
A Retrospective View of the Triple-Negative Breast Cancer Microenvironment: Novel Markers, Interactions, and Mechanisms of Tumor-Associated Components Using Public Single-Cell RNA-Seq Datasets


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
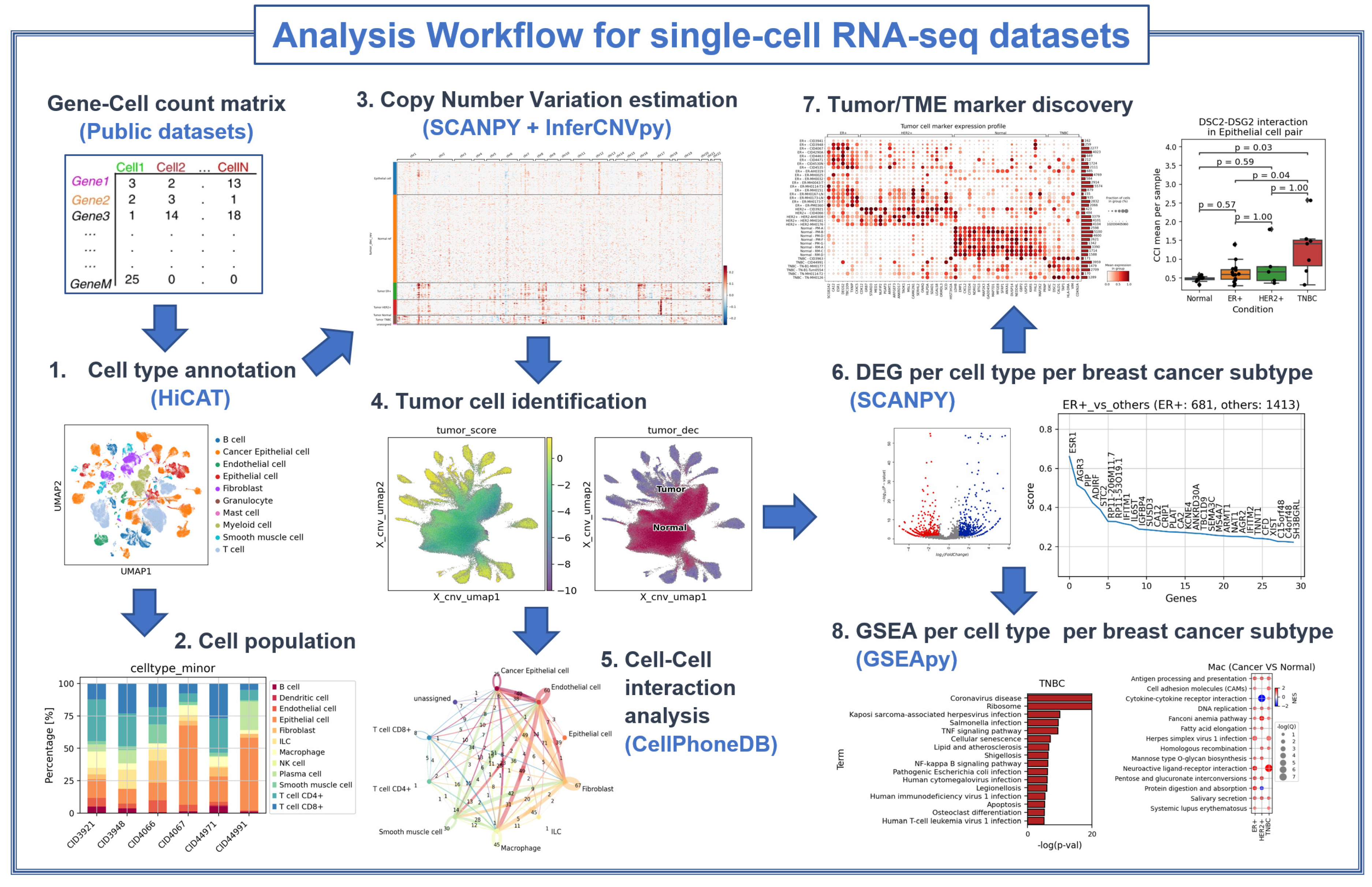
2.1. Datasets Used
2.2. Cell Type Identification
2.3. Tumor Cell Identification
- Perform dimension reduction of CNV patterns using principal component analysis with 15 components to obtain PCA-transform CNV patterns.
