
Chromatin Profiles Are Prognostic of Clinical Response to Bortezomib-Containing Chemotherapy in Pediatric Acute Myeloid Leukemia: Results from the COG AAML1031 Trial

 , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Population
2.2. RPPA Methodology
2.3. ATAC-seq
2.4. Cell Lines and shRNA Knockdown Cells
2.5. RNA-seq
2.6. Statistical Analysis
3. Results
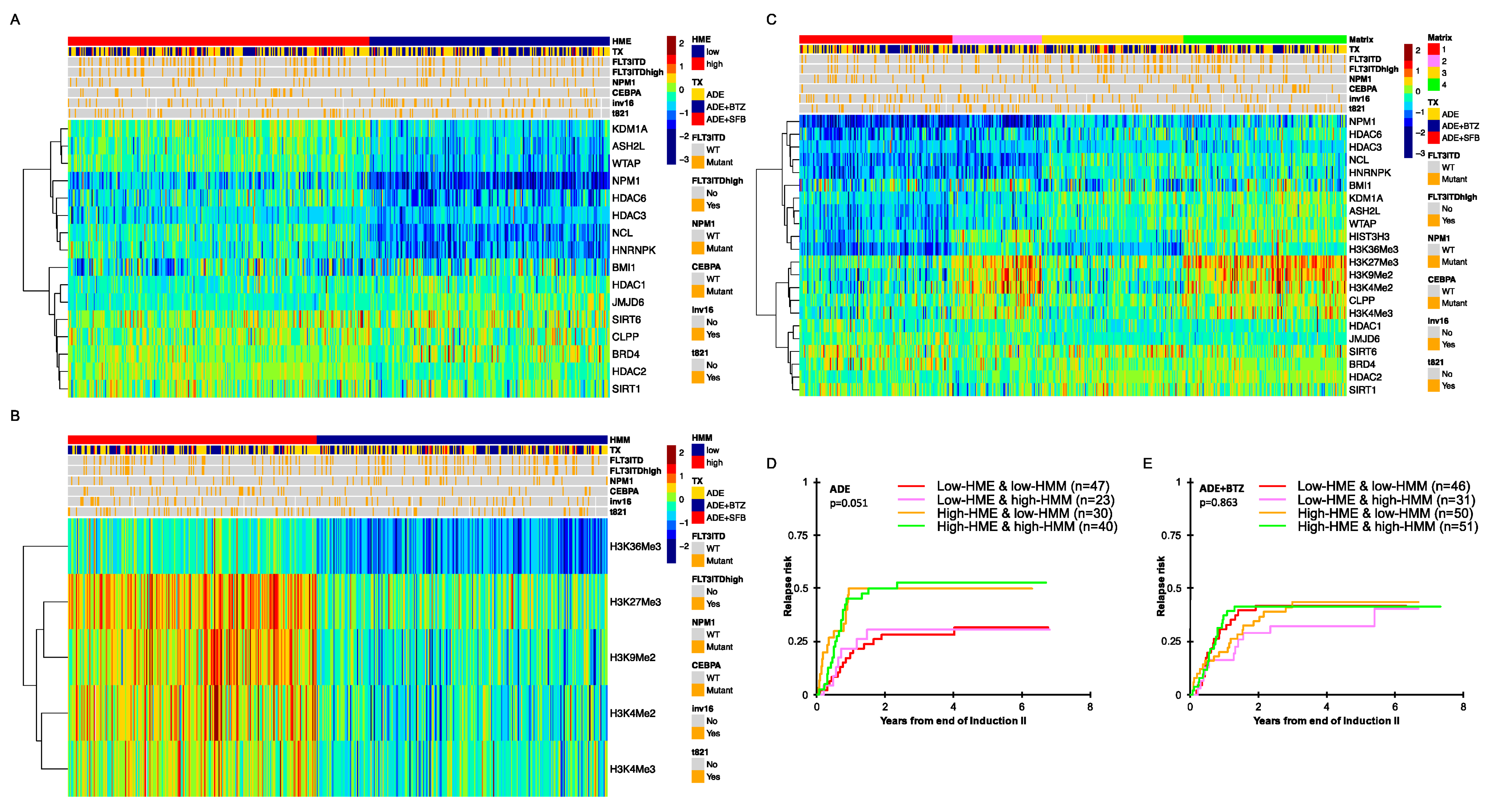
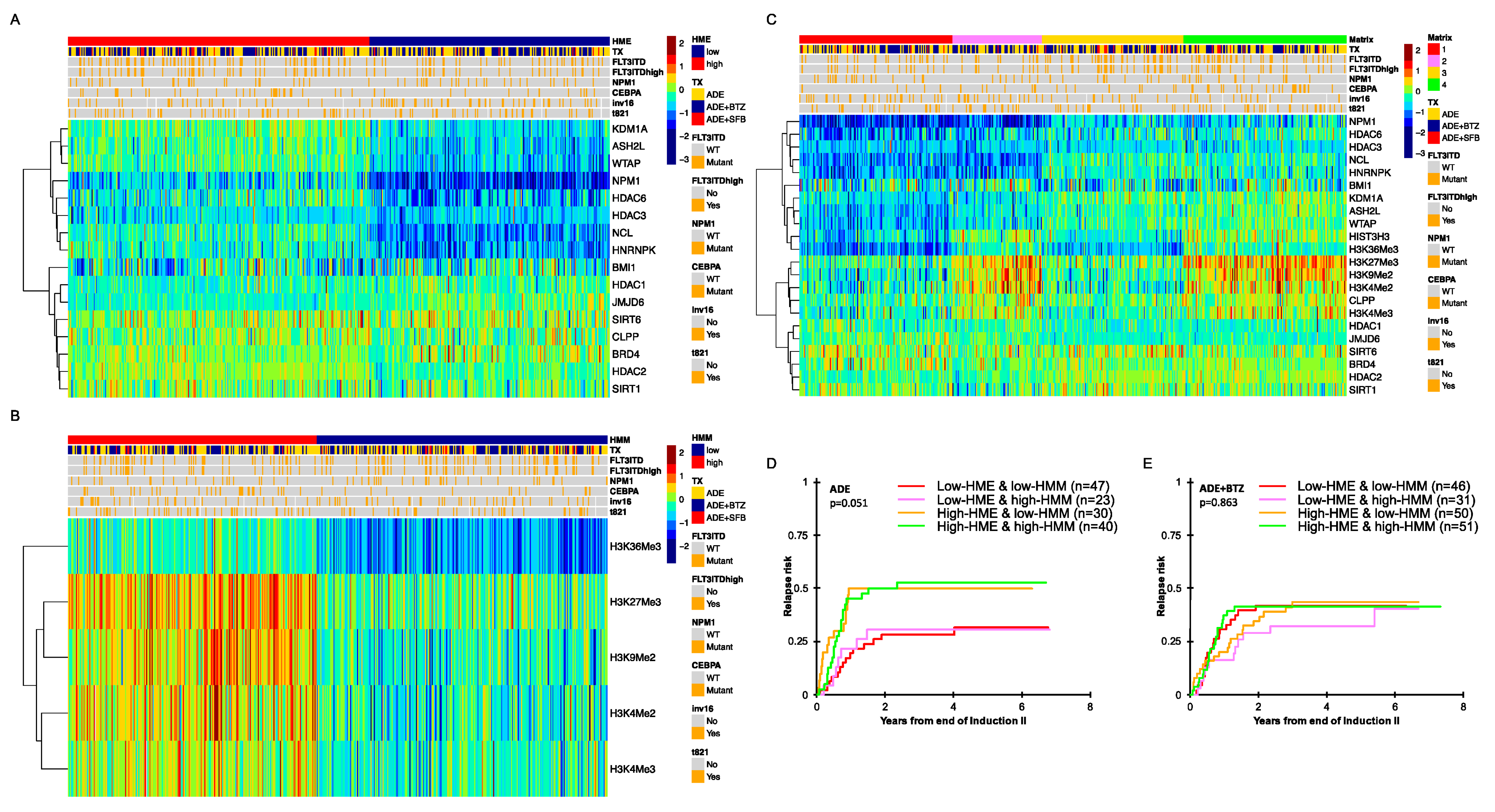
3.1. Histone- and Chromatin-Modifying Protein Expressions Are Heterogeneous among De Novo Pediatric AML
