Recent Advances in Therapeutic Strategies to Improve Colorectal Cancer Treatment


Abstract
Simple Summary
Abstract
1. Introduction
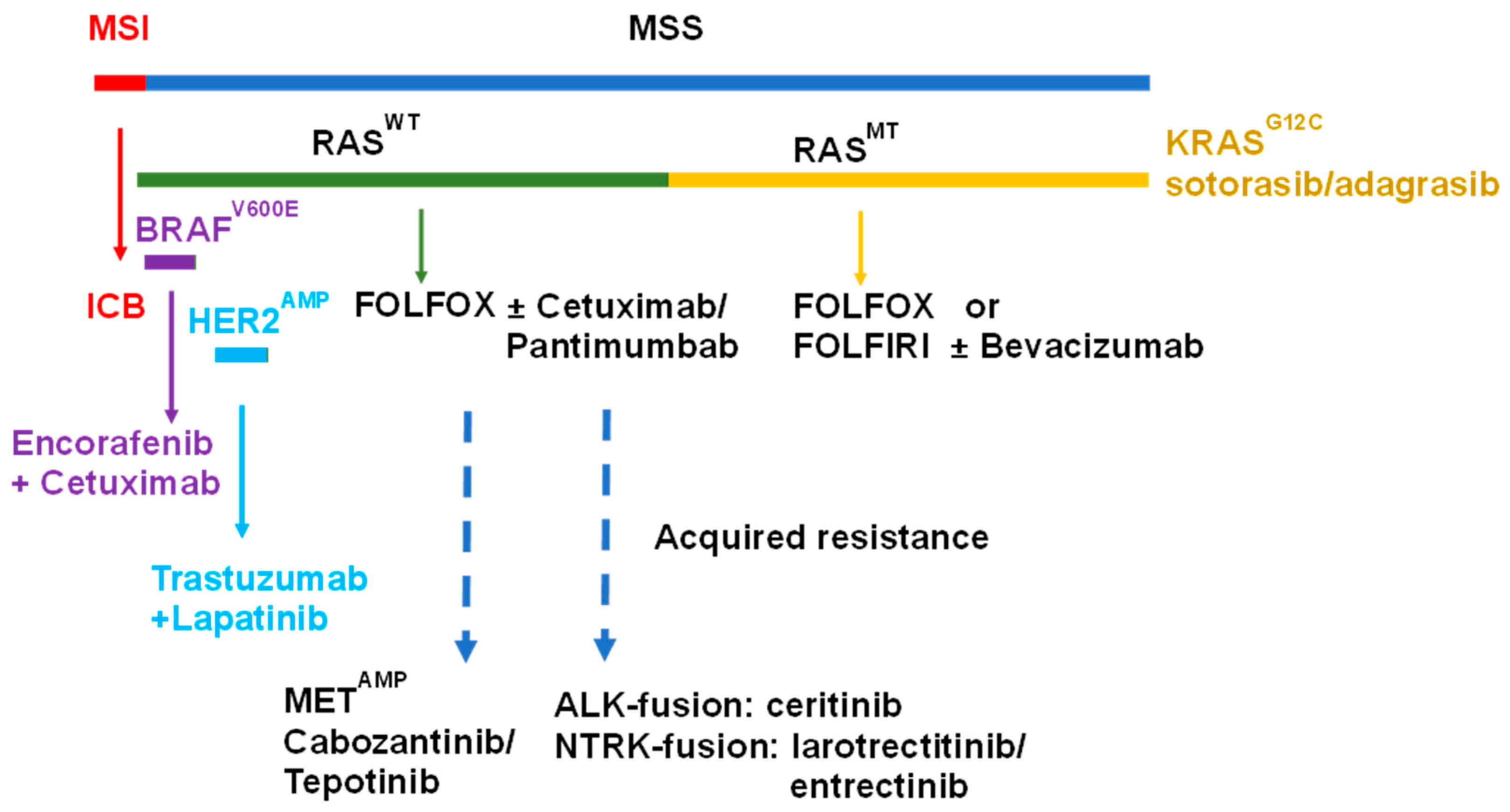
2. Biomarker-Based Stratification of mCRC
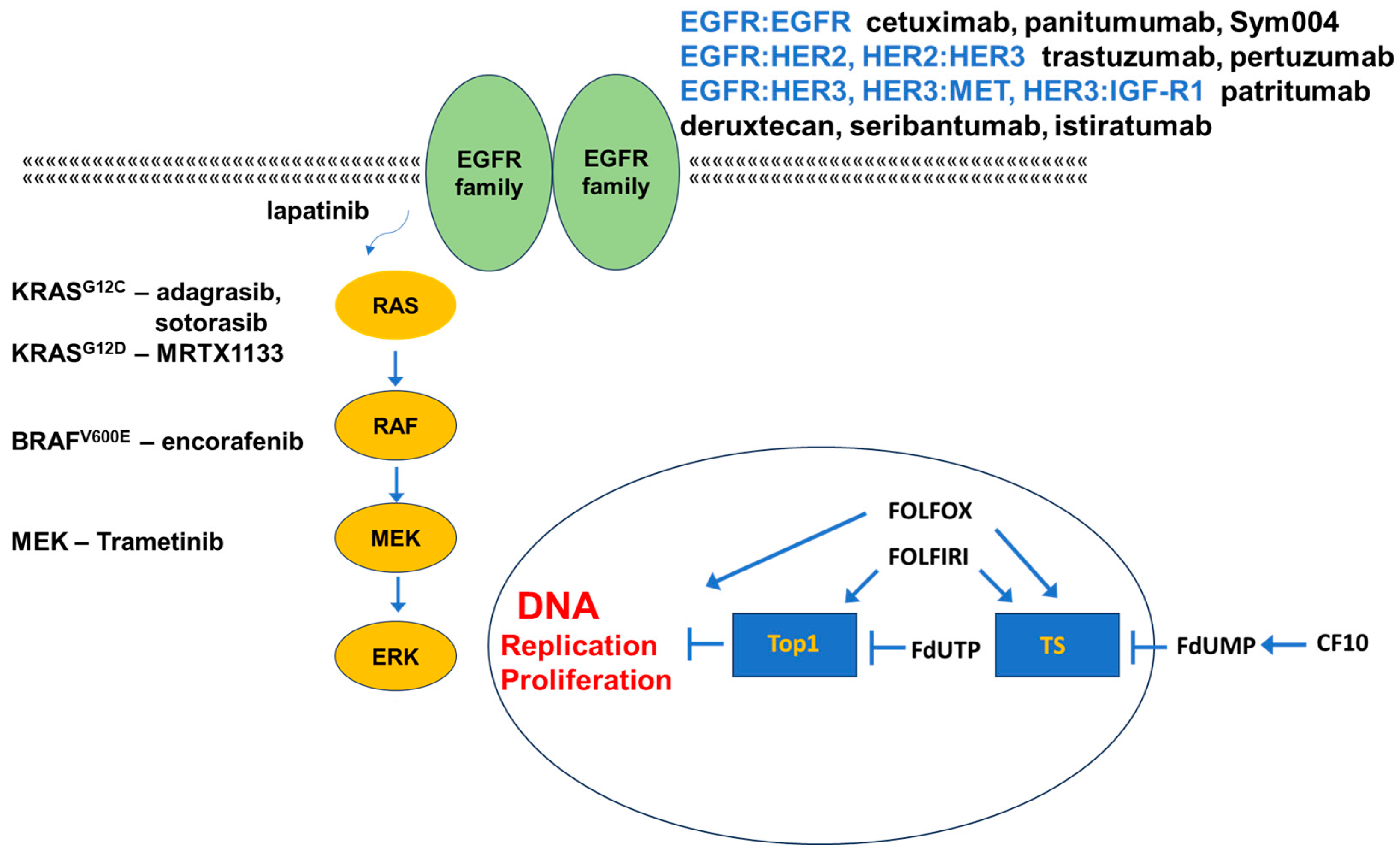
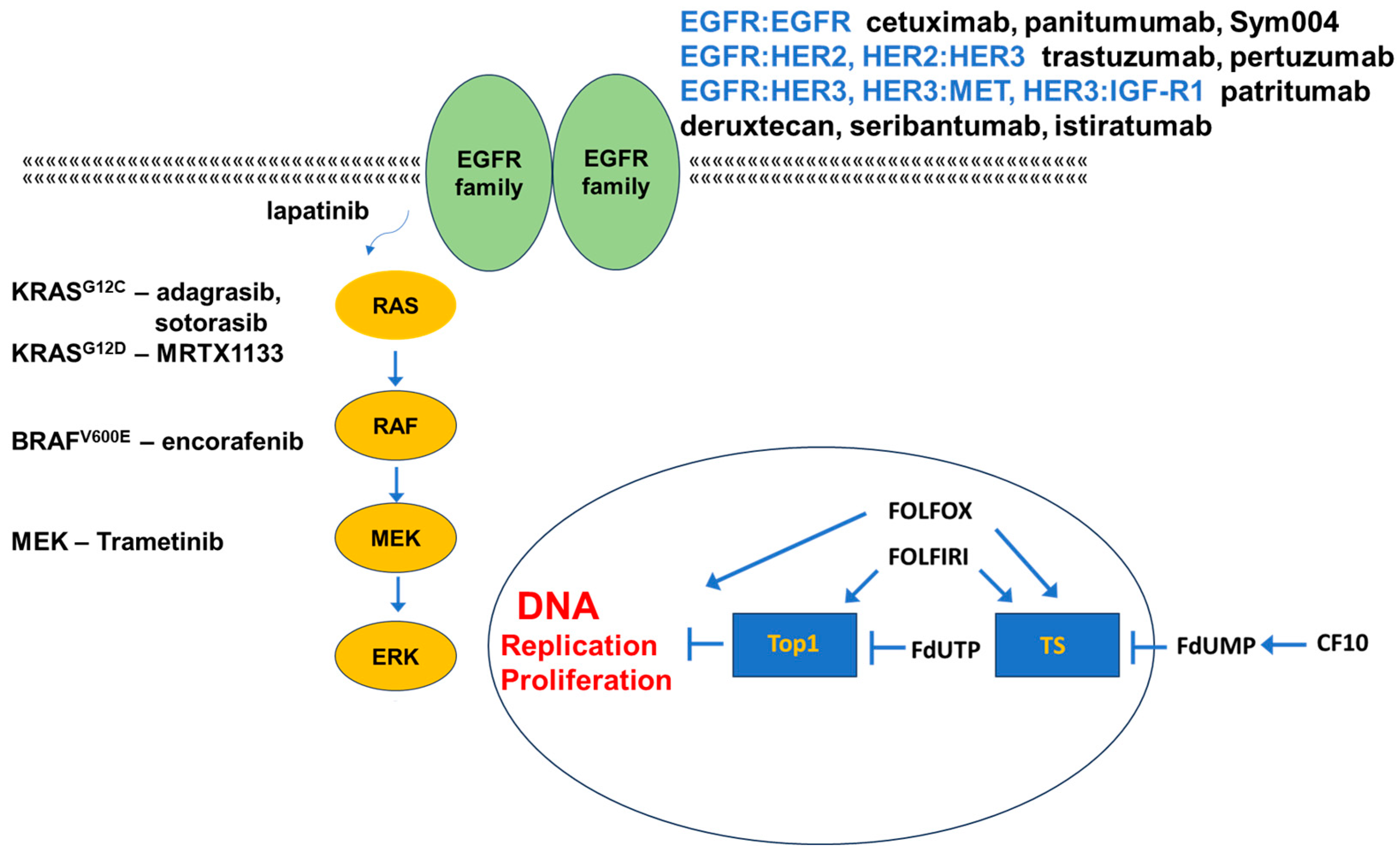
3. Targeting EGFR in mCRC
3.1. Targeting EGFR
3.2. Targeting Other EGFR-Family Members
3.3. Alternative Targets in EGFR-Resistant mCRC
4. Targeting Mutant KRAS
5. Targeting Mutant BRAF
6. Alternative Targets of Activated EGFR
7. Wnt-Pathway Targeting
8. Targeting VEGFR
9. FP-Based Chemotherapy in CRC
9.1. Adjuvant Chemotherapy in Primary Colorectal Cancer
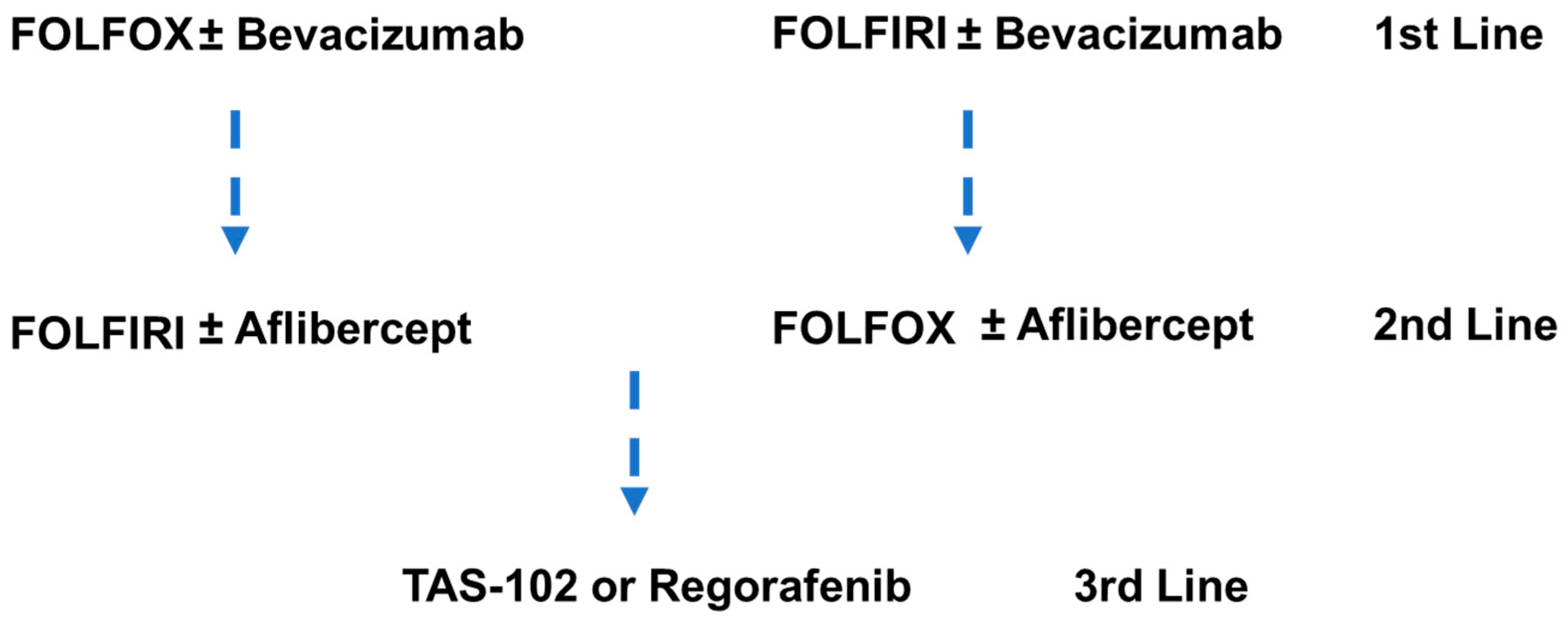
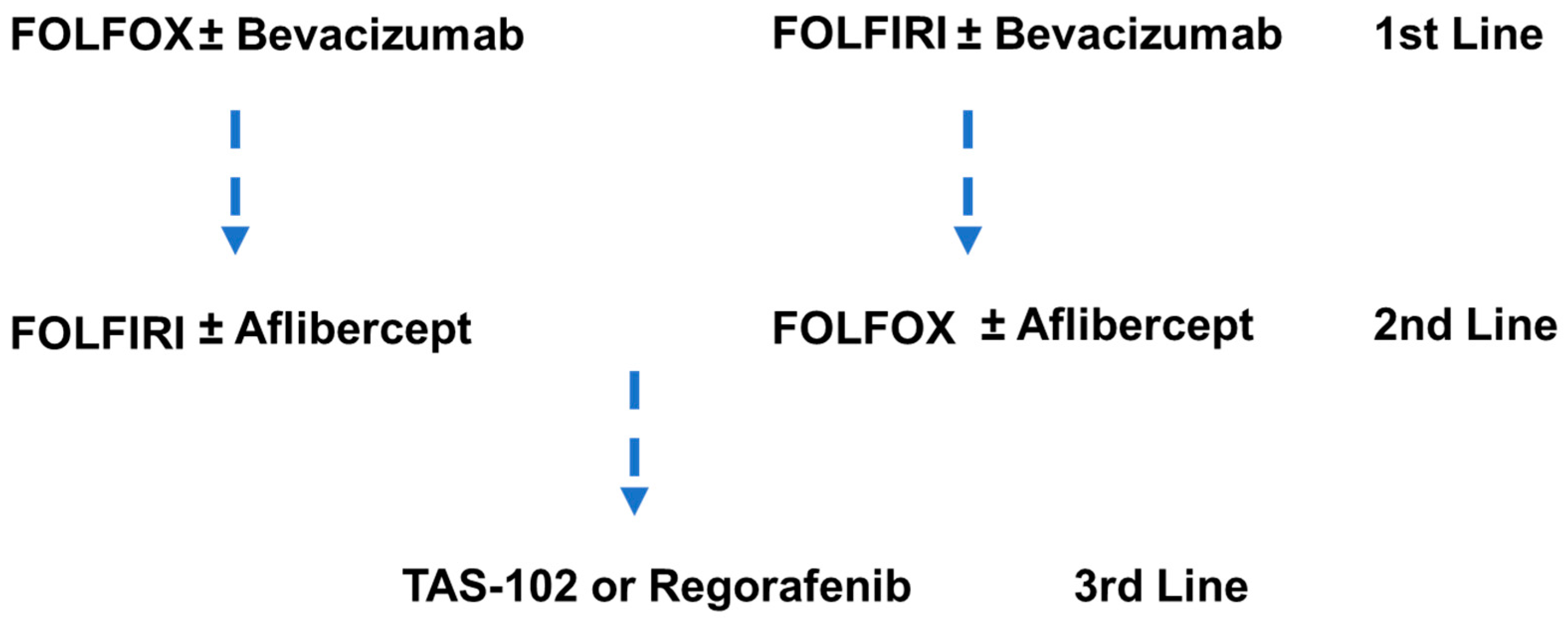
9.2. Chemotherapy in Metastatic Colorectal Cancer
10. Immunotherapy Approaches for mCRC
11. Conclusions and Future Perspectives
Funding
Data Availability Statement
Conflicts of Interest
References
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Kusnik, A.; Renjithlal, S.L.M.; Chodos, A.; Shanmukhappa, S.C.; Eid, M.M.; Renjith, K.M.; Alweis, R. Trends in Colorectal Cancer Mortality in the United States, 1999–2020. Gastroenterol. Res. 2023, 16, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Kasi, P.M.; Shahjehan, F.; Cochuyt, J.J.; Li, Z.; Colibaseanu, D.T.; Merchea, A. Rising Proportion of Young Individuals with Rectal and Colon Cancer. Clin. Color. Cancer 2019, 18, e87–e95. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, F.; Ou, F.S.; Qu, X.; Zemla, T.J.; Niedzwiecki, D.; Tam, R.; Mahajan, S.; Goldberg, R.M.; Bertagnolli, M.M.; Blanke, C.D.; et al. Mutational Analysis of Patients with Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J. Clin. Oncol. 2019, 37, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lan, Z.; Liao, W.; Horner, J.W.; Xu, X.; Liu, J.; Yoshihama, Y.; Jiang, S.; Shim, H.S.; Slotnik, M.; et al. Histone demethylase KDM5D upregulation drives sex differences in colon cancer. Nature 2023, 619, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, S.; Liu, Y.; Zhang, C.; Li, H.; Lai, B. Metastatic patterns and survival outcomes in patients with stage IV colon cancer: A population-based analysis. Cancer Med. 2020, 9, 361–373. [Google Scholar] [CrossRef]
- Gmeiner, W.H.; Okechukwu, C.C. Review of 5-FU resistance mechanisms in colorectal cancer: Clinical significance of attenuated on-target effects. Cancer Drug Resist. 2023, 6, 257–272. [Google Scholar] [CrossRef]