- Use PCA-transform CNV patterns to construct neighbor graph with 15 neighbors per cell (kneighbors_graph method in the scikit-learn package).
- The neighbor graph was used to perform clustering via the Louvain algorithm with a resolution of 2. (We used the Louvain algorithm in the scikit-network package.)
- An aggregated adjacency matrix among clusters was obtained.
- The clusters for which the percentage of normal reference cells was greater than 20% were set as seed clusters. The other clusters are set “unprocessed”.
- We chose a cluster in the unprocessed pool. If its connectivity with the seed clusters was highest among others and greater than the minimum connectivity within the seed clusters, we added it to the seed clusters. This step is repeated until there are no unprocessed clusters that have connectivity higher than or equal to the threshold.
- Initial decision: Initially, those in the seed clusters were considered normal, while those in the unprocessed ones were considered tumors.
- With the initial decision, we obtained a Gaussian mixture model (GMM) using those marked as normal. (GaussianMixture method in the scikit-learn package).
- The negative GMM scores were then computed using the “score_samples” method to weigh the CNV scores obtained using InferCNVpy.
- A bimodal approximation of the weighted CNV scores was used to make the final decision.
2.4. Differentially Expressed Gene Analysis
- For a specific cell type (target cell type), we first selected samples for which the number of cells of that type was larger than the lower cutoff for the number of target cells, e.g., 200.
- The upper cutoff for the number of cells was set to twice the minimum number of target cells among the selected samples.
- For samples with more target cells than the upper cutoff, we randomly selected cells up to the upper cutoff.
2.5. Selection of Marker Genes
- nonzero fraction in target ≥ 0.5
- nonzero fraction in reference < 0.3
2.6. Gene Set Enrichment Analysis
2.7. Cell-to-Cell Interaction Analysis
- For each cell type, cell–cell interactions were inferred separately for each sample. We excluded samples that had fewer than 40 cells.
- We filtered out by the p value cutoff of 0.05 and mean cutoff of 0.1.
- For each subtype (condition), we collected the cell–cell interactions if they occurred in at least 75% of the samples under the same conditions.
3. Results
3.1. Cell Type Population Shows High Heterogeneity in Immune Cell Proportion Even within a Subtype, Indicating That Immune Reaction Is Not Subtype-Specific
3.2. CNV-Based Tumor Cell Identification Results Also Show High Heterogeneity, Especially in the TNBC Subtype
3.3. Subtype-Specific Markers of Cancer Epithelial Cells Include Novel TNBC Markers
3.4. Markers of Other TME Components Show Clear Differences from Normal Tissue and Heterogeneity among Samples in Immune Cell Reaction
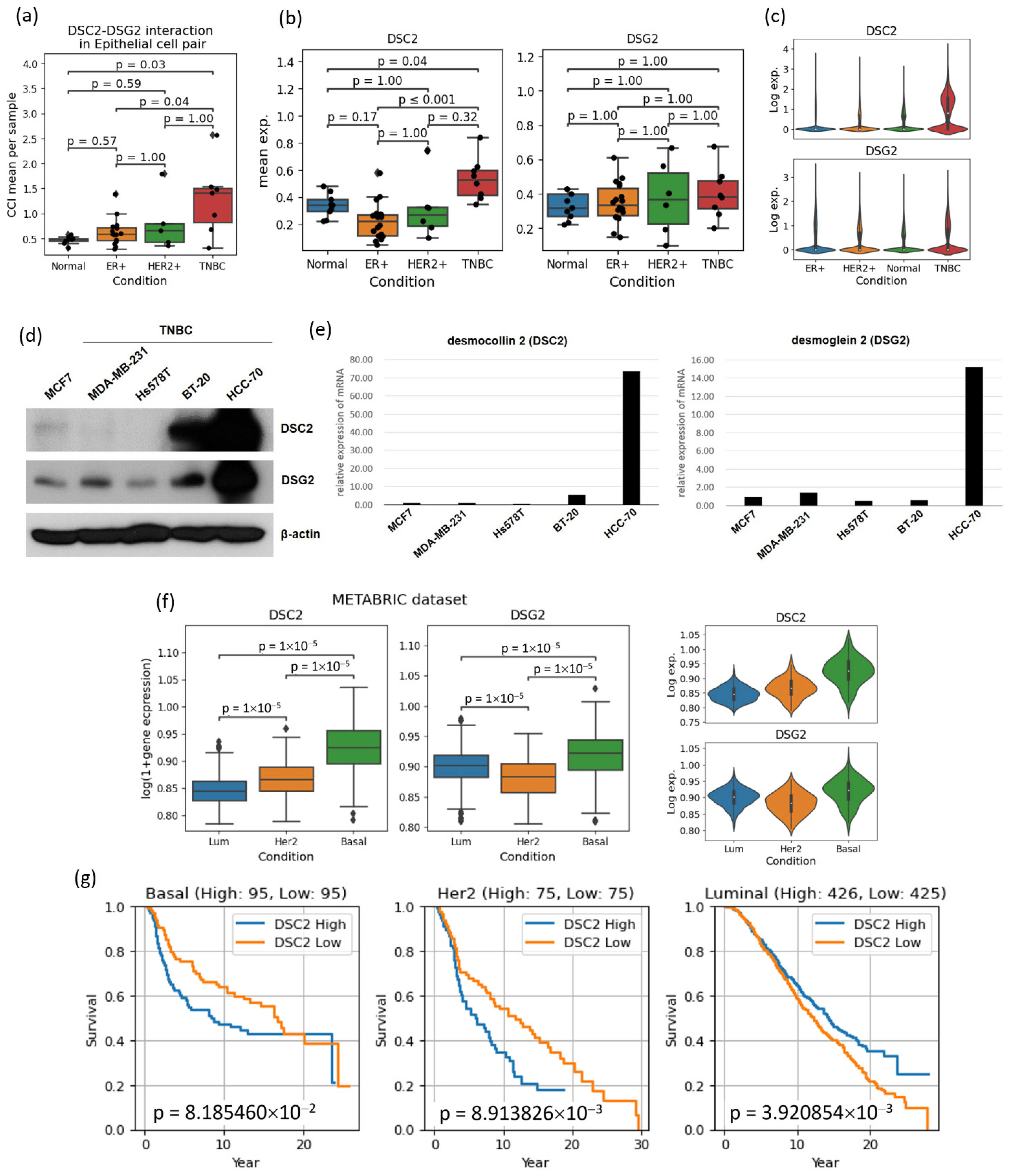