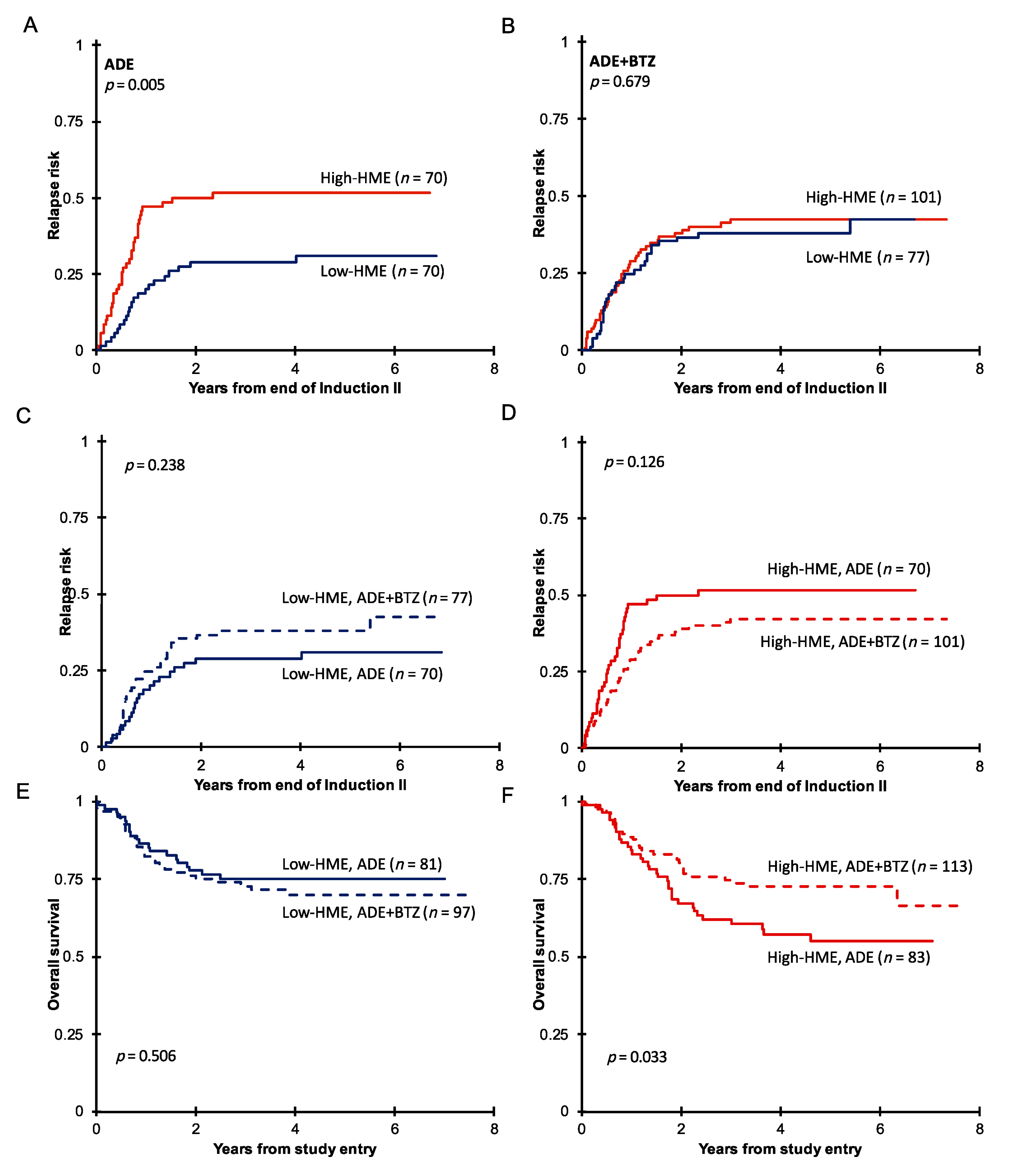
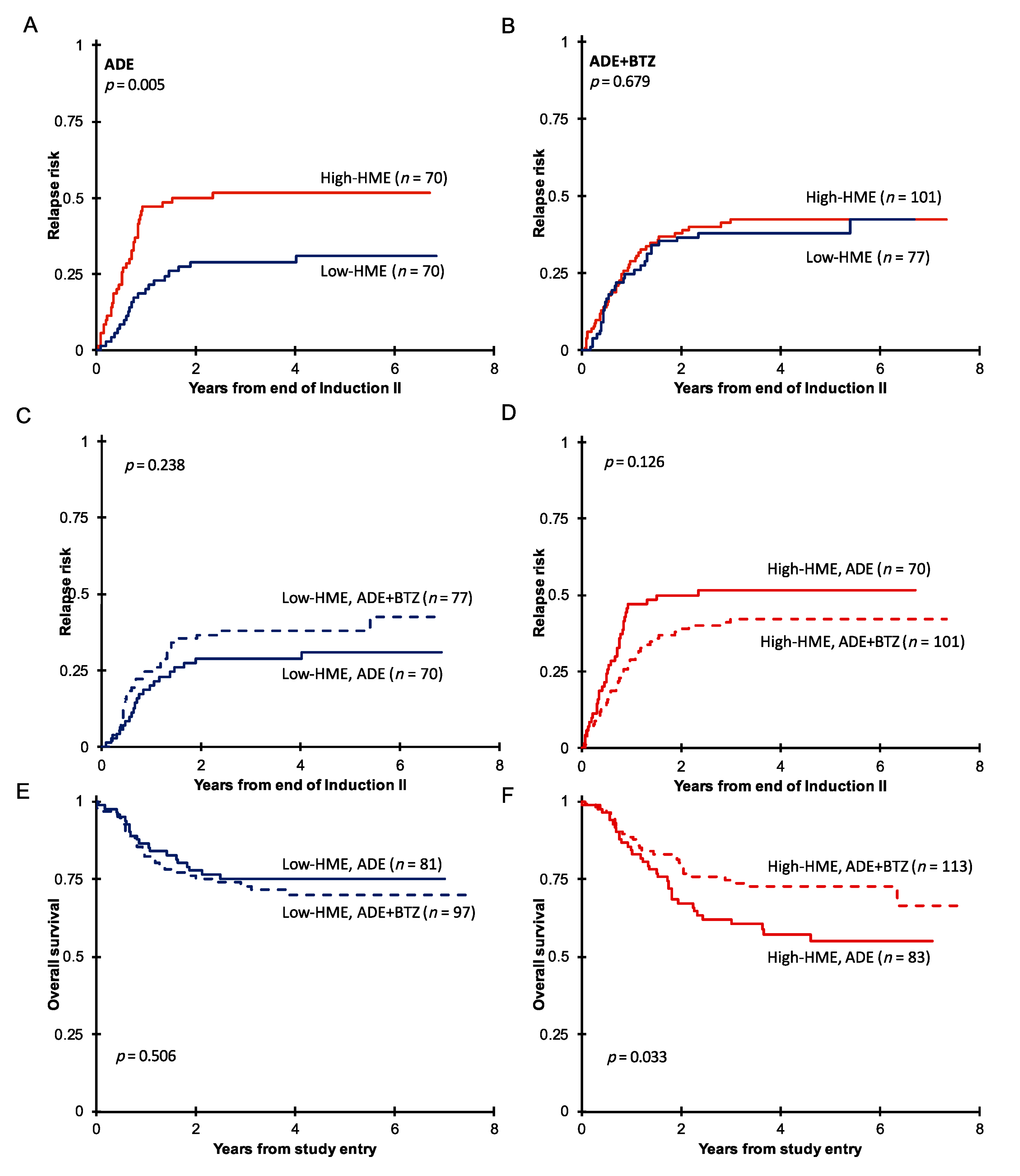
3.2. Pediatric AML with High HME Levels Benefitted from Bortezomib-Containing Chemotherapy
3.3. High-HME Proteomic Profile Is an Independent Adverse Prognostic Factor for Relapse in ADE-Treated De Novo Pediatric AML
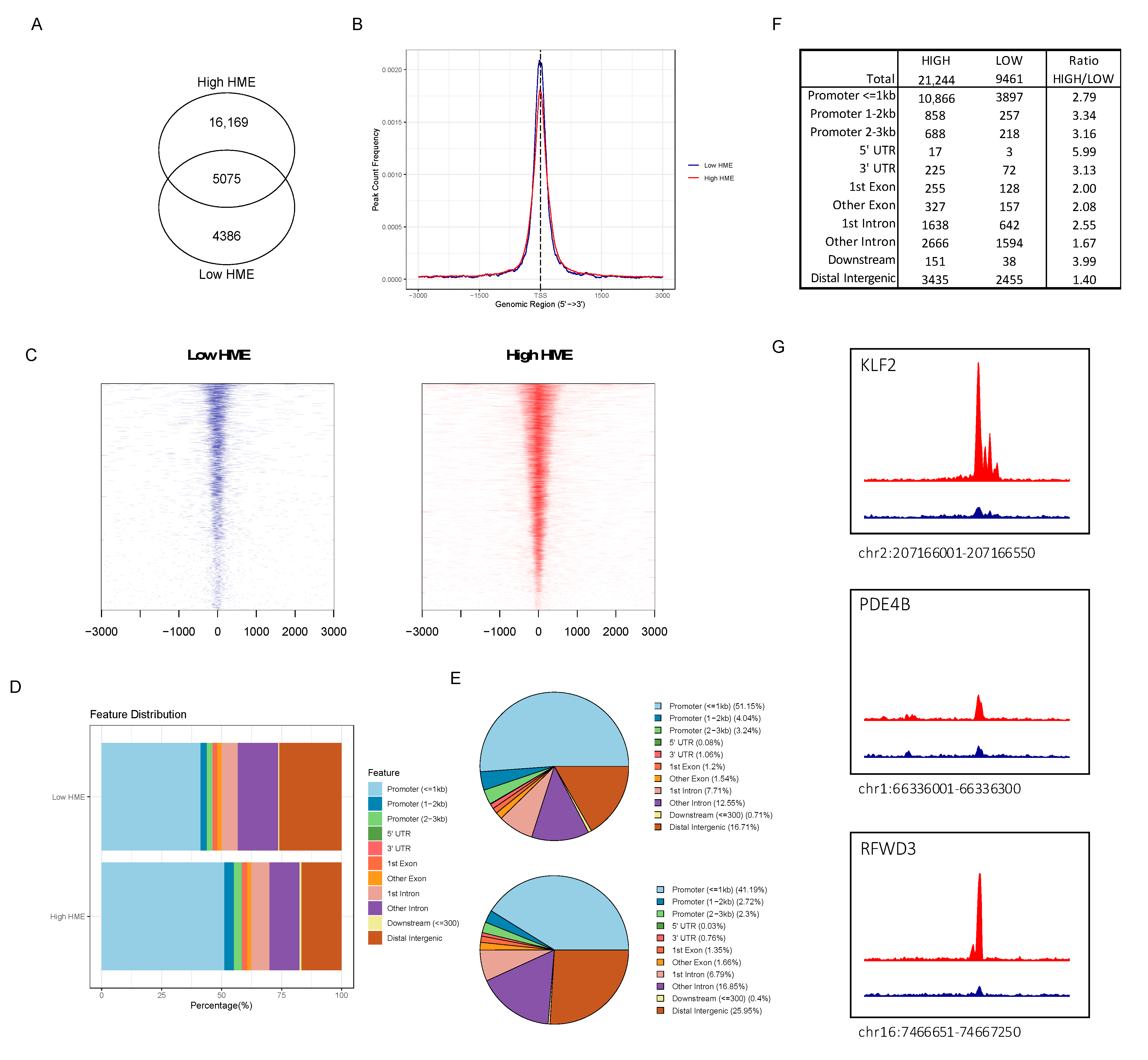
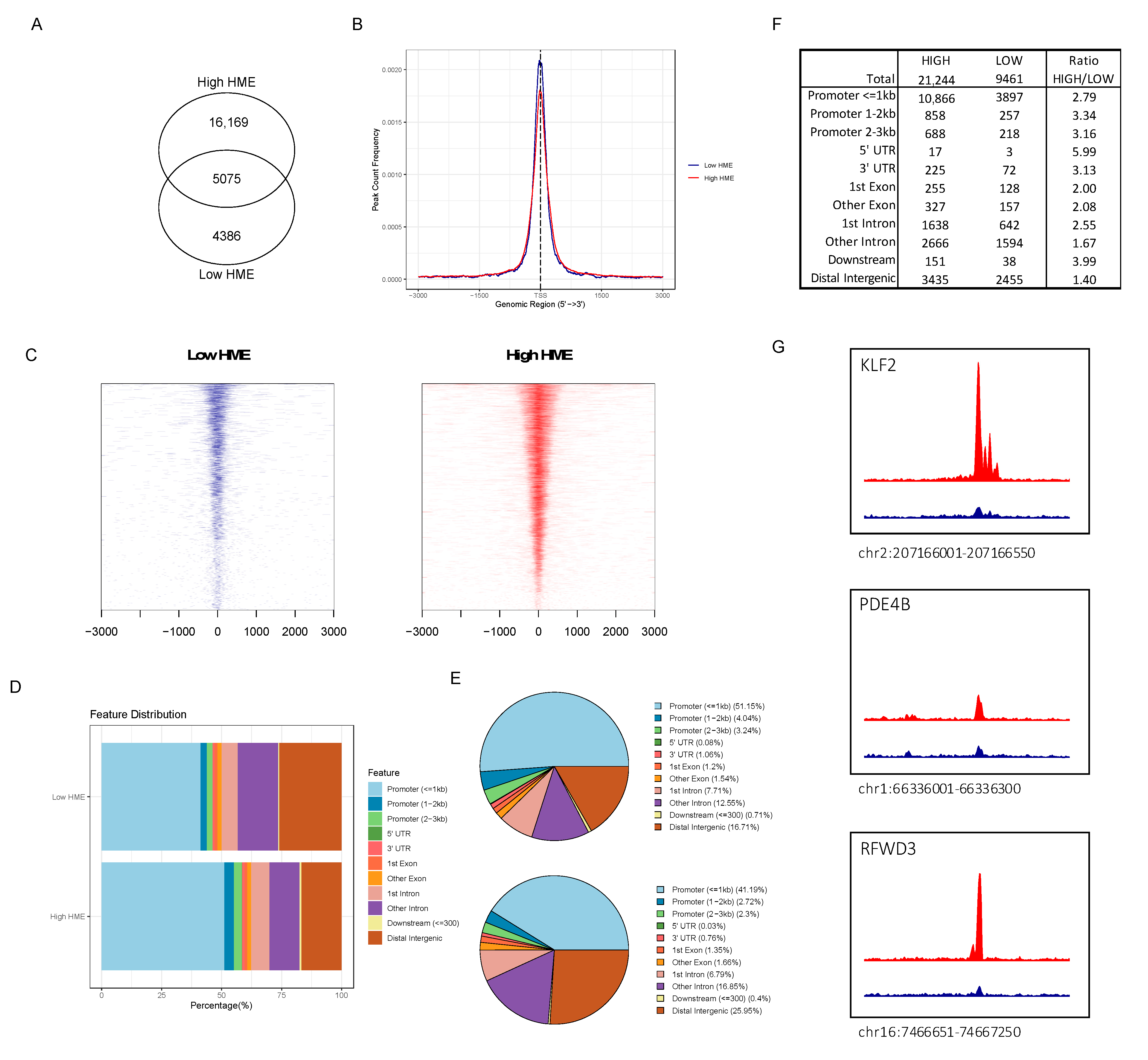
3.4. ATAC-seq Reveals Higher Chromatin Accessibility in Patients with More Activated HME
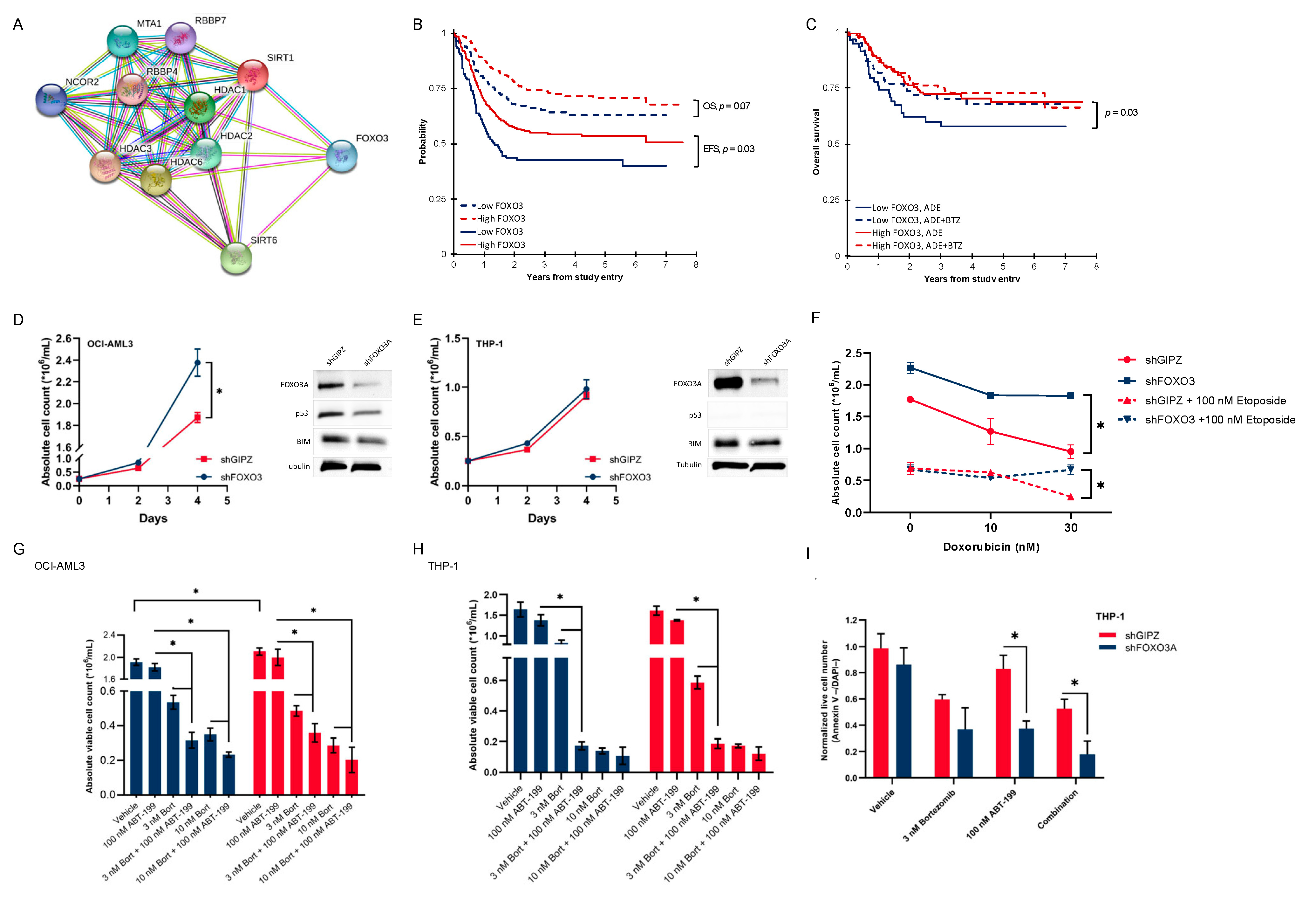
