
Emerging Therapies in Kirsten Rat Sarcoma Virus (+) Non-Small-Cell Lung Cancer



Abstract
Simple Summary
Abstract
1. Introduction
2. KRAS Biology
2.1. Structure and Signaling Pathways
2.2. Cooperation of KRAS and WNT Pathways
2.3. Mutations
2.4. Comutations
2.5. Why Was the KRAS Mutation Undruggable?
3. Direct Targeting of KRAS
3.1. Sotorasib (AMG510)
3.2. Adagrasib (MRTX849)
3.3. Novel Direct Inhibitors of KRAS G12C
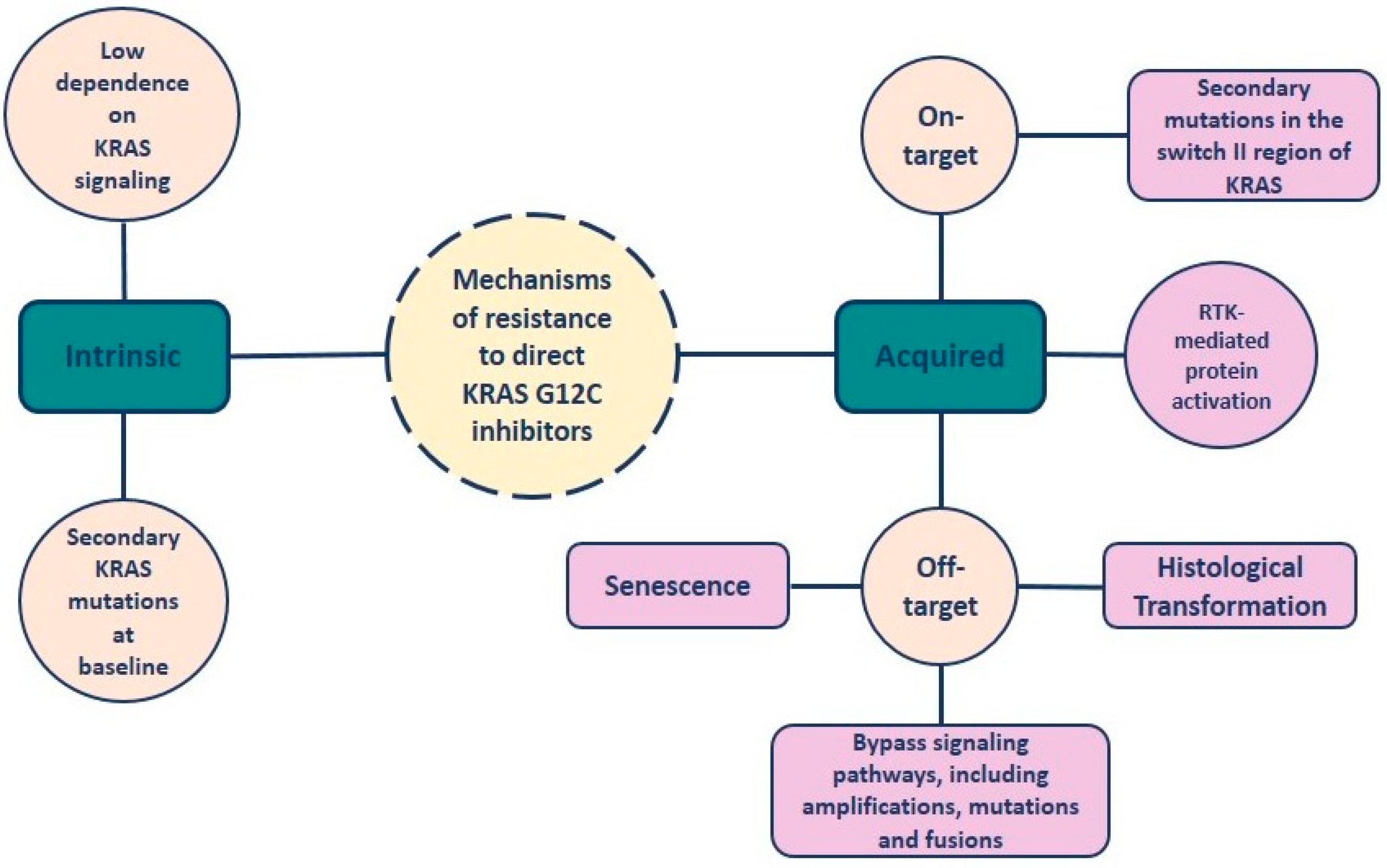
4. Resistance to KRAS-G12C Inhibitors
5. Indirect Inhibition of KRAS
5.1. Inhibitors of the Upstream Signaling Pathway
- EGFR inhibitors
- SHP2 and SOS1 inhibitors
5.2. Inhibitors of the Downstream Signaling Pathway
- MAPK pathway (RAF/MEK/ERK)
- PI3K/AKT/mTOR pathway
5.3. FAK Inhibitors
5.4. DDR1 Inhibitors
5.5. YB-1 Inhibitors
5.6. HSP90 Inhibitors
5.7. CDK 4/6 Inhibitors
6. Platinum-Based Chemotherapy in KRAS-Mutated NSCLC
7. Immunotherapy in KRAS-Mutated NSCLC
8. KRAS-G12V and -G12D Mutations
9. Other Emerging Therapies
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ganti, A.K.; Klein, A.B.; Cotarla, I.; Seal, B.; Chou, E. Update of Incidence, Prevalence, Survival, and Initial Treatment in Patients With Non–Small Cell Lung Cancer in the US. JAMA Oncol. 2021, 7, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung Cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Gomatou, G.; Syrigos, N.; Kotteas, E. Osimertinib Resistance: Molecular Mechanisms and Emerging Treatment Options. Cancers 2023, 15, 841. [Google Scholar] [CrossRef] [PubMed]
- Tsamis, I.; Gomatou, G.; Chachali, S.P.; Trontzas, I.P.; Patriarcheas, V.; Panagiotou, E.; Kotteas, E. BRAF/MEK inhibition in NSCLC: Mechanisms of resistance and how to overcome it. Clin. Transl. Oncol. 2023, 25, 10–20. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: Guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J. Mol. Diagn. 2018, 20, 129–159. [Google Scholar] [CrossRef]
- Ho, C.; Tong, K.M.; Ramsden, K.; Ionescu, D.; Laskin, J. Histologic classification of non-small-cell lung cancer over time: Reducing the rates of not-otherwise-specified. Curr. Oncol. 2015, 22, 164–170. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The frequency of Ras mutations in cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Tsongalis, P.D.; Peterson, J.D.; de Abreu, F.B.; Black, C.C.; Gutmann, E.J.; Liu, X.; Tafe, L.J.; Amos, C.I.; Tsongalis, G.J. The potential utility of re-mining results of somatic mutation testing: KRAS status in lung adenocarcinoma. Cancer Genet. 2016, 209, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Chang, E.H.; Gonda, M.A.; Ellis, R.W.; Scolnick, E.M.; Lowy, D.R. Human genome contains four genes homologous to transforminggenes of Harvey and Kirsten murine sarcoma viruses. Proc. Natl. Acad. Sci. USA 1982, 79, 4848–4852. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS targeted therapies: Is the undruggable drugged? Nat Rev Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F. Ras Proteins: Different Signals from Different Locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–385. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; To, M.D. The Genetics and Biology of KRAS in Lung Cancer. Chin. J. Cancer. 2013, 32, 63–70. [Google Scholar] [CrossRef]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer. 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Tímár, J. The clinical relevance of KRAS gene mutation in non-small-cell lung cancer. Curr. Opin. Oncol. 2014, 26, 138–144. [Google Scholar] [CrossRef]
- Barbacid, M. ras genes. Annu. Rev. Biochem. 1987, 56, 779–827. [Google Scholar] [CrossRef]
- Pantsar, T. The Current Understanding of KRAS Protein Structure and Dynamics. Comput. Struct. Biotechnol. J. 2019, 18, 189–198. [Google Scholar] [CrossRef]
- PNAS. Drugging an Undruggable Pocket on KRAS. Available online: https://www.pnas.org/doi/10.1073/pnas.1904529116 (accessed on 26 February 2024).