3.5. Cell–Cell Interactions Analysis Identified Novel TNBC-Specific Interactions Including DSC2-DSG2, Suggesting That TNBC Tumor Cells Are More Tightly Aggregated
3.6. Gene Set Enrichment Analysis Shows Subtype-Specific Cell State Changes in Various TME Components
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lüönd, F.; Tiede, S.; Christofori, G. Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during malignant progression. Brit J. Cancer 2021, 125, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thurlimann, B.; Senn, H.J.; Panel, m. Strategies for subtypes-dealing with the diversity of breast cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Orrantia-Borunda, E.; Anchondo-Nunez, P.; Acuna-Aguilar, L.E.; Gomez-Valles, F.O.; Ramirez-Valdespino, C.A. Subtypes of Breast Cancer. In Breast Cancer; Mayrovitz, H.N., Ed.; Exon publications: Brisbane, AU, Australia, 2022. [Google Scholar] [CrossRef]
- Emens, L.A.; Davidson, N.E. Trastuzumab in breast cancer. Oncology 2004, 18, 1117–1128; discussion 1112–1131, 1118–1137. [Google Scholar] [PubMed]
- Ishii, K.; Morii, N.; Yamashiro, H. Pertuzumab in the treatment of HER2-positive breast cancer: An evidence-based review of its safety, efficacy, and place in therapy. Core Evid. 2019, 14, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Wang, P.; He, S.; Zhu, J.; Shi, Y.; Wang, J. Progress and Prospect of Immunotherapy for Triple-Negative Breast Cancer. Front. Oncol. 2022, 12, 919072. [Google Scholar] [CrossRef]
- Qiu, D.; Zhang, G.; Yan, X.; Xiao, X.; Ma, X.; Lin, S.; Wu, J.; Li, X.; Wang, W.; Liu, J.; et al. Prospects of Immunotherapy for Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 797092. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 610303. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, L.; Zhang, Y.; Li, Z.; Siemers, N.; Zhang, Z. Insights Gained from Single-Cell Analysis of Immune Cells in the Tumor Microenvironment. Annu. Rev. Immunol. 2021, 39, 583–609. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.W.; Xiong, F.; Guo, C.; Tang, Y.Y.; Zhou, Y.J.; Liao, Q.J.; et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Canc Res. 2021, 40, 81. [Google Scholar] [CrossRef]
- Ren, L.; Li, J.; Wang, C.; Lou, Z.; Gao, S.; Zhao, L.; Wang, S.; Chaulagain, A.; Zhang, M.; Li, X.; et al. Single cell RNA sequencing for breast cancer: Present and future. Cell Death Discov. 2021, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, T.; Jin, Y.; Huang, B.; Zhang, Y. Progress and Clinical Application of Single-Cell Transcriptional Sequencing Technology in Cancer Research. Front. Oncol. 2020, 10, 593085. [Google Scholar] [CrossRef]
- Lee, J.; Kim, M.; Kang, K.; Yang, C.S.; Yoon, S. Hierarchical cell-type identifier accurately distinguishes immune-cell subtypes enabling precise profiling of tissue microenvironment with single-cell RNA-sequencing. Brief. Bioinform. 2023, 24. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Fang, Z.; Liu, X.; Peltz, G. GSEApy: A comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 2023, 39, btac757. [Google Scholar] [CrossRef]