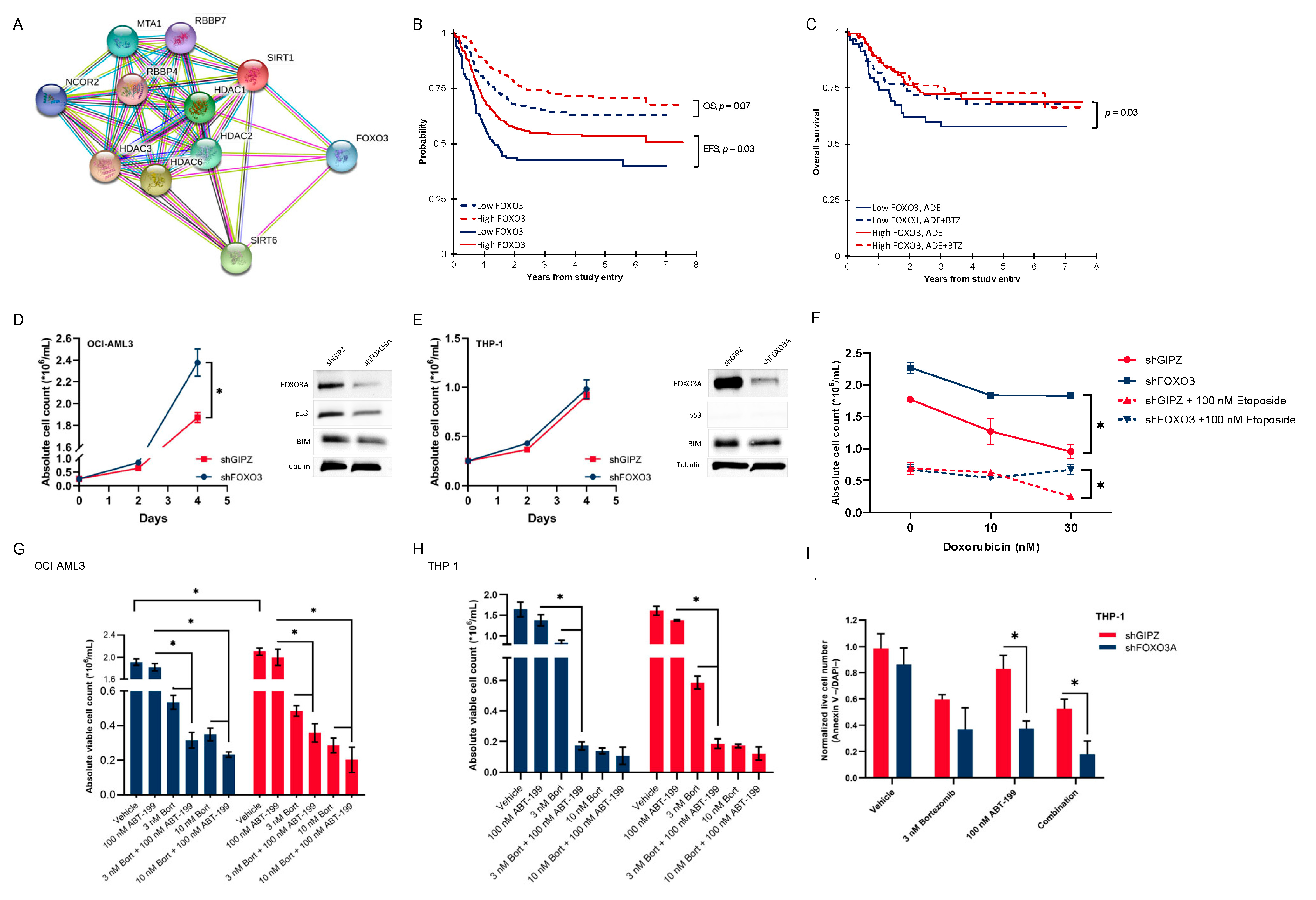
3.5. Activated Histone Deacetylase Proteins Associated with Loss of Transcription Factor FOXO3
3.6. Pediatric AML Patients with Low FOXO3 Levels Are Potential Candidates for Bortezomib Addition
3.7. Low-FOXO3 Cells Show Resistance to Doxorubicin and Etoposide In Vitro
3.8. In Vitro Proteasome and bcl-2 Inhibition Are More than Additive, and This Effect Is Dependent on the FOXO3 Protein Expression
3.9. FOXO3 Negatively Correlates with the Vimentin Expression on the mRNA and Protein Levels
3.10. Characterization of an Open Chromatin Signature for High-HME Patients That Responded to Bortezomib-Containing Chemotherapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Additional Information
Appendix A.1.1. Patient Population
Appendix A.1.2. Sample Processing
Appendix A.1.3. RPPA Methodology
Appendix A.1.4. RPPA Quality Control
Appendix A.1.5. shRNA Knockdown Cells
Appendix A.1.6. Cell Treatment and Cytotoxicity
Appendix A.1.7. ATAC Library Preparation and Sequencing
Appendix A.1.8. ATAC-seq Data Analysis
Appendix A.1.9. Characterization of the Open Chromatin Signature for High-HME Patients That Responded to Bortezomib-Containing Chemotherapy
- -
- DA regions that are associated with the same outcome regardless of the treatment include the following:
- ○
- HIGH-ADEB-RESP vs. HIGH-ADE-RESP.
- -
- DA regions that are associated with the same treatment but with different outcomes include the following:
- ○
- HIGH-ADEB-RESP vs. HIGH-ADEB-NONRESP;
- ○
- LOW-ADEB-NONRESP vs. LOW-ADEB-RESP.