- Maertens, O.; Cichowski, K. An Expanding Role for RAS GTPase Activating Proteins (RAS GAPs) in Cancer. Adv. Biol. Regul. 2014, 55, 1–14. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Dance, M.; Montagner, A.; Salles, J.-P.; Yart, A.; Raynal, P. The Molecular Functions of Shp2 in the Ras/Mitogen-Activated Protein Kinase (ERK1/2) Pathway. Cell. Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS proteins and their regulators in human disease. Cell. 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Warren, G.W.; Cummings, K.M. Tobacco and lung cancer: Risks, trends, and outcomes in patients with cancer. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Jones, J.C.; Farias, V.D.A.; Mackeyev, Y.; Singh, P.K.; Quiñones-Hinojosa, A.; Krishnan, S. Identification of Synergistic Drug Combinations to Target KRAS-Driven Chemoradioresistant Cancers Utilizing Tumoroid Models of Colorectal Adenocarcinoma and Recurrent Glioblastoma. Front. Oncol. 2022, 12, 840241. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Xu, J.; Bian, X.; Wu, J.L.; Kang, W.; Qian, Y.; Li, W.; Chen, H.; Gou, H.; Liu, D.; et al. In Colorectal Cancer Cells With Mutant KRAS, SLC25A22-Mediated Glutaminolysis Reduces DNA Demethylation to Increase WNT Signaling, Stemness, and Drug Resistance. Gastroenterology 2020, 159, 2163–2180.e6. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Z.; Zhao, L.; Li, L.; Zuo, W.; Han, W.Z.A.L. High Expression of RAD51 Promotes DNA Damage Repair and Survival in KRAS-Mutant Lung Cancer Cells. BMB Rep. 2019, 52, 151–156. [Google Scholar] [CrossRef]
- Yang, L.; Shen, C.; Estrada-Bernal, A.; Robb, R.; Chatterjee, M.; Sebastian, N.; Webb, A.; Mo, X.; Chen, W.; Krishnan, S.; et al. Oncogenic KRAS Drives Radioresistance Through Upregulation of NRF2- 53BP1-Mediated non-Homologous End-Joining Repair. Nucleic Acids Res. 2021, 49, 11067–11082. [Google Scholar] [CrossRef]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular epidemiology of EGFR and KRAS mutations in 3026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS mutant cancers. Clin Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [PubMed]
- Herdeis, L.; Gerlach, D.; McConnell, D.B.; Kessler, D. Stopping the beating heart of cancer: KRAS reviewed. Curr. Opin. Struct. Biol. 2021, 71, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Judd, J.; Abdel Karim, N.; Khan, H.; Naqash, A.R.; Baca, Y.; Xiu, J.; VanderWalde, A.M.; Mamdani, H.; Raez, L.E.; Nagasaka, M.; et al. Characterization of KRAS Mutation Subtypes in Non–Small Cell Lung Cancer. Mol. Cancer Ther. 2021, 20, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Carbone, D.P.; Garassino, M.; Barlesi, F. Targeting KRAS in Non-Small-Cell Lung Cancer: Recent Progress and New Approaches. Ann. Oncol. 2021, 32, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and outcomes of patients with metastatic KRAS-mutant lung adenocarcinomas: The lung cancer mutation consortium experience. J. Thorac. Oncol. 2019, 14, 876–889. [Google Scholar] [CrossRef]
- Dearden, S.; Stevens, J.; Wu, Y.L.; Blowers, D. Mutation incidence and coincidence in non small-cell lung cancer: Meta-analyses by ethnicity and histology (mutMap). Ann. Oncol. 2013, 24, 2371–2376. [Google Scholar] [CrossRef]
- Yu, H.A.; Sima, C.S.; Shen, R.; Kass, S.; Gainor, J.; Shaw, A.; Hames, M.; Iams, W.; Aston, J.; Lovly, C.M.; et al. Prognostic Impact of KRAS Mutation Subtypes in 677 Patients with Metastatic Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Goulding, R.E.; Chenoweth, M.; Carter, G.C.; Boye, M.E.; Sheffield, K.M.; John, W.J.; Leusch, M.S.; Muehlenbein, C.E.; Li, L.; Jen, M.-H.; et al. KRAS Mutation as a Prognostic Factor and Predictive Factor in Advanced/Metastatic Non-Small Cell Lung Cancer: A Systematic Literature Review and Meta-Analysis. Cancer Treat. Res. Commun. 2020, 24, 100200. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Ito, K.; Hayashi, Y.; Kimura, R.; Tan, T.Z.; Yamaguchi, R.; Ebi, H. Epithelial-to-Mesenchymal transition is a cause of both intrinsic and acquired resistance to KRAS G12C inhibitor in KRAS G12C-mutant non-small cell lung cancer. Clin. Cancer Res. 2020, 26, 5962–5973. [Google Scholar] [CrossRef]
- West, H.J.; McCleland, M.; Cappuzzo, F.; Reck, M.; Mok, T.S.; Jotte, R.M.; Nishio, M.; Kim, E.; Morris, S.; Zou, W.; et al. Clinical efficacy of atezolizumab plus bevacizumab and chemotherapy in KRAS-mutated non-small cell lung cancer with STK11, KEAP1, or TP53 comutations: Subgroup results from the phase III IMpower150 trial. J. Immunother. Cancer. 2022, 10, e003027. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, D.; Mazzotta, M.; Scalera, S.; Terrenato, I.; Sperati, F.; D’Ambrosio, L.; Pallocca, M.; Corleone, G.; Krasniqi, E.; Pizzuti, L.; et al. KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann. Oncol. 2020, 31, 1746–1754. [Google Scholar] [CrossRef]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the clinical development candidate MRTX849, a covalent KRAS(G12C) inhibitor for the treatment of cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef]