- Wu, X.Q.; Lu, W.P.; Zhang, W.X.; Zhang, D.N.; Mei, H.T.; Zhang, M.F.; Cui, Y.J.; Zhuo, Z.L. Integrated analysis of single-cell RNA-seq and bulk RNA-seq unravels the heterogeneity of cancer-associated fibroblasts in TNBC. Aging 2023, 15, 12674–12697. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fang, Y.; Bao, L.L.; Wu, F.; Wang, S.L.; Hao, S.Y. Intercellular Communication Reveals Therapeutic Potential of Epithelial-Mesenchymal Transition in Triple-Negative Breast Cancer. Biomolecules 2022, 12, 1478. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Pal, B.; Chen, Y.S.; Vaillant, F.; Capaldo, B.D.; Joyce, R.; Song, X.Y.; Bryant, V.L.; Penington, J.S.; Di Stefano, L.; Ribera, N.T.; et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. Embo J. 2021, 40, e107333. [Google Scholar] [CrossRef]
- Gray, G.K.; Li, C.M.; Rosenbluth, J.M.; Selfors, L.M.; Girnius, N.; Lin, J.R.; Schackmann, R.C.J.; Goh, W.L.; Moore, K.; Shapiro, H.K.; et al. A human breast atlas integrating single-cell proteomics and transcriptomics. Dev. Cell 2022, 57, 1400–1420.e7. [Google Scholar] [CrossRef]
- Kim, H.; Lee, J.; Kang, K.; Yoon, S. MarkerCount: A stable, count-based cell type identifier for single-cell RNA-seq experiments. Comput. Struct. Biotechnol. J. 2022, 20, 3120–3132. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Coyington, K.R.; Contreras, A.; Fuqua, S.A.W.; Sayage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Lacle, M.M.; Moelans, C.B.; Kornegoor, R.; van der Pol, C.; Witkamp, A.J.; van der Wall, E.; Rueschoff, J.; Buerger, H.; van Diest, P.J. Chromosome 17 copy number changes in male breast cancer. Cell Oncol. 2015, 38, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Garrido, M.A.; Rodriguez, T.; Zinchenko, S.; Maleno, I.; Ruiz-Cabello, F.; Concha, A.; Olea, N.; Garrido, F.; Aptsiauri, N. HLA class I alterations in breast carcinoma are associated with a high frequency of the loss of heterozygosity at chromosomes 6 and 15. Immunogenetics 2018, 70, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Bueno-Fortes, S.; Berral-Gonzalez, A.; Sanchez-Santos, J.M.; Martin-Merino, M.; De Las Rivas, J. Identification of a gene expression signature associated with breast cancer survival and risk that improves clinical genomic platforms. Bioinform. Adv. 2023, 3, vbad037. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, Q.; Chen, H.; Hu, L.; Negi, H.; Zheng, Y.; Ahmed, Y.; Wu, Z.; Li, D. Binding of anterior gradient 2 and estrogen receptor-alpha: Dual critical roles in enhancing fulvestrant resistance and IGF-1-induced tumorigenesis of breast cancer. Cancer Lett. 2016, 377, 32–43. [Google Scholar] [CrossRef]
- Obacz, J.; Sommerova, L.; Sicari, D.; Durech, M.; Avril, T.; Iuliano, F.; Pastorekova, S.; Hrstka, R.; Chevet, E.; Delom, F.; et al. Extracellular AGR3 regulates breast cancer cells migration via Src signaling. Oncol. Lett. 2019, 18, 4449–4456. [Google Scholar] [CrossRef]