References
- de Rooij, J.; Zwaan, C.; van den Heuvel-Eibrink, M. Pediatric AML: From Biology to Clinical Management. J. Clin. Med. 2015, 4, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2019, 24, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.J.M.; De Moerloose, B.; Dworzak, M.; Gibson, B.E.S.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [PubMed]
- Hoff, F.W.; Hu, C.W.; Qiu, Y.; Ligeralde, A.; Yoo, S.Y.; Mahmud, H.; De Bont, E.S.J.M.; Qutub, A.A.; Horton, T.M.; Kornblau, S.M. Recognition of recurrent protein expression patterns in pediatric acute myeloid leukemia identified new therapeutic targets. Mol. Cancer Res. 2018, 16, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, A.D.; de Bont, E.; Kornblau, S.M. Targeted therapy in acute myeloid leukemia: Current status and new insights from a proteomic perspective. Expert Rev. Proteom. 2020, 17, 1–10. [Google Scholar] [CrossRef]
- McCann, T.S.; Tansey, W.P. Functions of the Proteasome on Chromatin. Biomolecules 2014, 4, 1026–1044. [Google Scholar] [CrossRef] [PubMed]
- Catic, A.; Suh, C.Y.; Hill, C.T.; Daheron, L.; Henkel, T.; Orford, K.W.; Dombkowski, D.M.; Liu, T.; Liu, X.S.; Scadden, D.T. Genome-wide map of nuclear protein degradation shows NCoR1 turnover as a key to mitochondrial gene regulation. Cell 2013, 155, 1380–1395. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Ezhkova, E.; Li, B.; Pattenden, S.G.; Tansey, W.P.; Workman, J.L. The proteasome regulatory particle alters the SAGA coactivator to enhance its interactions with transcriptional activators. Cell 2005, 123, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kwak, J.; Kim, M.; Lee, D. Separation of a functional deubiquitylating module from the SAGA complex by the proteasome regulatory particle. Nat. Commun. 2013, 4, 2641. [Google Scholar] [CrossRef]
- Kinyamu, H.K.; Bennett, B.D.; Bushel, P.R.; Archer, T.K. Proteasome inhibition creates a chromatin landscape favorable to RNA Pol II processivity. J. Biol. Chem. 2020, 295, 1271–1287. [Google Scholar] [CrossRef]
- Leshchenko, V.V.; Kuo, P.Y.; Jiang, Z.; Weniger, M.A.; Overbey, J.; Dunleavy, K.; Wilson, W.H.; Wiestner, A.; Parekh, S. Harnessing Noxa demethylation to overcome Bortezomib resistance in mantle cell lymphoma. Oncotarget 2015, 6, 27332. [Google Scholar] [CrossRef]
- Kikuchi, J.; Wada, T.; Shimizu, R.; Izumi, T.; Akutsu, M.; Mitsunaga, K.; Noborio-Hatano, K.; Nobuyoshi, M.; Ozawa, K.; Kano, Y.; et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood 2010, 116, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Horton, T.; Gannavarapu, A.; Blaney, S.; D’Argenio, D.; Plon, S.; Berg, S. Bortezomib interactions with chemotherapy agents in acute leukemia in vitro. Cancer Chemother. Pharmacol. 2006, 58, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Minderman, H.; Zhou, Y.; O’Loughlin, K.L.; Baer, M.R. Bortezomib activity and in vitro interactions with anthracyclines and cytarabine in acute myeloid leukemia cells are independent of multidrug resistance mechanisms and p53 status. Cancer Chemother. Pharmacol. 2007, 60, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Attar, E.C.; Johnson, J.L.; Amrein, P.C.; Lozanski, G.; Wadleigh, M.; DeAngelo, D.J.; Kolitz, J.E.; Powell, B.L.; Voorhees, P.; Wang, E.S.; et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J. Clin. Oncol. 2013, 31, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Attar, E.C.; De Angelo, D.J.; Supko, J.G.; D’Amato, F.; Zahrieh, D.; Sirulnik, A.; Wadleigh, M.; Ballen, K.K.; McAfee, S.; Miller, K.B.; et al. Phase I and pharmacokinetic study of bortezomib in combination with idarubicin and cytarabine in patients with acute myelogenous leukemia. Clin. Cancer Res. 2008, 14, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Horton, T.M.; Perentesis, J.P.; Gamis, A.S.; Alonzo, T.A.; Gerbing, R.B.; Ballard, J.; Adlard, K.; Howard, D.S.; Smith, F.O.; Jenkins, G.; et al. A Phase 2 study of bortezomib combined with either idarubicin/cytarabine or cytarabine/etoposide in children with relapsed, refractory or secondary acute myeloid leukemia: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 1754–1760. [Google Scholar] [CrossRef]
- Aplenc, R.; Meshinchi, S.; Sung, L.; Alonzo, T.; Choi, J.; Fisher, B.; Gerbing, R.; Hirsch, B.; Horton, T.M.; Kahwash, S.; et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: A report from the Children’s Oncology Group. Haematologica 2020, 105, 1879–1886. [Google Scholar] [CrossRef]
- Hoff, F.W.; van Dijk, A.D.; Qiu, Y.H.; Ruvolo, P.P.; Gerbing, R.B.; Leonti, A.R.; Jenkins, G.N.; Gamis, A.S.; Aplenc, R.; Kolb, E.A.; et al. Heat Shock Factor 1 (HSF1-pSer326) Predicts Response to Bortezomib-Containing Chemotherapy in Pediatric AML: A COG Study. Blood 2021, 137, 1050–1060. [Google Scholar] [CrossRef]
- van Dijk, A.D.; Hu, C.W.; de Bont, E.S.J.M.; Qiu, Y.; Hoff, F.W.; Yoo, S.Y.; Coombes, K.R.; Qutub, A.A.; Kornblau, S.M. Histone Modification Patterns Using RPPA-Based Profiling Predict Outcome in Acute Myeloid Leukemia Patients. Proteomics 2018, 18, 1700379. [Google Scholar] [CrossRef]
- Hoff, F.W.; Hu, C.W.; Qiu, Y.; Ligeralde, A.; Yoo, S.K.; Scheurer, M.E.; De Bont, E.S.J.M.; Qutub, A.A.; Kornblau, S.M.; Horton, T.M. Recurrent patterns of protein expression signatures in pediatric acute lymphoblastic leukemia: Recognition and therapeutic guidance. Mol. Cancer Res. 2018, 16, 1263–1274. [Google Scholar] [CrossRef]