- Goebel, L.; Müller, M.P.; Goody, R.S.; Rauh, D. KRasG12C inhibitors in clinical trials: A short historical perspective. RSC Med. Chem. 2020, 11, 760–770. [Google Scholar] [CrossRef]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The Path to the Clinic: A Comprehensive Review on Direct KRASG12C Inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- FDA. FDA Approves First Targeted Therapy for Lung Cancer Mutation Previously Considered Resistant to Drug Therapy. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-targeted-therapy-lung-cancer-mutation-previously-considered-resistant-drug (accessed on 26 February 2024).
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Skoulidis, F.; Govindan, R.; Velcheti, V.; Li, B.; Besse, B.; Dy, G.; Kim, D.; Schuler, M.; Vincent, M.; et al. P52.03 Efficacy of Sotorasib in KRAS p.G12C-Mutated NSCLC with Stable Brain Metastases: A Post-Hoc Analysis of CodeBreak 100. J. Thorac. Oncol. 2021, 16, S1123. [Google Scholar] [CrossRef]
- Skoulidis, F.; Schuler, M.; Wolf, J.; Barlesi, F.; Price, T.; Dy, G.; Govindan, R.; Borghaei, H.; Falchook, G.; Li, B.; et al. Genomic profiles and potential determinants of response and resistance in KRAS p/G12C-mutated NSCLC treated with sotorasib. J. Thorac. Oncol. 2020, 16, S929–S930. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2021, 28, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Spira, A.; Besse, B.; Wolf, J.; Skoulidis, F.; Borghaei, H.; Goto, K.; Park, K.; Griesinger, F.; Felip, E.; et al. CodeBreak 200: A phase III multicenter study of sotorasib (AMG 510), a KRAS (G12C) inhibitor, versus docetaxel in patients with previously treated advanced non small cell lung cancer (NSCLC) harboring KRAS p. G12C mutation. Ann. Oncol. 2020, 31, S894–S895. [Google Scholar] [CrossRef]
- Johnson, M.L.; De Langen, J.; Waterhouse, D.M.; Mazieres, J.; Dingemans, A.C.; Mountzios, G.; Pless, M.; Wolf, J.; Schuler, M.; Lena, H.; et al. Sotorasib versus Docetaxel for Previously Treated Non-Small Cell Lung Cancer with KRASG12C Mutation: CodeBreaK 200 Phase III Study. ESMO Congress 2022, LBA10. Available online: https://dailyreporter.esmo.org/esmo-congress-2022/news/sotorasib-improves-pfs-versus-docetaxel-in-patients-with-pre-treated-kras-g12c-mutated-nsclc (accessed on 26 February 2024).
- Lumakras®/Lumykras®(Sotorasib) Demonstrates Superior Progression-Free Survival over Docetaxel in First Positive Phase 3 Trial of a Kras G12c Inhibitor in Non-Small Cell Lung Cancer. Available online: https://www.prnewswire.com/news-releases/lumakraslumykras-sotorasib-demonstrates-superior-progression-free-survival-over-docetaxel-in-first-positive-phase-3-trial-of-a-kras-g12c-inhibitor-in-non-small-cell-lung-cancer-301621632.html (accessed on 26 February 2024).
- Hong, D.S.; Strickler, J.H.; Fakih, M.; Falchook, G.S.; Li, B.T.; Durm, G.A.; Burns, T.F.; Ramalingam, S.S.; Goldberg, S.B.; Frank, R.C.; et al. Trial in progress: A phase 1b study of sotorasib, a specific and irreversible KRAS G12C inhibitor, as monotherapy in non-small cell lung cancer (NSCLC) with brain metastasis and in combination with other anticancer therapies in advanced solid tumors (CodeBreaK 101). J. Clin. Oncol. 2021, 39, TPS2669. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS G12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.A.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRASG12C Solid Tumors (KRYSTAL1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Ou, S.H.I.; Johnson, M.L.; Christensen, J.; Velastegui, K.; Potvin, D.; Faltaos, D.; Chao, R.C. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. J. Clin. Oncol. 2019, 37, TPS3161. [Google Scholar] [CrossRef]
- Jänne, P.A.; Papadopoulos, K.; Rybkin, I.; Johnson, M. A Phase 1 Clinical Trial Evaluating the Pharmacokinetics (PK), Safety, and Clinical Activity of MRTX849, a Mutant-Selective Small Molecule KRAS G12C Inhibitor, in Advanced Solid Tumors. Available online: https://www.mirati.com/wp-content/uploads/AACR-NCI-EORTC-Clinical-Data-Presentation_Janne_October-2019-1-1.pdf (accessed on 26 February 2024).
