; Golán, I.; SDadras, M.; Mezheyeuski, A.; Bellomo, C.; Tzavlaki, K.; Morén, A.; Carreras-Puigvert, J.; Caja, L. NOX4 regulates TGFβ-induced proliferation and self-renewal in glioblastoma stem cells. Mol. Oncol. 2022, 16, 1891–1912. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.Z.; Liu, Q.; Wang, X.G.; Xu, G.Z.; Zhao, T.; Lou, M.Q. Hypoxia-induced USP22-BMI1 axis promotes the stemness and malignancy of glioma stem cells via regulation of HIF-1α. Life Sci. 2020, 247, 117438. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Gong, S.; Liao, B.; Pan, J.; Wang, J.; Zou, D.; Zhao, L.; Xiong, S.; Deng, Y.; Yan, Q.; et al. HIF1α/HIF2α induces glioma cell dedifferentiation into cancer stem cells through Sox2 under hypoxic conditions. J. Cancer. 2022, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Masiero, M.; Banham, A.H.; Harris, A.L. The notch ligand JAGGED1 as a target for anti-tumor therapy. Front. Oncol. 2014, 4, 254. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer. 2015, 14, 28. [Google Scholar] [CrossRef]
- Yi, L.; Zhou, X.; Li, T.; Liu, P.; Hai, L.; Tong, L.; Ma, H.; Tao, Z.; Xie, Y.; Zhang, C.; et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J. Exp. Clin. Cancer Res. 2019, 38, 339. [Google Scholar] [CrossRef]
- Hung, H.C.; Liu, C.C.; Chuang, J.Y.; Su, C.L.; Gean, P.W. Inhibition of Sonic Hedgehog Signaling Suppresses Glioma Stem-Like Cells Likely Through Inducing Autophagic Cell Death. Front. Oncol. 2020, 10, 1233. [Google Scholar] [CrossRef]
- Agrawal, K.; Chauhan, S.; Kumar, D. Expression analysis and regulation of GLI and its correlation with stemness and metabolic alteration in human brain tumor. 3 Biotech 2023, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luan, Y.; Ma, K.; Zhang, Z.; Liu, Y.; Chen, X. ISL1 Promotes Human Glioblastoma-Derived Stem Cells’ Self-Renewal by Activation of Sonic Hedgehog/GLI1 Function. Stem Cells Dev. 2022, 31, 258–268. [Google Scholar] [CrossRef]
- Wu, A.T.H.; Huang, H.S.; Wen, Y.T.; Lawal, B.; Mokgautsi, N.; Huynh, T.T.; Hsiao, M.; Wei, L. A Preclinical Investigation of GBM-N019 as a Potential Inhibitor of Glioblastoma via Exosomal mTOR/CDK6/STAT3 Signaling. Cells 2021, 10, 2391. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Vander Vorst, K.; Dreyer, C.A.; Konopelski, S.E.; Lee, H.; Ho, H.H.; Carraway, K.L., 3rd. Wnt/PCP Signaling Contribution to Carcinoma Collective Cell Migration and Metastasis. Cancer Res. 2019, 79, 1719–1729. [Google Scholar] [CrossRef]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef] [PubMed]
- Alkailani, M.I.; Aittaleb, M.; Tissir, F. WNT signaling at the intersection between neurogenesis and brain tumorigenesis. Front. Mol. Neurosci. 2022, 15, 1017568. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, L.V.; Tejeda-Muñoz, N.; De Robertis, E.M. Cell Biology of Canonical Wnt Signaling. Annu. Rev. Cell Dev. Biol. 2021, 37, 369–389. [Google Scholar] [CrossRef]
- Ghosh, N.; Hossain, U.; Mandal, A.; Sil, P.C. The Wnt signaling pathway: A potential therapeutic target against cancer. Ann. N. Y. Acad. Sci. 2019, 1443, 54–74. [Google Scholar] [CrossRef]