- Tibes, R.; Qiu, Y.; Lu, Y.; Hennessy, B.; Andreeff, M.; Mills, G.B.; Kornblau, S.M. Reverse phase protein array: Validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 2006, 5, 2512–2521. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Tibes, R.; Qiu, Y.H.; Chen, W.; Kantarjian, H.M.; Andreeff, M.; Coombes, K.R.; Mills, G.B. Functional proteomic profiling of AML predicts response and survival. Blood 2009, 113, 154–164. [Google Scholar] [CrossRef]
- Smith, J.L.; Ries, R.E.; Santaguida, M.T.; Gekas, C.; Alonzo, T.A.; Gerbing, R.B.; Gamis, A.S.; Aplenc, R.; Kolb, E.A.; Hylkema, T.; et al. Comprehensive Transcriptome Profiling of Cryptic CBFA2T3-GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options—A COG and Target Pediatric AML Study. Blood 2018, 132 (Suppl. S1), 881. [Google Scholar] [CrossRef]
- Agresti, A. An Introduction to Categorical Data Analysis, 3rd ed.; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Kruskal, W.H.; Wallis, W.A. Use of Ranks in One-Criterion Variance Analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Kalbfleisch, J.D.; Prentice, R.L. The Statistical Analysis of Failure Time Data; Wiley: New York, NY, USA, 1980. [Google Scholar]
- Cox, D.R. Regression Models and Life-Tables. J. R. Stat. Soc. Ser. B (Methodol.) 1972, 34, 187–220. [Google Scholar] [CrossRef]
- Hu, C.W.; Kornblau, S.M.; Slater, J.H.; Qutub, A.A. Progeny Clustering: A Method to Identify Biological Phenotypes. Sci. Rep. 2015, 5, 12894. [Google Scholar] [CrossRef]
- Aplenc, R.; Meshinchi, S.; Sung, L.; Alonzo, T.A.; Pollard, J.; Gerbing, R.; Raimondi, S.C.; Hirsch, B.A.; Loken, M.R.; Winter, L.; et al. The Addition of Bortezomib to Standard Chemotherapy for Pediatric Acute Myeloid Leukemia Has Increased Toxicity without Therapeutic Benefit: A Report from the Children’s Oncology Group. Blood 2016, 128, 899. [Google Scholar] [CrossRef]
- Wang, F.; Chan, C.H.; Chen, K.; Guan, X.; Lin, H.K.; Tong, Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene 2012, 31, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.M.; Park, S.H.; Tsai, W.B.; Wang, S.Y.; Ikeda, M.A.; Berek, J.S.; Chen, D.J.; Hu, M.C.-T. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nat. Commun. 2012, 3, 1000. [Google Scholar] [CrossRef]
- Renault, V.M.; Thekkat, P.U.; Hoang, K.L.; White, J.L.; Brady, C.A.; Kenzelmann Broz, D.; Venturelli, O.S.; Johnson, T.M.; Oskoui, P.R.; Xuan, Z.; et al. The pro-longevity gene FoxO3 is a direct target of the p53 tumor suppressor. Oncogene 2011, 30, 3207–3221. [Google Scholar] [CrossRef]
- Gomes, A.R.; Brosens, J.J.; Lam, E.W. Resist or die: FOXO transcription factors determine the cellular response to chemotherapy. Cell Cycle 2008, 7, 3133–3136. [Google Scholar] [CrossRef]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 170. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Law, C.W.; Alhamdoosh, M.; Su, S.; Smyth, G.K.; Ritchie, M.E. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 2016, 5, 1408. [Google Scholar] [CrossRef]
- Ampuja, M.; Rantapero, T.; Rodriguez-Martinez, A.; Palmroth, M.; Alarmo, E.L.; Nykter, M.; Kallioniemi, A. Integrated RNA-seq and DNase-seq analyses identify phenotype-specific BMP4 signaling in breast cancer. BMC Genom. 2017, 18, 68. [Google Scholar] [CrossRef]
- Lara-Astiaso, D.; Weiner, A.; Lorenzo-Vivas, E.; Zaretsky, I.; Jaitin, D.A.; David, E.; Keren-Shaul, H.; Mildner, A.; Winter, D.; Jung, S.; et al. Chromatin state dynamics during blood formation. Science 2014, 345, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Villanueva, J.F.; Díaz-Molina, R.; García-González, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [PubMed]
- Dolfi, S.C.; Chan, L.L.; Qiu, J.; Tedeschi, P.M.; Bertino, J.R.; Hirshfield, K.M.; Oltvai, Z.N.; Vazquez, A. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 2013, 1, 20. [Google Scholar] [CrossRef]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef]
- Borjan, B.; Kern, J.; Steiner, N.; Gunsilius, E.; Wolf, D.; Untergasser, G. Spliced XBP1 Levels Determine Sensitivity of Multiple Myeloma Cells to Proteasome Inhibitor Bortezomib Independent of the Unfolded Protein Response Mediator GRP78. Front. Oncol. 2020, 9, 1530. [Google Scholar] [CrossRef]
- Minami, J.; Suzuki, R.; Mazitschek, R.; Gorgun, G.; Ghosh, B.; Cirstea, D.; Hu, Y.; Mimura, N.; Ohguchi, H.; Cottini, F.; et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2014, 28, 680–689. [Google Scholar] [CrossRef]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. BBA Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef]
- Gao, C.; Xiao, G.; Hu, J. Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell Biosci. 2014, 4, 13. [Google Scholar] [CrossRef]
- Dai, Y.; Guo, X.; Yang, C. Effect of bortezomib on proliferation and apoptosis of myeloma cells by activating Wnt/β-catenin signaling pathway. Oncol. Lett. 2020, 20, 1295–1299. [Google Scholar] [CrossRef] [PubMed]
- Horton, T.M.; Hoff, F.W.; van Dijk, A.; Jenkins, G.N.; Morrison, D.; Bhatla, T.; Hogan, L.; Romanos-Sirakis, E.; Meyer, J.; Carroll, W.L.; et al. The effects of sample handling on proteomics assessed by reverse phase protein arrays (RPPA): Functional proteomic profiling in leukemia. J. Proteom. 2020, 233, 104046. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Broad Institute. Picard Tools. Available online: https://broadinstitute.github.io/picard/ (accessed on 1 May 2021).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Reske, J.J.; Wilson, M.R.; Chandler, R.L. ATAC-seq normalization method can significantly affect differential accessibility analysis and interpretation. Epigenetics Chromatin 2020, 13, 22. [Google Scholar] [CrossRef]