- Fry, E.A.; Taneja, P.; Inoue, K. Clinical applications of mouse models for breast cancer engaging HER2/neu. Integr. Cancer Sci. Ther. 2016, 3, 593–603. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Winter, M.; Meignan, S.; Volkel, P.; Angrand, P.O.; Chopin, V.; Bidan, N.; Toillon, R.A.; Adriaenssens, E.; Lagadec, C.; Le Bourhis, X. Vimentin Promotes the Aggressiveness of Triple Negative Breast Cancer Cells Surviving Chemotherapeutic Treatment. Cells 2021, 10, 1504. [Google Scholar] [CrossRef] [PubMed]
- Alnuaimi, A.R.; Nair, V.A.; Malhab, L.J.B.; Abu-Gharbieh, E.; Ranade, A.V.; Pintus, G.; Hamad, M.; Busch, H.; Kirfel, J.; Hamoudi, R.; et al. Emerging role of caldesmon in cancer: A potential biomarker for colorectal cancer and other cancers. World J. Gastrointest. Oncol. 2022, 14, 1637–1653. [Google Scholar] [CrossRef]
- Reimer, F.; Bryan, S.; Legler, K.; Karn, T.; Eppenberger-Castori, S.; Matschke, J.; Pereira-Veiga, T.; Wikman, H.; Witzel, I.; Muller, V.; et al. The role of the desmosomal protein desmocollin 2 in tumour progression in triple negative breast cancer patients. Cancer Cell Int. 2023, 23, 47. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Feng, X.; Wang, Y.; Li, Q.; Li, X. Dysregulation of tumour microenvironment driven by circ-TPGS2/miR-7/TRAF6/NF-kappaB axis facilitates breast cancer cell motility. Autoimmunity 2021, 54, 284–293. [Google Scholar] [CrossRef]
- Gromova, I.; Gromov, P.; Honma, N.; Kumar, S.; Rimm, D.; Talman, M.L.; Wielenga, V.T.; Moreira, J.M. High level PHGDH expression in breast is predominantly associated with keratin 5-positive cell lineage independently of malignancy. Mol. Oncol. 2015, 9, 1636–1654. [Google Scholar] [CrossRef]
- Liu, R.Z.; Graham, K.; Glubrecht, D.D.; Germain, D.R.; Mackey, J.R.; Godbout, R. Association of FABP5 expression with poor survival in triple-negative breast cancer: Implication for retinoic acid therapy. Am. J. Pathol. 2011, 178, 997–1008. [Google Scholar] [CrossRef]
- Han, D.; Wang, L.; Chen, B.; Zhao, W.; Liang, Y.; Li, Y.; Zhang, H.; Liu, Y.; Wang, X.; Chen, T.; et al. USP1-WDR48 deubiquitinase complex enhances TGF-beta induced epithelial–mesenchymal transition of TNBC cells via stabilizing TAK1. Cell Cycle 2021, 20, 320–331. [Google Scholar] [CrossRef]
- Pernicone, N.; Peretz, L.; Grinshpon, S.; Listovsky, T. MDA-MB-157 Cell Line Presents High Levels of MAD2L2 and Dysregulated Mitosis. Anticancer. Res. 2020, 40, 5471–5480. [Google Scholar] [CrossRef]