- He, L.; Zhou, H.; Zeng, Z.; Yao, H.; Jiang, W.; Qu, H. Wnt/β-catenin signaling cascade: A promising target for glioma therapy. J. Cell Physiol. 2019, 234, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Kafka, A.; Bačić, M.; Tomas, D.; Žarković, K.; Bukovac, A.; Njirić, N.; Mrak, G.; Krsnik, Ž.; Pećina-Šlaus, N. Different behaviour of DVL1, DVL2, DVL3 in astrocytoma malignancy grades and their association to TCF1 and LEF1 upregulation. J. Cell Mol. Med. 2019, 23, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Tomas, D.; Marković, L.; Okštajner, P.K.; Sukser, V.; Krušlin, B. Wnt signaling transcription factors TCF-1 and LEF-1 are upregulated in malignant astrocytic brain tumors. Histol. Histopathol. 2014, 29, 1557–1564. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Varošanec, A.M.; Marković, L.; Krsnik, Ž.; Njirić, N.; Mrak, G. Expression patterns of Wnt signaling component, secreted frizzled-related protein 3 in astrocytoma and glioblastoma. Mol. Med. Rep. 2016, 13, 4245–4251. [Google Scholar] [CrossRef] [PubMed]
- Kafka, A.; Tomas, D.; Lechpammer, M.; Gabud, T.; Pažanin, L.; Pećina-Šlaus, N. Expression Levels and Localizations of DVL3 and sFRP3 in Glioblastoma. Dis. Markers 2017, 2017, 9253495. [Google Scholar] [CrossRef] [PubMed]
- Kafka, A.; Karin-Kujundžić, V.; Šerman, L.; Bukovac, A.; Njirić, N.; Jakovčević, A.; Pećina-Šlaus, N. Hypermethylation of Secreted Frizzled Related Protein 1 gene promoter in different astrocytoma grades. Croat. Med. J. 2018, 59, 213–223. [Google Scholar] [CrossRef]
- Brlek, P.; Kafka, A.; Bukovac, A.; Pećina-Šlaus, N. Integrative cBioPortal Analysis Revealed Molecular Mechanisms That Regulate EGFR-PI3K-AKT-mTOR Pathway in Diffuse Gliomas of the Brain. Cancers 2021, 13, 3247. [Google Scholar] [CrossRef]
- Guan, R.; Zhang, X.; Guo, M. Glioblastoma stem cells and Wnt signaling pathway: Molecular mechanisms and therapeutic targets. Chin. Neurosurg. J. 2020, 6, 25. [Google Scholar] [CrossRef]
- Du, L.; Lee, J.H.; Jiang, H.; Wang, C.; Wang, S.; Zheng, Z.; Shao, F.; Xu, D.; Xia, Y.; Li, J.; et al. β-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J. Exp. Med. 2020, 217, e20191115. [Google Scholar] [CrossRef]
- Pu, P.; Zhang, Z.; Kang, C.; Jiang, R.; Jia, Z.; Wang, G.; Jiang, H. Downregulation of Wnt2 and beta-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2009, 16, 351–361. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. WNT signaling and cancer stemness. Essays Biochem. 2022, 15, EBC20220016. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.T.; Lee, H.T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Suwala, A.K.; Hanaford, A.; Kahlert, U.D.; Maciaczyk, J. Clipping the Wings of Glioblastoma: Modulation of WNT as a Novel Therapeutic Strategy. J. Neuropathol. Exp. Neurol. 2016, 75, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Tompa, M.; Nagy, A.; Komoly, S.; Kalman, B. Wnt pathway markers in molecular subgroups of glioblastoma. Brain Res. 2019, 1718, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Hong, M.; Do, I.G.; Ha, S.Y.; Lee, D.; Suh, Y.L. Wnt5a, Ryk and Ror2 expression in glioblastoma subgroups. Pathol. Res. Pract. 2015, 211, 963–972. [Google Scholar] [CrossRef]
- Frumento, D.; Grossi, G.; Falesiedi, M.; Musumeci, F.; Carbone, A.; Schenone, S. Small Molecule Tyrosine Kinase Inhibitors (TKIs) for Glioblastoma Treatment. Int. J. Mol. Sci. 2024, 25, 1398. [Google Scholar] [CrossRef]