- Wilson, P.M.; Danenberg, P.V.; Johnston, P.G.; Lenz, H.J.; Ladner, R.D. Standing the test of time: Targeting thymidylate biosynthesis in cancer therapy. Nat. Rev. Clin. Oncol. 2014, 11, 282–298. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Punt, C.J.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2016, 14, 235–246. [Google Scholar] [CrossRef]
- Zeineddine, F.A.; Zeineddine, M.A.; Yousef, A.; Gu, Y.; Chowdhury, S.; Dasari, A.; Huey, R.W.; Johnson, B.; Kee, B.; Lee, M.S.; et al. Survival improvement for patients with metastatic colorectal cancer over twenty years. NPJ Precis. Oncol. 2023, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Koroukian, S.M.; Booker, B.D.; Vu, L.; Schumacher, F.R.; Rose, J.; Cooper, G.S.; Selfridge, J.E.; Markt, S.C. Receipt of Targeted Therapy and Survival Outcomes in Patients with Metastatic Colorectal Cancer. JAMA Netw. Open 2023, 6, e2250030. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Bonazzina, E.; Amatu, A.; Tosi, F.; Bencardino, K.; Gori, V.; Massihnia, D.; Cipani, T.; Spina, F.; Ghezzi, S.; et al. The Evolutionary Landscape of Treatment for BRAF(V600E) Mutant Metastatic Colorectal Cancer. Cancers 2021, 13, 137. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Ivey, G.D.; Johnston, F.M.; Azad, N.S.; Christenson, E.S.; Lafaro, K.J.; Shubert, C.R. Current Surgical Management Strategies for Colorectal Cancer Liver Metastases. Cancers 2022, 14, 1063. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Goel, A.; Chung, D.C. Pathways of Colorectal Carcinogenesis. Gastroenterology 2020, 158, 291–302. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarro, L.; Couturier, D.L.; Liu, L.; Schneider, M.; et al. A pan-cancer compendium of chromosomal instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef]
- Serebriiskii, I.G.; Connelly, C.; Frampton, G.; Newberg, J.; Cooke, M.; Miller, V.; Ali, S.; Ross, J.S.; Handorf, E.; Arora, S.; et al. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat. Commun. 2019, 10, 3722. [Google Scholar] [CrossRef]