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in non-small-cell lung cancer harboring a KRASG12C mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Spira, A.I.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. KRYSTAL-1: Activity and safety of adagrasib (MRTX849) in patients with advanced/metastatic non–small cell lung cancer (NSCLC) harboring a KRASG12C mutation. J. Clin. Oncol. 2022, 40, 9002. [Google Scholar] [CrossRef]
- New Drug Application for Adagrasib Accepted by FDA for KRAS G12C+ NSCLC. Available online: https://www.cancernetwork.com/view/new-drug-application-for-adagrasib-accepted-by-fda-for-kras-g12c-nsclc (accessed on 26 February 2024).
- Mirati Therapeutics Inc. A Randomized Phase 3 Study of MRTX849 versus Docetaxel in Patients with Previously Treated Non-Small Cell Lung Cancer with KRAS G12C Mutation. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04685135 (accessed on 26 February 2024).
- Mok, T.S.K.; Lawler, W.E.; Shum, M.K.; Dakhil, S.R.; Spira, A.I.; Barlesi, F.; Reck, M.; Garassino, M.C.; Spigel, D.R.; Alvarez, D.; et al. KRYSTAL-12: A randomized phase 3 study of adagrasib (MRTX849) versus docetaxel in patients (pts) with previously treated non-small-cell lung cancer (NSCLC) with KRASG12C mutation. J. Clin. Oncol. 2021, 39, TPS9129. [Google Scholar] [CrossRef]
- Riely, G.J.; Ou, S.I.; Rybkin, I.; Spira, A.; Papadopoulos, K.; Sabari, J.; Johnson, M.; Heist, R.; Bazhenova, L.; Barve, M.; et al. 99O_PR KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thoracic Oncol. 2021, 16, S751–S752. [Google Scholar] [CrossRef]
- Evidence of Antitumor Effect with GDC-6036 Monotherapy in KRAS G12C+ NSCLC Revealed at 2022 WCLC. Available online: https://www.cancernetwork.com/view/evidence-of-antitumor-effect-with-gdc-6036-monotherapy-in-kras-g12c-nsclc-revealed-at-2022-wclc (accessed on 26 February 2024).
- Sacher, A.; Patel, M.R.; Miller, W.H.; Desai, J.; Garralda, E.; Bowyer, S.; Kim, T.; De Miguel, M.; Falcon, A.; Krebs, M.; et al. OA03.04 Phase IA Study to Evaluate GDC-6036 Monotherapy in Patients with Non-small Cell Lung Cancer (NSCLC) with KRAS G12C Mutation. J. Thorac. Oncol. 2022, 17, S8–S9. [Google Scholar] [CrossRef]
- Luo, J.; Ostrem, J.; Pellini, B.; Imbody, D.; Stern, Y.; Solanki, H.S.; Haura, E.B.; Villaruz, L.C. Overcoming KRAS-mutant lung cancer. Am. Soc. Clin. Oncol. Educ. Book 2022, 41, 1–11. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Jiao, D.; Yang, S. Overcoming resistance to drugs targeting KRASG12C mutation. Innovation 2020, 1, 100035. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Kordiak, J.; Szemraj, J.; Grabska-Kobylecka, I.; Bialasiewicz, P.; Braun, M.; Kordek, R.; Nowak, D. Intratumor heterogeneity and tissue distribution of KRAS mutation in non-small cell lung cancer: Implications for detection of mutated KRAS oncogene in exhaled breath condensate. J. Cancer. Res. Clin. Oncol. 2019, 145, 241–251. [Google Scholar] [CrossRef]
- Blaquier, J.B.; Cardona, A.F.; Recondo, G. Resistance to KRAS (G12C) Inhibitors in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 787585. [Google Scholar] [CrossRef]
- Désage, A.-L.; Léonce, C.; Swalduz, A.; Ortiz-Cuaran, S. Targeting KRAS Mutant in Non-Small Cell Lung Cancer: Novel Insights Into Therapeutic Strategies. Front. Oncol. 2022, 12, 796832. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Suda, K.; Fujino, T.; Ohara, S.; Hamada, A.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Arita, T.; et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J. Thorac. Oncol. 2021, 16, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid nonuniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical pathway inhibition overcomes adaptive feedback resistance to KRAS(G12C) inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Xue, J.Y.; Lito, P. Targeting KRAS (G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell 2020, 183, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Aguilar, A.; Pedraz, C.; Chaib, I. KRAS inhibitors, approved. Nat. Cancer 2021, 2, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Potugari, B.; Nguyen, A.; Sukari, A.; Azmi, A.S.; Ou, S.-H.I. KRAS Inhibitors- yes but what next? Direct targeting of KRAS vaccines, adoptive T cell therapy and beyond. Cancer Treat. Rev. 2021, 101, 102309. [Google Scholar] [CrossRef]