- Shi, C.; Yang, X.; Bu, X.; Hou, N.; Chen, P. Alpha B-crystallin promotes the invasion and metastasis of colorectal cancer via epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2017, 489, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Yang, X.; Wang, X.; Zhao, L.; Zou, L.; Yang, Q.; Hou, Z.; Tan, J.; Zhang, H.; et al. GABRP sustains the stemness of triple-negative breast cancer cells through EGFR signaling. Cancer Lett. 2021, 514, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Wu, Y.; Liu, Z.; Yu, Y.; Sun, S.; Guo, M.; Sun, B.; Huang, C. CDKN2A-mediated molecular subtypes characterize the hallmarks of tumor microenvironment and guide precision medicine in triple-negative breast cancer. Front. Immunol. 2022, 13, 970950. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Kim, Y.; Lee, S.B.; Oh, C.W.; Lee, E.J.; Ko, J.Y.; Park, J.H. Autophagy inhibits cancer stemness in triple-negative breast cancer via miR-181a-mediated regulation of ATG5 and/or ATG2B. Mol. Oncol. 2022, 16, 1857–1875. [Google Scholar] [CrossRef]
- Zhou, G.; Yang, L.; Gray, A.; Srivastava, A.K.; Li, C.; Zhang, G.; Cui, T. The role of desmosomes in carcinogenesis. Onco Targets Ther. 2017, 10, 4059–4063. [Google Scholar] [CrossRef] [PubMed]
- Ahram, M.; Abdullah, M.S.; Mustafa, S.A.; Alsafadi, D.B.; Battah, A.H. Androgen downregulates desmocollin-2 in association with induction of mesenchymal transition of breast MDA-MB-453 cancer cells. Cytoskeleton 2021, 78, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, J.S.; Han, J.W. Analysis of Tumor Microenvironment Heterogeneity among Breast Cancer Subtypes to Identify Subtype-Specific Signatures. Genes 2023, 14, 44. [Google Scholar] [CrossRef]
- Valenza, C.; Salimbeni, B.T.; Santoro, C.; Trapani, D.; Antonarelli, G.; Curigliano, G. Tumor Infiltrating Lymphocytes across Breast Cancer Subtypes: Current Issues for Biomarker Assessment. Cancers 2023, 15, 767. [Google Scholar] [CrossRef]
- Chimento, A.; D’Amico, M.; Pezzi, V.; De Amicis, F. Notch Signaling in Breast Tumor Microenvironment as Mediator of Drug Resistance. Int. J. Mol. Sci. 2022, 23, 6296. [Google Scholar] [CrossRef]
- Akil, A.; Gutiérrez-García, A.K.; Guenter, R.; Rose, J.B.; Beck, A.W.; Chen, H.; Ren, B. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front. Cell Dev. Biol. 2021, 9, 642352. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Najor, N.A.; Green, K.J. Desmosomes: Regulators of cellular signaling and adhesion in epidermal health and disease. Cold Spring Harb. Perspect. Med. 2014, 4, a015297. [Google Scholar] [CrossRef] [PubMed]
- Chidgey, M.; Dawson, C. Desmosomes: A role in cancer? Br. J. Cancer 2007, 96, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.; Hatzfeld, M.; Keil, R. Desmosomes as Signaling Hubs in the Regulation of Cell Behavior. Front. Cell Dev. Biol. 2021, 9, 745670. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.H.; Chen, M.C.; Tsai, Y.P.; Tan, G.Y.T.; Hsu, P.H.; Jeng, Y.M.; Tsai, Y.F.; Yang, M.H.; Hwang-Verslues, W.W. Interplay between desmoglein2 and hypoxia controls metastasis in breast cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2014408118. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Duran, I.; Lee, J.L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.Z.; et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef]