- Jiang, H.; Grenley, M.O.; Bravo, M.J.; Blumhagen, R.Z.; Edgar, B.A. EGFR/Ras/MAPK signaling mediates adult midgut epithelial homeostasis and regeneration in Drosophila. Cell Stem Cell 2011, 8, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Puccini, A.; Tie, J. Redefining Colorectal Cancer by Tumor Biology. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Tanjak, P.; Chaiboonchoe, A.; Suwatthanarak, T.; Acharayothin, O.; Thanormjit, K.; Chanthercrob, J.; Suwatthanarak, T.; Wannasuphaphol, B.; Chumchuen, K.; Suktitipat, B.; et al. The KRAS-Mutant Consensus Molecular Subtype 3 Reveals an Immunosuppressive Tumor Microenvironment in Colorectal Cancer. Cancers 2023, 15, 1098. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen, S.; Corchete, L.A.; Gervas, R.; Rodriguez, A.; Garcia, M.; Alcazar, J.A.; Garcia, J.; Bengoechea, O.; Munoz-Bellvis, L.; Sayagues, J.M.; et al. Prognostic implications of EGFR protein expression in sporadic colorectal tumors: Correlation with copy number status, mRNA levels and miRNA regulation. Sci. Rep. 2020, 10, 4662. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Mota, J.M.; Collier, K.A.; Barros Costa, R.L.; Taxter, T.; Kalyan, A.; Leite, C.A.; Chae, Y.K.; Giles, F.J.; Carneiro, B.A. A comprehensive review of heregulins, HER3, and HER4 as potential therapeutic targets in cancer. Oncotarget 2017, 8, 89284–89306. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, A.; Kinehara, Y.; Kijima, R.; Tanaka, M.; Ninomiya, R.; Jokoji, R.; Tachibana, I. Metastatic Lung Tumors from Colorectal Cancer with EGFR Mutations That Responded to Osimertinib. Intern. Med. 2023, 62, 769–773. [Google Scholar] [CrossRef]
- Li, Q.H.; Wang, Y.Z.; Tu, J.; Liu, C.W.; Yuan, Y.J.; Lin, R.; He, W.L.; Cai, S.R.; He, Y.L.; Ye, J.N. Anti-EGFR therapy in metastatic colorectal cancer: Mechanisms and potential regimens of drug resistance. Gastroenterol. Rep. 2020, 8, 179–191. [Google Scholar] [CrossRef]
- Liu, T.; Jiang, S.; Teng, X.; Zhong, L.; Liu, M.; Jin, Y.; Dong, M. A comparison of panitumumab and cetuximab in the treatment of KRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Immunopharmacol. Immunotoxicol. 2023, 45, 1–9. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Lach, K.; Hsu, L.I.; Siadak, M.; Stecher, M.; Ward, J.; Beckerman, R.; Strickler, J.H. Impact of Anti-EGFR Therapies on HER2-Positive Metastatic Colorectal Cancer: A Systematic Literature Review and Meta-Analysis of Clinical Outcomes. Oncologist 2023, 28, 885–893. [Google Scholar] [CrossRef]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Cora, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef]