- Knight, T.; Irving, J.A. Ras/Raf/MEK/ERK Pathway Activation in Childhood Acute Lymphoblastic Leukemia and Its Therapeutic Targeting. Front. Oncol. 2014, 4, 160. [Google Scholar] [CrossRef]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Cseh, B.; Doma, E.; Baccarini, M. “RAF” neighborhood: Protein-protein interaction in the Raf/Mek/Erk pathway. FEBS Lett. 2014, 588, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Capelletto, E.; Bironzo, P.; Denis, L.; Koustenis, A.; Bungaro, M.; Novello, S. Single agent VS6766 or VS-6766 plus defactinib in KRAS-mutant nonsmall-cell lung cancer: The RAMP-202 phase II trial. Future Oncol. 2022, 18, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Cucurull, M.; Notario, L.; Sanchez-Cespedes, M.; Hierro, C.; Estival, A.; Carcereny, E.; Saigí, M. Targeting KRAS in Lung Cancer Beyond KRAS G12C Inhibitors: The Immune Regulatory Role of KRAS and Novel Therapeutic Strategies. Front. Oncol. 2022, 11, 793121. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose escalation study of BKM120, an oral pan Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Morrison, G.; Dean, B.; Hop, C.; Tobler, L.; Percey, S.; Meng, M.; Reuschel, S.; West, D.; Holden, S.; et al. A solid phase extraction-liquid chromatographic-tandem mass spectrometry method for determination of concentrations of GDC-0941, a small molecule class I phosphatidylinositide 3-kinase inhibitor, to support clinical development. J. Pharm. Biomed. Anal. 2012, 61, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Rodon, J.; Bedell, C.; Kwak, E.L.; Baselga, J.; Braña, I.; Pandya, S.S.; Scheffold, C.; Laird, A.D.; Nguyen, L.T.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 233–245. [Google Scholar] [CrossRef]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin. Cancer Res. 2019, 25, 796–807. [Google Scholar] [CrossRef]
- Konstantinidou, G.; Ramadori, G.; Torti, F.; Kangasniemi, K.; Ramirez, R.E.; Cai, Y.; Behrens, C.; Dellinger, M.T.; Brekken, R.A.; Wistuba, I.I.; et al. RHOAFAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov. 2013, 3, 444–457. [Google Scholar] [CrossRef]
- Gerber, D.E.; Camidge, D.R.; Morgensztern, D.; Cetnar, J.; Kelly, R.J.; Ramalingam, S.S.; Spigel, D.R.; Jeong, W.; Scaglioni, P.P.; Zhang, S.; et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer 2020, 139, 60–67. [Google Scholar] [CrossRef]
- Altaf, R.; Ilyas, U.; Ma, A.; Shi, M. Identification and validation of differentially expressed genes for targeted therapy in NSCLC using integrated bioinformatics analysis. Front. Oncol. 2023, 13, 1206768. [Google Scholar] [CrossRef]
- Yang, S.H.; Baek, H.A.; Lee, H.J.; Park, H.S.; Jang, K.Y.; Kang, M.J.; Lee, D.G.; Lee, Y.C.; Moon, W.S.; Chung, M.J. Discoidin domain receptor 1 is associated with poor prognosis of non-small cell lung carcinomas. Oncol. Rep. 2010, 24, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Khozooei, S.; Veerappan, S.; Toulany, M. YB-1 activating cascades as potential targets in KRAS-mutated tumors. Strahlenther Onkol. 2023, 199, 1110–1127. [Google Scholar] [CrossRef] [PubMed]
- Maier, E.; Attenberger, F.; Tiwari, A.; Lettau, K.; Rebholz, S. Dual Targeting of Y-Box Binding Protein-1 and Akt Inhibits Proliferation and Enhances the Chemosensitivity of Colorectal Cancer Cells. Cancers 2019, 11, 562. [Google Scholar] [CrossRef]
- Zuehlke, A.D.; Moses, M.A.; Neckers, L. Heat shock protein 90: Its inhibition and function. Philos. Trans. R Soc. Lond. B Biol. Sci. 2018, 373, 20160527. [Google Scholar] [CrossRef]
- Socinski, M.A.; Goldman, J.; El-Hariry, I.; Koczywas, M.; Vukovic, V.; Horn, L.; Paschold, E.; Salgia, R.; West, H.; Sequist, L.V.; et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 3068–3077. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Fennell, D.A.; Kovcin, V.; Ciuleanu, T.E.; Ramlau, R.; Kowalski, D.; Schenker, M.; Yalcin, I.; Teofilovici, F.; Vukovic, V.M.; et al. Randomized Phase III Study of Ganetespib, a Heat Shock Protein 90 Inhibitor, With Docetaxel Versus Docetaxel in Advanced Non-Small Cell Lung Cancer (GALAXY-2). J. Clin. Oncol. 2020, 38, 613–622. [Google Scholar] [CrossRef]
- Gutierrez, M.; Guo, R.; Giaccone, G.; Liu, S.V.; Hao, Z.; Hilton, C.; Hinson, J.M., Jr.; Kris, M.G.; Orlemans, E.O.; Drilon, A. Phase 1 multicenter study of the HSP90 inhibitor SNX-5422 plus carboplatin and paclitaxel in patients with lung cancers. Lung Cancer 2021, 162, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef]
- Goldman, J.W.; Mazieres, J.; Barlesi, F.; Dragnev, K.H.; Koczywas, M.; Göskel, T.; Cortot, A.B.; Girard, N.; Wesseler, C.; Bischoff, H.; et al. A randomized phase III study of abemaciclib versus erlotinib in patients with stage IV non-small cell lung cancer with a detectable KRAS mutation who failed prior platinum-based therapy: JUNIPER. Front. Oncol. 2020, 10, 578756. [Google Scholar] [CrossRef] [PubMed]