- Low, J.J.W.; Sulaiman, S.A.; Johdi, N.A.; Abu, N. Immunomodulatory effects of extracellular vesicles in glioblastoma. Front. Cell Dev. Biol. 2022, 10, 996805. [Google Scholar] [CrossRef]
- Yang, W.; Li, Y.; Gao, R.; Xiu, Z.; Sun, T. Mhc class I dysfunction of glioma stem cells escapes from ctl-mediated immune response Via activation of Wnt/Beta-catenin signaling pathway. Oncogene 2020, 39, 1098–1111. [Google Scholar] [CrossRef]
- Menna, G.; Manini, I.; Cesselli, D.; Skrap, M.; Olivi, A.; Ius, T.; Della Pepa, G.M. Immunoregulatory effects of glioma-associated stem cells on the glioblastoma peritumoral microenvironment: A differential PD-L1 expression from core to periphery? Neurosurg. Focus 2022, 52, E4. [Google Scholar] [CrossRef]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; De Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro Oncol. 2017, 19 (Suppl. S3), iii21. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022, 24, 1935–1949. [Google Scholar] [CrossRef] [PubMed]
- Porčnik, A.; Novak, M.; Breznik, B.; Majc, B.; Hrastar, B.; Šamec, N.; Zottel, A.; Jovčevska, I.; Vittori, M.; Rotter, A.; et al. TRIM28 Selective Nanobody Reduces Glioblastoma Stem Cell Invasion. Molecules 2021, 26, 5141. [Google Scholar] [CrossRef] [PubMed]
- Zottel, A.; Jovčevska, I.; Šamec, N.; Mlakar, J.; Šribar, J.; Križaj, I.; Skoblar Vidmar, M.; Komel, R. Anti-vimentin, anti-TUFM, anti-NAP1L1 and anti-DPYSL2 nanobodies display cytotoxic effect and reduce glioblastoma cell migration. Ther. Adv. Med. Oncol. 2020, 12, 1758835920915302. [Google Scholar] [CrossRef] [PubMed]
- Rehman, F.U.; Liu, Y.; Zheng, M.; Shi, B. Exosomes based strategies for brain drug delivery. Biomaterials 2023, 293, 121949. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.F.; Hossain, A.; Momin, E.N.; Hasan, I.; Singh, S.; Adachi, S.; Gumin, J.; Ledbetter, D.; Yang, J.; Long, L.; et al. Tumor-specific polycistronic miRNA delivered by engineered exosomes for the treatment of glioblastoma. Neuro Oncol. 2024, 26, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, J.; Liu, X.; Liu, L.; Huang, S.; Wu, A.; Xu, Z.; Zhang, X.; Li, Z.; Ni, F.; et al. Glioma stem cell-derived exosomes induce the transformation of astrocytes via the miR-3065-5p/DLG2 signaling axis. Glia 2024, 72, 857–871. [Google Scholar] [CrossRef]
- Bassoy, E.Y.; Kasahara, A.; Chiusolo, V.; Jacquemin, G.; Boydell, E.; Zamorano, S.; Riccadonna, C.; Pellegatta, S.; Hulo, N.; Dutoit, V.; et al. ER–mitochondria contacts control surface glycan expression and sensitivity to killer lymphocytes in glioma stem-like cells. EMBO J. 2017, 36, 1493–1512. [Google Scholar] [CrossRef]
- Turos-Cabal, M.; Sánchez-Sánchez, A.M.; Puente-Moncada, N.; Herrera, F.; Rodriguez-Blanco, J.; Antolin, I.; Alvarez-Vega, M.A.; Rodríguez, C.; Martín, V. Endoplasmic reticulum regulation of glucose metabolism in glioma stem cells. Int. J. Oncol. 2024, 64, 1. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Signaling Pathway | Gene for Signaling Component | Frequency of Gene Alteration * |
|---|---|---|
| RTK/RAS/PI3K | EGFR, | 29.04% |
| PDGFRA, | 8.89% | |
| MET, | 4.15% | |