- Tevaarwerk, A.J.; Kolesar, J.M. Lapatinib: A small-molecule inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases used in the treatment of breast cancer. Clin. Ther. 2009, 31 Pt 2, 2332–2348. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Nakamura, Y.; Okamoto, W.; Kato, T.; Esaki, T.; Kato, K.; Komatsu, Y.; Yuki, S.; Masuishi, T.; Nishina, T.; Ebi, H.; et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: A phase 2 trial. Nat. Med. 2021, 27, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, E.; Cervera, P.; Andre, T.; Cohen, R. Targeting HER2 in colorectal cancer. Bull. Cancer 2023, 110, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Ledel, F.; Hallstrom, M.; Ragnhammar, P.; Ohrling, K.; Edler, D. HER3 expression in patients with primary colorectal cancer and corresponding lymph node metastases related to clinical outcome. Eur. J. Cancer 2014, 50, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Kilroy, M.K.; Park, S.; Feroz, W.; Patel, H.; Mishra, R.; Alanazi, S.; Garrett, J.T. HER3 Alterations in Cancer and Potential Clinical Implications. Cancers 2022, 14, 6174. [Google Scholar] [CrossRef] [PubMed]
- Rathore, M.; Zhang, W.; Wright, M.; Bhattacharya, R.; Fan, F.; Vaziri-Gohar, A.; Winter, J.; Wang, Z.; Markowitz, S.D.; Willis, J.; et al. Liver Endothelium Promotes HER3-Mediated Cell Survival in Colorectal Cancer with Wild-Type and Mutant KRAS. Mol. Cancer Res. 2022, 20, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Gandullo-Sanchez, L.; Ocana, A.; Pandiella, A. HER3 in cancer: From the bench to the bedside. J. Exp. Clin. Cancer Res. 2022, 41, 310. [Google Scholar] [CrossRef] [PubMed]
- Koyama, K.; Ishikawa, H.; Abe, M.; Shiose, Y.; Ueno, S.; Qiu, Y.; Nakamaru, K.; Murakami, M. Patritumab deruxtecan (HER3-DXd), a novel HER3 directed antibody drug conjugate, exhibits in vitro activity against breast cancer cells expressing HER3 mutations with and without HER2 overexpression. PLoS ONE 2022, 17, e0267027. [Google Scholar] [CrossRef]
- Azadi, A.; Golchini, A.; Delazar, S.; Abarghooi Kahaki, F.; Dehnavi, S.M.; Payandeh, Z.; Eyvazi, S. Recent Advances on Immune Targeted Therapy of Colorectal Cancer Using bi-Specific Antibodies and Therapeutic Vaccines. Biol. Proced. Online 2021, 23, 13. [Google Scholar] [CrossRef]
- Lucas, L.M.; Dwivedi, V.; Senfeld, J.I.; Cullum, R.L.; Mill, C.P.; Piazza, J.T.; Bryant, I.N.; Cook, L.J.; Miller, S.T.; Lott, J.H.t.; et al. The Yin and Yang of ERBB4: Tumor Suppressor and Oncoprotein. Pharmacol. Rev. 2022, 74, 18–47. [Google Scholar] [CrossRef]