- Passiglia, F.; Cappuzzo, F.; Alabiso, O.; Bettini, A.C.; Bidoli, P.; Chiari, R.; Defferrari, C.; Delmonte, A.; Finocchiaro, G.; Francini, G.; et al. Efficacy of nivolumab in pre-treated nonsmall-cell lung cancer patients harbouring KRAS mutations. Br. J. Cancer 2019, 120, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Dai, Y.; Zhu, L.; Wang, C.; Fei, X. Poor response to platinum-based chemotherapy is associated with KRAS mutation and concomitant low expression of BRAC1 and TYMS in NSCLC. J. Inter. Med. Res. 2016, 44, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Jiang, T.; Li, X.; Zhao, C.; Zhang, L. Characterization of distinct types of KRAS mutation and its impact on first line platinum based chemotherapy in Chinese patients with advanced non small cell lung cancer. Oncol. Let. 2017, 14, 6525–6532. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous nonsmall-cell lung cancer. N. Engl. J Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicenter randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Schrock, A.B.; Fabrizio, D.; Frampton, G.M.; Sun, J.; He, J.; Gowen, K.; Johnson, M.L.; Bauer, T.M.; Kalemkerian, G.P.; et al. Total mutation burden (TMB) in lung cancer (LC) and relationship with response to PD-1/PD-L1 targeted therapies. J. Clin. Oncol. 2016, 34, 9017. [Google Scholar] [CrossRef]
- Kadara, H.; Choi, M.; Zhang, J.; Parra, E.R.; Rodriguez-Canales, J.; Gaffney, S.G.; Zhao, Z.; Behrens, C.; Fujimoto, J.; Chow, C.; et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann. Oncol. 2017, 28, 75–82. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS Mutation-Induced Upregulation of PD-L1 Mediates Immune Escape in Human Lung Adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The Superior Efficacy of Anti-PD1/PD-L1 Immunotherapy in KRAS-Mutant Non-Small Cell Lung Cancer That Correlates with an Inflammatory Phenotype and Increased Immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef] [PubMed]
- D’Incecco, A.; Andreozzi, M.; Ludovini, V.; Rossi, E.; Capodanno, A.; Landi, L.; Tibaldi, C.; Minuti, G.; Salvini, J.; Coppi, E.; et al. PD-1 and PD-L1 Expression in Molecularly Selected Non-Small-Cell Lung Cancer Patients. Br. J. Cancer 2015, 112, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; El-Refai, S.M.; Sha, W.; Puccini, A.; Grothey, A.; George, T.J.; Hwang, J.J.; O’Neil, B.; Barrett, A.S.; Kadakia, K.C.; et al. Landscape of KRASG12C, Associated Genomic Alterations, and Interrelation With Immuno-Oncology Biomarkers in KRASMutated Cancers. JCO Precis. Oncol. 2022, 6, e2100245. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.; Lopes, G.; Kowalski, D.; Kasahara, K.; Wu, Y.-L.; De Castro, G.; Cho, B.; Turna, H.; Cristescu, R.; Aurora-Garg, D.; et al. LBA4 Association of KRAS mutational status with response to pembrolizumab monotherapy given as first-line therapy for PD-L1-positive advanced non-squamous NSCLC in Keynote-042. Ann. Oncol. 2019, 30, xi63–xi64. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zheng, S.; Wang, Z.; Wang, S.; Wang, X.; Yang, L.; Xu, H.; Cao, Z.; Feng, X.; Xue, Q.; et al. KRAS-G12D Mutation Drives Immune Suppression and the Primary Resistance of Anti-PD-1/PD-L1 Immunotherapy in Non-Small Cell Lung Cancer. Cancer Commun. 2022, 42, 828–847. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Alessi, J.V.; Elkrief, A.; Wang, X.; Cortellini, A.; Li, Y.Y.; Vaz, V.R.; Gupta, H.; Pecci, F.; Barrichello, A.; et al. Dissecting the Clinicopathologic, Genomic, and Immunophenotypic Correlates of KRASG12D-Mutated Non-Small-Cell Lung Cancer. Ann. Oncol. 2022, 33, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Xu, B.; Zhang, H.; Fang, S. Lung Adenocarcinoma Patients with KEAP1 Mutation Harboring Low Immune Cell Infiltration and Low Activity of Immune Environment. Thorac. Cancer 2021, 12, 2458–2467. [Google Scholar] [CrossRef]
- Ricciuti, B.; Arbour, K.C.; Lin, J.J.; Vajdi, A.; Vokes, N.; Hong, L.; Zhang, J.; Tolstorukov, M.Y.; Li, Y.Y.; Spurr, L.F.; et al. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J. Thorac. Oncol. 2022, 17, 399–410. [Google Scholar] [CrossRef]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Assoun, S.; Theou-Anton, N.; Nguenang, M.; Cazes, A.; Danel, C.; Abbar, B.; Pluvy, J.; Gounant, V.; Khalil, A.; Namour, C.; et al. Association of TP53 Mutations with Response and Longer Survival under Immune Checkpoint Inhibitors in Advanced Non Small-Cell Lung Cancer. Lung Cancer 2019, 132, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Briere, D.M.; Li, S.; Calinisan, A.; Sudhakar, N.; Aranda, R.; Hargis, L.; Peng, D.H.; Deng, J.; Engstrom, L.D.; Hallin, J.; et al. The KRAS (G12C) Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy. Mol. Cancer Ther. 2021, 20, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Falchook, G.S.; Durm, G.A.; Burns, T.F.; Skoulidis, F.; Ramalingam, S.S.; Spira, A.; Bestvina, C.M.; Goldberg, S.B.; Veluswamy, R.; et al. OA03.06 CodeBreaK 100/101: First Report of Safety/Efficacy of Sotorasib in Combination with Pembrolizumab or Atezolizumab in Advanced KRAS p.G12C NSCLC. J. Thorac. Oncol. 2022, 17, S10–S11. [Google Scholar] [CrossRef]
- Mirati Therapeutics Inc. A Phase 2 Trial of MRTX849 Monotherapy and in Combination with Pembrolizumab in Patients with Advanced Non-Small Cell Lung Cancer with KRAS G12C Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT04613596 (accessed on 26 February 2024).