- Joshi, R.S.; Kanugula, S.S.; Sudhir, S.; Pereira, M.P.; Jain, S.; Aghi, M.K. The Role of Cancer-Associated Fibroblasts in Tumor Progression. Cancers 2021, 13, 1399. [Google Scholar] [CrossRef]
- Feng, B.; Wu, J.; Shen, B.; Jiang, F.; Feng, J. Cancer-associated fibroblasts and resistance to anticancer therapies: Status, mechanisms, and countermeasures. Cancer Cell Int. 2022, 22, 166. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue Type | Num. Cells | Num. Genes | Num. Samples | |
|---|---|---|---|---|
| GSE176078 | Tumor | 100,064 | 29,733 | 26 (12, 5, 9, 0, 0) |
| GSE161529 | Tumor/Normal | 428,024 | 33,538 | 62 (20, 6, 8, 4, 24) |
| GSE180878 | Normal | 52,681 | 20,437 | 16 (0, 0, 0, 0, 16) |
| Gene | Description | Reference |
|---|---|---|
| VIM | Treatment-induced upregulation of VIM is associated with increased invasion in breast cancer cells; silencing it reduces metastasis, suggesting therapeutic promise. | [35] |
| CALD1 | Upregulated CALD1 gene expression in breast cancer is linked to aggressive tumor traits, such as heightened growth rate and cell motility, fostering carcinogenesis via epithelial–mesenchymal transition induction. | [36] |
| DSC2 | In epithelial cells, DSC2 encodes desmocollin protein, which is closely linked to heart disease and plays a crucial role in cell adhesion. However, its overexpression in tumors increases cell cohesion, hindering chemotherapy diffusion and promoting tumor growth. This is particularly associated with poor prognosis in aggressive subtypes like HER2-positive and TNBC. | [37] |
| TPGS2 | TPGS2 is pivotal in modulating the tumor microenvironment (TME) and fostering metastasis in breast cancer via the circ-TPGS2-mediated miR-7/TRAF6/NF-κB signaling cascade, creating a feedback loop that enhances the pro-metastatic impact of circ-TPGS2. | [38] |
| PHGDH | Overexpression of PHGDH enzyme is a hallmark of cancer cells, linked to accelerated cell division and proliferation, with ongoing research aiming to develop targeted therapies; in TNBC, it tends to be upregulated, often associated with CK5-positive cell lineage across diverse tissues and cancer types. | [39] |
| FABP5 | Within breast tissue, FABP5 facilitates fatty acid delivery, supporting cancer cell proliferation; its upregulation in prostate and breast cancer cells, notably in HR-negative breast cancer with poor prognosis, correlates with EpCAM upregulation, driven by c-Myc to promote tumor cell survival and progression. | [40] |
| USP1 | Mutations in USP1 can disrupt DNA repair regulation, potentially leading to cancer development; meanwhile, the USP1/WDR48 complex mediates TGF-β signaling, enhancing TNBC cell mobility and EMT. | [41] |
| MAD2L2 | Dysregulation or mutation of MAD2L2, involved in error detection and regulation of cell division, impacts tumor development and growth, drawing attention as a potential therapeutic target. Particularly, its overexpression in MDA-MB-157 TNBC cells correlates with enhanced cell division promotion compared to other breast cancer subtypes. | [42] |