- Loupakis, F.; Cremolini, C.; Fioravanti, A.; Orlandi, P.; Salvatore, L.; Masi, G.; Schirripa, M.; Di Desidero, T.; Antoniotti, C.; Canu, B.; et al. EGFR ligands as pharmacodynamic biomarkers in metastatic colorectal cancer patients treated with cetuximab and irinotecan. Target. Oncol. 2014, 9, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Salazar, R.; Tabernero, J. Overcoming Resistance to Anti-EGFR Therapy in Colorectal Cancer. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e149–e156. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ji, Q.; Li, Q. Resistance to anti-EGFR therapies in metastatic colorectal cancer: Underlying mechanisms and reversal strategies. J. Exp. Clin. Cancer Res. 2021, 40, 328. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Brand, T.M.; Starr, M.M.; Li, C.; Huppert, E.J.; Luthar, N.; Pedersen, M.W.; Horak, I.D.; Kragh, M.; Wheeler, D.L. Sym004, a novel EGFR antibody mixture, can overcome acquired resistance to cetuximab. Neoplasia 2013, 15, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Siravegna, G.; Mussolin, B.; Kearns, J.D.; Wolf, B.B.; Misale, S.; Lazzari, L.; Bertotti, A.; Trusolino, L.; Adjei, A.A.; et al. MM-151 overcomes acquired resistance to cetuximab and panitumumab in colorectal cancers harboring EGFR extracellular domain mutations. Sci. Transl. Med. 2016, 8, 324ra314. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.J.; Basu Mallick, A.; Dotan, E.; Cohen, S.J.; Gold, P.J.; Hochster, H.S.; Subramaniam, S.; Barzi, A.; Watts, G.S.; Blatchford, P.J.; et al. A Phase II Study Investigating Cabozantinib in Patients with Refractory Metastatic Colorectal Cancer (AGICC 17CRC01). Cancer Res. Commun. 2022, 2, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Van Cutsem, E.; Prausova, J.; Isambert, N.; Neuzillet, C.; Siena, S.; Pietrantonio, F.; Falk, S.; Wainberg, Z.A.; Raghav, K.P.S.; et al. Tepotinib plus cetuximab in patients (pts) with RAS/BRAF wild-type left-sided metastatic colorectal cancer (mCRC) and acquired resistance to anti-EGFR antibody therapy due to MET amplification (METamp). J. Clin. Oncol. 2021, 39, TPS149. [Google Scholar] [CrossRef]
- Jia, J.; Morse, M.A.; Nagy, R.J.; Lanman, R.B.; Strickler, J.H. Cell-Free DNA Profiling to Discover Mechanisms of Exceptional Response to Cabozantinib Plus Panitumumab in a Patient with Treatment Refractory Metastatic Colorectal Cancer. Front. Oncol. 2018, 8, 305. [Google Scholar] [CrossRef]