- Xie, M.; Xu, X.; Fan, Y. KRAS-mutant non-small cell lung cancer: An emerging promisingly treatable subgroup. Front. Oncol. 2021, 11, 672612. [Google Scholar] [CrossRef]
- Renaud, S.; Seitlinger, J.; Falcoz, P.E.; Schaeffer, M.; Voegeli, A.C.; Legrain, M.; Beau-Faller, M.; Massard, G. Specific KRAS amino acid substitutions and EGFR mutations predict site-specific recurrence and metastasis following non-small-cell lung cancer surgery. Br. J. Cancer 2016, 115, 346–353. [Google Scholar] [CrossRef]
- Mellema, W.W.; Masen-Poos, L.; Smit, E.F.; Hendriks, L.E.; Aerts, J.G.; Termers, A.; Goosens, M.J.; Smit, H.J.; Heuvel, M.M.v.D.; van der Wekken, A.J.; et al. Comparison of clinical outcome after first-line platinum based chemotherapy in different types of KRAS mutated advanced non-small-cell lung cancer. Lung Cancer 2015, 90, 249–254. [Google Scholar] [CrossRef]
- Garassino, M.C.; Marabese, M.; Rusconi, P.; Rulli, E.; Martelli, O.; Farina, G.; Scanni, A.; Broggini, M. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in nonsmall-cell lung cancer. Ann. Oncol. 2011, 22, 235–237. [Google Scholar] [CrossRef]
- Pan, L.N.; Ma, Y.F.; Li, Z.; Hu, J.A.; Xu, Z.H. KRAS G12V mutation upregulates PD-L1 expression via TGF-β/EMT signaling pathway in human nonsmall-cell lung cancer. Cell Biol. Int. 2021, 45, 795–803. [Google Scholar] [CrossRef]
- Koltun, E.; Cregg, J.; Rice, M.A.; Whalen, D.M.; Freilich, R.; Jiang, J.; Hansen, R.; Bermingham, A.; Knox, J.E.; Dinglasan, J.; et al. Abstract 1260: First-in-Class, Orally Bioavailable KRASG12V(ON) Tri-Complex Inhibitors, as Single Agents and in Combinations, Drive Profound Anti-Tumor Activity in Preclinical Models of KRASG12V Mutant Cancers. Cancer Res. 2021, 81, 1260. [Google Scholar] [CrossRef]
- Gao, G.; Liao, W.; Ma, Q.; Zhang, B.; Chen, Y.; Wang, Y. KRAS G12D mutation predicts lower TMB and drives immune suppression in lung adenocarcinoma. Lung Cancer 2020, 149, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS (G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted degradation of oncogenic KRAS G12C by VHL-recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- A Study of mRNA-5671/V941 as Monotherapy and in Combination with Pembrolizumab (V941-001). Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03948763 (accessed on 26 February 2024).
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Bartolacci, C.; Andreani, C.; Vale, G.; Berto, S.; Melegari, M. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat. Comm. 2022, 13, 4327. [Google Scholar] [CrossRef] [PubMed]
- Bortoletto, A.S.; Parchem, R.J. KRAS Hijacks the miRNA Regulatory Pathway in Cancer. Cancer Res. 2023, 83, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- McLoed, A.G.; Sherrill, T.P.; Cheng, D.S.; Han, W.; Saxon, J.A.; Gleaves, L.A.; Wu, P.; Polosukhin, V.V.; Karin, M.; Yull, F.E.; et al. Neutrophil-Derived IL-1β Impairs the Efficacy of NF-κB Inhibitors against Lung Cancer. Cell Rep. 2016, 16, 120–132. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Deng, S.; Clowers, M.J.; Velasco, W.V.; Ramos-Castaneda, M.; Moghaddam, S.J. Understanding the Complexity of the Tumor Microenvironment in K-ras Mutant Lung Cancer: Finding an Alternative Path to Prevention and Treatment. Front. Oncol. 2019, 9, 1556. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Trial | Phase | KRAS Inhibitor | Combinations | Status/Outcomes | References |
|---|---|---|---|---|---|
| CodeBreak 100 NCT03600883 | I/II | sotorasib | monotherapy | mPFS 6.8 months mOS 12.5 months FDA Approval | [52,53,56] |
| CodeBreak 200 NCT04303780 | III | sotorasib | monotherapy vs. docetaxel | mPFS 5.6 months vs. 4.5 months (HR 0.66) | [57,58,59] |
| CodeBreak 201 NCT04933695 | II | sotorasib | monotherapy first line (st. IV, TPS <1% and/or comut. STK11) | ongoing | [52] |
| CodeBreak 101 NCT04185883 | Ib/II | sotorasib | EGFR inh, SHP2 inh, mTOR inh, CDK inh. Chemotherapy, ICIs, etc. | Ongoing | [60] |
| KRYSTAL-1 NCT03785249 | I/II | adagrasib | monotherapy/afatinib, pembrolizumab, cetuximab | mPFS 6.5 months mOS 12.6 months FDA Approval | [62,63,64,67] |
| KRYSTAL-12 NCT04685135 | III | adagrasib | monotherapy vs. docetaxel | ongoing | [68,69] |
| KRYSTAL-7 NCT04613596 | II/III | adagrasib | pembrolizumab/pembrolizumab + adagrasib vs. pembrolizumab + chemotherapy (first line) | ongoing | [70] |
| KRYSTAL-14 NCT04975256 | I/Ib | adagrasib | BI 1701963 (SOS inhibitor) | completed | |
| KRYSTAL-2 NCT04330664 | I/II | adagrasib | TNO155 (SHP2 inhibitor) | ongoing | |
| KRYSTAL-16 NCT05178888 | I/Ib | adagrasib | palbociclib (CDK4/6 inh.) | ongoing | |
| NCT04449874 | I | GDC-6036 | monotherapy/ bevacizumab, atezolizumab, cetuximab, erlotinib, etc. | ongoing | [71,72] |
| NCT04585035 | I/II | D-1553 | monotherapy/other agents | ongoing | [73] |