| FGFR2, | 2.27% | |
| FGFR3, | 2.81% | |
| PTEN | 41.82% | |
| p53 | TP53, | 43.47% |
| MDM1, | 2.39% | |
| MDM2, | 5.84% | |
| MDM4, | 4.46% | |
| CDKN2A | 33.02% | |
| RB | RB1, | 11,27% |
| CDK4, | 10.78% | |
| CDK6 | 2.68% | |
| NOTCH | NOTCH1 (NICD), | 5.87% |
| NOTCH2 | 2.47% | |
| NOTCH3 | 3.12% | |
| CXCR4 | 0.54% | |
| WNT | CTNNB1, | 0.93% |
| LEF1, | 0.54% | |
| TCF1 (HNF1A) | 1.4% | |
| DVL1, | 1.74% | |
| DVL2, | 1.63% | |
| DVL3, | 2.39% | |
| SFRP1, | 0.33% | |
| SFRP3 (FRZB), | 0.54% | |
| GSK3β | 0.93% | |
| TERT | 25.62% | |
| Hypoxia | HIF1A, | 1.2% |
| HIF2A (EPAS1) | 0.72% | |
| SHH | GLI2, | 0.92% |
| GLI3, | 5.74% | |
| ISL1 | 0.65% | |
| TGF-β | PDGFB, | 1.41% |
| NF-κB (RELA,NFKB), | 0.65% | |
| NODAL, | 0.82% | |
| SMAD2, | 0.76% | |
| SMAD3, | 0.82% | |
| SOX4, | 0.82% | |
| SOX2, | 4.12% | |
| LIF, | 0.6% | |
| BMP7 | 0.6% | |
| JAK-STAT | STAT3 | 1.05% |
| AKT | AKT1, | 1.27% |
| AKT2, | 1.44% | |
| AKT3 | 1.27% |
| Signaling Component | Signaling Inhibitor | Phase/Identification Number/Status |
|---|---|---|
| EGFR | Epitinib | I/NCT03231501/unknown status |
| BDTX-1535 | I-II/NCT05256290/recruiting | |
| WEE1 | Adavosertib (AZD1775) | 0-I/NCT02207010/completed |
| I/NCT01849146/active, not recruiting | ||
| ALK, IGFR1, FAK | Ceritinib (LKD378) | 0-I/NCT02605746/completed |
| TRK, ALK, ROS1 | Entrectinib (Rxdx-101) | I-II/NCT02650401/active, not recruiting |
| CXCR4, MMP2 and MMP9 | USL311 | I-II/NCT02765165/terminated |
| 0-I/NCT03526822/recruiting | ||
| VEGFR, PDGFR, FGFR, Src | Ponatinib | II/NCT02478164/completed |
| VXM01 | I-II/NCT03750071/active, not recruiting | |
| NEO212 | I-II/NCT06047379/recruiting | |
| Axitinib | II/NCT01562197/completed | |
| Bevacizumab and BKM120 | I-II/NCT01349660/completed with results | |
| II/NCT01743950/recruiting | ||
| I-II/NCT06011109/recruiting | ||
| Erdafitinib | II/NCT05859334/recruiting | |
| VEGFR, TIE-2, PDGFR, FGFR, KIT, RET, RAF | Regorafenib | II/NCT02926222/completed |
| Anlotinib | I-II/NCT04004975/unknown status | |
| II/NCT04547855 unknown status | ||
| Src, VEGFR, c-MET | Dasatinib | I-II/NCT00892177/completed |
| I/NCT01744652/completed | ||
| I/NCT05432518/recruiting | ||
| APL-101 | II/NCT03175224/recruiting | |
| BTK, Bruton’s tyrosine Kinase | Ibrutinib | I/NCT05106296/recruiting |
| STAT3 | WP1066 | I/NCT01904123/completed |
| II/NCT05879250/not yet recruiting | ||
| AKT | Nelfinavir | I/NCT00694837/completed |
| mTOR | RMC-5552 | NCT05557292/recruiting |
| GSK3beta | DSP-0390 | I/NCT05023551/active not recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pećina-Šlaus, N.; Hrašćan, R. Glioma Stem Cells—Features for New Therapy Design. Cancers 2024, 16, 1557. https://doi.org/10.3390/cancers16081557
Pećina-Šlaus N, Hrašćan R. Glioma Stem Cells—Features for New Therapy Design. Cancers. 2024; 16(8):1557. https://doi.org/10.3390/cancers16081557
Chicago/Turabian StylePećina-Šlaus, Nives, and Reno Hrašćan. 2024. "Glioma Stem Cells—Features for New Therapy Design" Cancers 16, no. 8: 1557. https://doi.org/10.3390/cancers16081557
APA StylePećina-Šlaus, N., & Hrašćan, R. (2024). Glioma Stem Cells—Features for New Therapy Design. Cancers, 16(8), 1557. https://doi.org/10.3390/cancers16081557