- Reidy, D.L.; Vakiani, E.; Fakih, M.G.; Saif, M.W.; Hecht, J.R.; Goodman-Davis, N.; Hollywood, E.; Shia, J.; Schwartz, J.; Chandrawansa, K.; et al. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 4240–4246. [Google Scholar] [CrossRef]
- Sclafani, F.; Kim, T.Y.; Cunningham, D.; Kim, T.W.; Tabernero, J.; Schmoll, H.J.; Roh, J.K.; Kim, S.Y.; Park, Y.S.; Guren, T.K.; et al. A Randomized Phase II/III Study of Dalotuzumab in Combination with Cetuximab and Irinotecan in Chemorefractory, KRAS Wild-Type, Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2015, 107, djv258. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Seufferlein, T. Regorafenib. Recent. Results Cancer Res. 2018, 211, 45–56. [Google Scholar] [CrossRef]
- Fakih, M.; Sandhu, J.; Lim, D.; Li, X.; Li, S.; Wang, C. Regorafenib, Ipilimumab, and Nivolumab for Patients with Microsatellite Stable Colorectal Cancer and Disease Progression with Prior Chemotherapy: A Phase 1 Nonrandomized Clinical Trial. JAMA Oncol. 2023, 9, 627–634. [Google Scholar] [CrossRef]
- Subbiah, V.; Khawaja, M.R.; Hong, D.S.; Amini, B.; Yungfang, J.; Liu, H.; Johnson, A.; Schrock, A.B.; Ali, S.M.; Sun, J.X.; et al. First-in-human trial of multikinase VEGF inhibitor regorafenib and anti-EGFR antibody cetuximab in advanced cancer patients. JCI Insight 2017, 2, e90380. [Google Scholar] [CrossRef]
- Morano, F.; Pietrantonio, F. Anti-Epidermal Growth Factor Receptor Maintenance Therapy in Metastatic Colorectal Cancer-Another Piece to the Puzzle. JAMA Netw. Open 2023, 6, e2333488. [Google Scholar] [CrossRef] [PubMed]
- Deming, D.A.; Cavalcante, L.L.; Lubner, S.J.; Mulkerin, D.L.; LoConte, N.K.; Eickhoff, J.C.; Kolesar, J.M.; Fioravanti, S.; Greten, T.F.; Compton, K.; et al. A phase I study of selumetinib (AZD6244/ARRY-142866), a MEK1/2 inhibitor, in combination with cetuximab in refractory solid tumors and KRAS mutant colorectal cancer. Investig. New Drugs 2016, 34, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Van Emburgh, B.O.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Siena, S.; Bardelli, A. Acquired resistance to EGFR-targeted therapies in colorectal cancer. Mol. Oncol. 2014, 8, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Clifton, K.; Rich, T.A.; Parseghian, C.; Raymond, V.M.; Dasari, A.; Pereira, A.A.L.; Willis, J.; Loree, J.M.; Bauer, T.M.; Chae, Y.K.; et al. Identification of Actionable Fusions as an Anti-EGFR Resistance Mechanism Using a Circulating Tumor DNA Assay. JCO Precis. Oncol. 