| KontRASt-01 NCT04699188 | I/II | JDQ443 | monotherapy/ TNO155, tislelizumab | ongoing | |
| NCT05009329 | I/II | JAB-21822 | monotherapy | ongoing | |
| NCT04956640 | I | LY3537982 | monotherapy/ abemaciclib, cetuximab, pembrolizumab, chemotherapy, etc. | ongoing | |
| NCT05462717 | I/Ib | RMC-6291 | monotherapy | ongoing |
| Trial | Phase | KRAS Inhibitor | Combinations | Status/ Outcomes | References |
|---|---|---|---|---|---|
| NCT03634982 | I | RMC-4630 (SHP2 Inhibitor) | monotherapy | completed | [87] |
| NCT03114319 | I | TNO155 (SHP2 Inhibitor) | monotherapy | ongoing | [87] |
| NCT04111458 | I | BI 1701963 (SOS1 Inhibitor) | monotherapy/ trametinib (MEK Inh.) | ongoing | [87] |
| NCT03284502 | I | belvarafenib (RAF Inh.) | cobimetinib, cetuximab | ongoing | [88,89,90,91] |
| NCT01859026 | I | binimetinib (MEK Inh.) | erlotinib | completed | [88,89,90,91] |
| NCT01986166 | I | cobimetinib (MEK Inh.) | duligotuzumab (EGFR/HER3 Inh.) | completed | [88,89,90,91] |
| RAMP203 NCT05074810 | I/II | Avutometinib [VS-6766] (RAF–MEK Inhibitor) | sotorasib | ongoing | [92] |
| RAMP204 NCT05375994 | I/II | Avutometinib [VS-6766] (RAF–MEK Inhibitor) | adagrasib | ongoing | [92] |
| NCT04418167 | I | JSI-1187-01 (ERK Inhibitor) | monotherapy/ dabrafenib (BRAF Inh.) | ongoing | |
| NCT04073680 | I/II | serabelisib (PI3K Inhibitor) | canagliflozin (SGLT2 Inh.) | completed | [94,95,96] |
| NCT04586270 | I | TAS0612 (AKT Inhibitor) | monotherapy | ongoing | [94,95,96] |
| TORCMEK NCT02583542 | I/II | AZD2014 (mTOR Inhibitor) | selumetinib (MEK Inh.) | completed | [94,95,96] |
| NCT01951690 | II | defactinib (FAK Inhibitor) | monotherapy | completed | [99] |
| GALAXY 2 NCT01798485 | III | ganetespib (HSP90 Inhibitor) | docetaxel vs. docetaxel alone | terminated (futility) | [106] |
| NCT01892046 | I | SNX-5422 (HSP90 Inhibitor) | chemotherapy | completed | [107] |
| Trial | Phase | KRAS Inhibitor | Combinations | Status/ Outcomes | References |
|---|---|---|---|---|---|
| NCT05737706 | I/II | MRTX1133 (KRAS-G12D Inh.) | monotherapy | ongoing | [141] |
| V941-001 NCT03948763 | I | V941 (mRNA cancer vaccine) | monotherapy/pembrolizumab | completed | [144] |
| NCT04735068 | II | hydroxychloroquine (autophagy inhibitor) | binimetinib (MEK Inh.) | ongoing | [145] |
| CANOPY-N NCT03968419 | II | canakinumab (anti-IL-1β agent) | monotherapy/pembrolizumab | terminated (low enrollment) | [149,150,151] |
| CHORUS NCT04905316 | II | canakinumab (anti-IL-1β agent) | durvalumab, chemoradiation | ongoing | [149,150,151] |
| CANOPY-2 NCT03626545 | III | canakinumab (anti-IL-1β agent) | docetaxel vs. placebo + docetaxel | terminated (lack of efficacy) | [149,150,151] |
| NCT04691817 | I/II | tecilizumab (IL-6 Inhibitor) | atezolizumab | ongoing | [149,150,151] |
| NCT04940299 | II | tecilizumab (IL-6 Inhibitor) | nivolumab—ipilimumab | ongoing | [149,150,151] |
| Pathway | Target | Inhibitor | References |
|---|---|---|---|
| Upstream signaling pathway | EGFR | erlotinib | [61] |
| SHP2 | RMC-4630 TNO-155 | [87] | |
| SOS 1 | BI 1701963 | [87] | |
| Downstream signaling pathway MAPK pathway (RAF–MEK–ERK) | RAF | belvarafenib | [88,89,90,91] |
| MEK RAF–MEK | binimetinib cobimetinib VS-6766 | [88,89,90,91] [92] | |
| ERK | JSI-1187-01 | ||
| Downstream signaling pathway PI3K–AKT–mTOR pathway | PI3K | BKM120 serabelisib XL147 | [94,95,96] |
| AKT | TAS0612 | [94,95,96] | |
| mTOR | AZD2014 | [94,95,96] | |
| Other pathways | FAK | defactinib | [99] |
| HSP90 | ganetespib SNX-5422 | [105,106] [107] | |
| CDK 4/6 | palbociclib | [74,108,109,110] | |
| DDR1 | |||
| YB-1—RSK | LJI308 | ||
| Immune system | ICIs | nivolumab atezolizumab | [113] [115] |
| Metabolic rewiring | autophagy | hydroxychloroquine | [145] |
| FASN | TVB-2640 | [146] | |
| Tumor microenvironment (TME) | IL-1β IL-6 | canakinumab tecilizumab | [148,149,150,151] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karachaliou, A.; Kotteas, E.; Fiste, O.; Syrigos, K. Emerging Therapies in Kirsten Rat Sarcoma Virus (+) Non-Small-Cell Lung Cancer. Cancers 2024, 16, 1447. https://doi.org/10.3390/cancers16081447
Karachaliou A, Kotteas E, Fiste O, Syrigos K. Emerging Therapies in Kirsten Rat Sarcoma Virus (+) Non-Small-Cell Lung Cancer. Cancers. 2024; 16(8):1447. https://doi.org/10.3390/cancers16081447
Chicago/Turabian StyleKarachaliou, Anastasia, Elias Kotteas, Oraianthi Fiste, and Konstantinos Syrigos. 2024. "Emerging Therapies in Kirsten Rat Sarcoma Virus (+) Non-Small-Cell Lung Cancer" Cancers 16, no. 8: 1447. https://doi.org/10.3390/cancers16081447
APA StyleKarachaliou, A., Kotteas, E., Fiste, O., & Syrigos, K. (2024). Emerging Therapies in Kirsten Rat Sarcoma Virus (+) Non-Small-Cell Lung Cancer. Cancers, 16(8), 1447. https://doi.org/10.3390/cancers16081447