| CRYAB | Highly expressed in several cancer types, including breast, cervical, and renal cell carcinoma, CRYAB binds to proteins, promoting cell survival and exerting a protective role. In breast cancer, it is associated with tumor invasion, metastasis, and prognosis, with breast epithelial cells expressing elevated CRYAB levels showing tumor-like growth patterns in vitro, implicating CRYAB in tumor formation. | [43] |
| GABRP | Recent research confirms activation of the EGFR signaling pathway in TNBC, where abundant GABRP in TNBC stem cells interacts with EGFR, sustaining its high expression levels and elevating chemoresistance. Inhibiting GABRP-induced EGFR signaling, which includes paclitaxel, doxorubicin, and cisplatin, holds promise for improving chemotherapy response. | [44] |
| CDKN2A | In a recent study of 587 TNBC patients, tumor samples were categorized into six subtypes, revealing abnormal amplification of the CDKN2A/B gene in the basal-like 1 type. Our findings indicate upregulated CDKN2A expression in TNBC patients, possibly due to defects in the CDKN2A gene itself, known as a tumor suppressor that halts the cell cycle during the G1 phase. | [6,45] |
| ATG5 | ATG5 and ATG2B are regulated by miR-181a, which weakens cancer stem cells and enhances autophagy in TNBC. This suggests ATG5′s role in suppressing cancer stem cells in TNBC, offering potential for TNBC treatment using compounds like curcumin. | [46] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Yang, W.; Hong, D.; Won, H.S.; Yoon, S. A Retrospective View of the Triple-Negative Breast Cancer Microenvironment: Novel Markers, Interactions, and Mechanisms of Tumor-Associated Components Using Public Single-Cell RNA-Seq Datasets. Cancers 2024, 16, 1173. https://doi.org/10.3390/cancers16061173
Kim M, Yang W, Hong D, Won HS, Yoon S. A Retrospective View of the Triple-Negative Breast Cancer Microenvironment: Novel Markers, Interactions, and Mechanisms of Tumor-Associated Components Using Public Single-Cell RNA-Seq Datasets. Cancers. 2024; 16(6):1173. https://doi.org/10.3390/cancers16061173
Chicago/Turabian StyleKim, Minsoo, Wonhee Yang, Dawon Hong, Hye Sung Won, and Seokhyun Yoon. 2024. "A Retrospective View of the Triple-Negative Breast Cancer Microenvironment: Novel Markers, Interactions, and Mechanisms of Tumor-Associated Components Using Public Single-Cell RNA-Seq Datasets" Cancers 16, no. 6: 1173. https://doi.org/10.3390/cancers16061173
APA StyleKim, M., Yang, W., Hong, D., Won, H. S., & Yoon, S. (2024). A Retrospective View of the Triple-Negative Breast Cancer Microenvironment: Novel Markers, Interactions, and Mechanisms of Tumor-Associated Components Using Public Single-Cell RNA-Seq Datasets. Cancers, 16(6), 1173. https://doi.org/10.3390/cancers16061173