2019, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Yakirevich, E.; Resnick, M.B.; Mangray, S.; Wheeler, M.; Jackson, C.L.; Lombardo, K.A.; Lee, J.; Kim, K.M.; Gill, A.J.; Wang, K.; et al. Oncogenic ALK Fusion in Rare and Aggressive Subtype of Colorectal Adenocarcinoma as a Potential Therapeutic Target. Clin. Cancer Res. 2016, 22, 3831–3840. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.W.; Ou, Q.; Wu, X.; Nagasaka, M.; Shao, Y.; Ou, S.I.; Yang, Y. NTRK fusion positive colorectal cancer is a unique subset of CRC with high TMB and microsatellite instability. Cancer Med. 2022, 11, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting KRAS in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 57. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Zhong, K.; Jiang, T.; Liu, Z.; Kwan, H.Y.; Su, T. The current understanding on the impact of KRAS on colorectal cancer. Biomed. Pharmacother. 2021, 140, 111717. [Google Scholar] [CrossRef] [PubMed]
- Nash, G.M.; Gimbel, M.; Shia, J.; Nathanson, D.R.; Ndubuisi, M.I.; Zeng, Z.S.; Kemeny, N.; Paty, P.B. KRAS mutation correlates with accelerated metastatic progression in patients with colorectal liver metastases. Ann. Surg. Oncol. 2010, 17, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Montes, A.; Alonso Orduna, V.; Asensio Martinez, E.; Rodriguez Salas, N.; Torres, E.; Cacho Lavin, D.; Rodriguez Alonso, R.M.; Falco, E.; Oliva, J.C.; Cirera, L.; et al. The Frequency of Specific KRAS Mutations, and Their Impact on Treatment Choice and Survival, in Patients with Metastatic Colorectal Cancer. Oncologist 2023, 28, e902–e909. [Google Scholar] [CrossRef]
- Bteich, F.; Mohammadi, M.; Li, T.; Bhat, M.A.; Sofianidi, A.; Wei, N.; Kuang, C. Targeting KRAS in Colorectal Cancer: A Bench to Bedside Review. Int. J. Mol. Sci. 2023, 24, 12030. [Google Scholar] [CrossRef]
- Appels, N.M.; Beijnen, J.H.; Schellens, J.H. Development of farnesyl transferase inhibitors: A review. Oncologist 2005, 10, 565–578. [Google Scholar] [CrossRef]
- Adjei, A.A.; Mauer, A.; Bruzek, L.; Marks, R.S.; Hillman, S.; Geyer, S.; Hanson, L.J.; Wright, J.J.; Erlichman, C.; Kaufmann, S.H.; et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2003, 21, 1760–1766. [Google Scholar] [CrossRef]