- Lun, A.T.L.; Smyth, G.K. Csaw: A Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 2015, 44, e45. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Hahne, F.; Ivanek, R. Statistical Genomics: Methods and Protocols; Springer: New York, NY, USA, 2016. [Google Scholar]
- Ritchie, M.E.; Smyth, G.K.; Phipson, B.; Wu, D.; Hu, Y.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients (n = 410) | % of Patients | C1 | C2 | C3 | C4 | p Value | |
|---|---|---|---|---|---|---|---|
| Total | 100% | 29% | 17% | 26% | 28% | - | |
| Sex | Female | 49% | 50% | 47% | 50% | 48% | 0.97 |
| Age (years) | <1 | 12% | 16% | 14% | 7% | 12% | 0.48 |
| 2 till 10 | 33% | 33% | 31% | 37% | 32% | ||
| >11 | 54% | 51% | 54% | 56% | 56% | ||
| Race | Black | 12% | 13% | 8% | 10% | 15% | 0.58 |
| Ethnicity | Hispanic | 17% | 18% | 19% | 16% | 16% | 0.93 |
| CNS status | 1 | 60% | 58% | 64% | 65% | 54% | 0.74 |
| 2 | 30% | 31% | 27% | 26% | 35% | ||
| 3 | 10% | 11% | 9% | 9% | 11% | ||
| WBC | >100,000 | 25% | 24% | 24% | 25% | 26% | 0.97 |
| Cytogenetics | Inv(16)/t(16;16) | 14% | 19% | 20% | 9% | 9% | 0.02 |
| t(8;21) | 16% | 14% | 17% | 11% | 21% | 0.23 | |
| Normal | 28% | 24% | 24% | 31% | 31% | 0.55 | |
| t(9;11)(p22;q23)/11q23 | 18% | 18% | 17% | 21% | 14% | 0.55 | |
| Monosomy −5, −7, or +8 | 8% | 7% | 6% | 14% | 4% | 0.04 | |
| Other | 17% | 18% | 16% | 13% | 21% | 0.46 | |
| FLT3-ITD | Mutant | 22% | 19% | 16% | 30% | 23% | 0.12 |
| High allelic ratio (>0.4) | 15% | 10% | 10% | 24% | 13% | 0.01 | |
| CEBPA | Mutant | 10% | 4% | 10% | 7% | 17% | 0.01 |
| NPM1 | Mutant | 11% | 13% | 10% | 9% | 12% | 0.84 |
| Treatment | ADE | 40% | 45% | 40% | 34% | 41% | 0.26 |
| ADE+BTZ | 51% | 49% | 56% | 52% | 50% | ||
| ADE+SFB †† | 9% | 7% | 4% | 14% | 9% | ||
| Risk stratification † | High risk | 30% | 28% | 20% | 41% | 27% | 0.02 |
| Complete | ADE patients (n = 164) | 85.4% | 88.7% | 82.1% | 83.3% | 85.1% | 0.84 |
| Remission | ADE+BTZ patients (n = 210) | 84.8% | 79.3% | 79.5% | 89.3% | 89.5% | 0.26 |
| Relapse | ADE patients (n = 140) | 34.8% | 26.4% | 25.0% | 41.7% | 44.7% | 0.13 |
| ADE+BTZ patients (n = 178) | 34.3% | 32.8% | 28.2% | 37.5% | 36.8% | 0.77 |
| Univariable | RR from End of Course 2 | OS from Study Entry | |||||||
|---|---|---|---|---|---|---|---|---|---|
| N | HR | 95%CI | p Value | N | HR | 95%CI | p Value | ||
| Matrix | C1 | 47 | 1 | 53 | 1 | ||||
| C2 | 23 | 1.04 | 0.43–2.54 | 0.928 | 28 | 1.01 | 0.40–2.52 | 0.990 | |
| C3 | 30 | 2.17 | 1.04–4.51 | 0.038 | 36 | 1.80 | 0.84–3.82 | 0.129 | |
| C4 | 40 | 2.14 | 1.11–4.13 | 0.023 | 47 | 1.88 | 0.94–3.75 | 0.075 | |
| HME | High | 70 | 1 | 83 | 1 | ||||
| Low | 70 | 0.47 | 0.28–0.80 | 0.005 | 81 | 0.54 | 0.31–0.94 | 0.030 | |
| HMM | High | 63 | 1 | 75 | 1 | ||||
| Low | 77 | 0.819 | 0.49–1.37 | 0.449 | 89 | 0.84 | 0.50–1.43 | 0.524 | |
| Multivariable | RR from End of Course 2 | OS from Study Entry | |||||||
|---|---|---|---|---|---|---|---|---|---|
| N | HR | 95%CI | p Value | N | HR | 95%CI | p Value | ||
| HME | High | 70 | 1 | 82 | 1 | ||||
| Low | 70 | 0.45 | 0.26–0.77 | 0.004 | 79 | 0.59 | 0.33–1.06 | 0.077 | |
| Age (year olds) | <1 | 15 | 2.44 | 1.15–5.16 | 0.020 | 19 | 2.70 | 1.20–6.08 | 0.017 |
| 2–10 | 47 | 1 | 51 | 1 | |||||
| >11 | 78 | 0.86 | 0.47–1.58 | 0.623 | 91 | 1.24 | 0.65–2.35 | 0.520 | |
| AAMl1031 | Low risk | 119 | 1 | 126 | 1 | ||||
| risk group definition | High risk | 21 | 0.79 | 0.35–1.79 | 0.577 | 35 | 2.96 | 1.66–5.27 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Dijk, A.D.; Hoff, F.W.; Qiu, Y.; Hubner, S.E.; Go, R.L.; Ruvolo, V.R.; Leonti, A.R.; Gerbing, R.B.; Gamis, A.S.; Aplenc, R.; et al. Chromatin Profiles Are Prognostic of Clinical Response to Bortezomib-Containing Chemotherapy in Pediatric Acute Myeloid Leukemia: Results from the COG AAML1031 Trial. Cancers 2024, 16, 1448. https://doi.org/10.3390/cancers16081448
van Dijk AD, Hoff FW, Qiu Y, Hubner SE, Go RL, Ruvolo VR, Leonti AR, Gerbing RB, Gamis AS, Aplenc R, et al. Chromatin Profiles Are Prognostic of Clinical Response to Bortezomib-Containing Chemotherapy in Pediatric Acute Myeloid Leukemia: Results from the COG AAML1031 Trial. Cancers. 2024; 16(8):1448. https://doi.org/10.3390/cancers16081448
Chicago/Turabian Stylevan Dijk, Anneke D., Fieke W. Hoff, Yihua Qiu, Stefan E. Hubner, Robin L. Go, Vivian R. Ruvolo, Amanda R. Leonti, Robert B. Gerbing, Alan S. Gamis, Richard Aplenc, and et al. 2024. "Chromatin Profiles Are Prognostic of Clinical Response to Bortezomib-Containing Chemotherapy in Pediatric Acute Myeloid Leukemia: Results from the COG AAML1031 Trial" Cancers 16, no. 8: 1448. https://doi.org/10.3390/cancers16081448
APA Stylevan Dijk, A. D., Hoff, F. W., Qiu, Y., Hubner, S. E., Go, R. L., Ruvolo, V. R., Leonti, A. R., Gerbing, R. B., Gamis, A. S., Aplenc, R., Kolb, E. A., Alonzo, T. A., Meshinchi, S., de Bont, E. S. J. M., Horton, T. M., & Kornblau, S. M. (2024). Chromatin Profiles Are Prognostic of Clinical Response to Bortezomib-Containing Chemotherapy in Pediatric Acute Myeloid Leukemia: Results from the COG AAML1031 Trial. Cancers, 16(8), 1448. https://doi.org/10.3390/cancers16081448