- Riely, G.J.; Johnson, M.L.; Medina, C.; Rizvi, N.A.; Miller, V.A.; Kris, M.G.; Pietanza, M.C.; Azzoli, C.G.; Krug, L.M.; Pao, W.; et al. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J. Thorac. Oncol. 2011, 6, 1435–1437. [Google Scholar] [CrossRef]
- Zhang, F.L.; Kirschmeier, P.; Carr, D.; James, L.; Bond, R.W.; Wang, L.; Patton, R.; Windsor, W.T.; Syto, R.; Zhang, R.; et al. Characterization of Ha-ras, N-ras, Ki-Ras4A, and Ki-Ras4B as in vitro substrates for farnesyl protein transferase and geranylgeranyl protein transferase type I. J. Biol. Chem. 1997, 272, 10232–10239. [Google Scholar] [CrossRef]
- Papke, B.; Murarka, S.; Vogel, H.A.; Martin-Gago, P.; Kovacevic, M.; Truxius, D.C.; Fansa, E.K.; Ismail, S.; Zimmermann, G.; Heinelt, K.; et al. Identification of pyrazolopyridazinones as PDEdelta inhibitors. Nat. Commun. 2016, 7, 11360. [Google Scholar] [CrossRef]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Reita, D.; Pabst, L.; Pencreach, E.; Guerin, E.; Dano, L.; Rimelen, V.; Voegeli, A.C.; Vallat, L.; Mascaux, C.; Beau-Faller, M. Direct Targeting KRAS Mutation in Non-Small Cell Lung Cancer: Focus on Resistance. Cancers 2022, 14, 1321. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; El-Refai, S.M.; Sha, W.; Puccini, A.; Grothey, A.; George, T.J.; Hwang, J.J.; O’Neil, B.; Barrett, A.S.; Kadakia, K.C.; et al. Landscape of KRAS(G12C), Associated Genomic Alterations, and Interrelation with Immuno-Oncology Biomarkers in KRAS-Mutated Cancers. JCO Precis. Oncol. 2022, 6, e2100245. [Google Scholar] [CrossRef] [PubMed]
- Patelli, G.; Tosi, F.; Amatu, A.; Mauri, G.; Curaba, A.; Patane, D.A.; Pani, A.; Scaglione, F.; Siena, S.; Sartore-Bianchi, A. Strategies to tackle RAS-mutated metastatic colorectal cancer. ESMO Open 2021, 6, 100156. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.G.; Kopetz, S.; Kuboki, Y.; Kim, T.W.; Munster, P.N.; Krauss, J.C.; Falchook, G.S.; Han, S.W.; Heinemann, V.; Muro, K.; et al. Sotorasib for previously treated colorectal cancers with KRAS(G12C) mutation (CodeBreaK100): A prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 2022, 23, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.I.; Janne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients with Advanced KRAS(G12C) Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef]
- Fakih, M.G.; Salvatore, L.; Esaki, T.; Modest, D.P.; Lopez-Bravo, D.P.; Taieb, J.; Karamouzis, M.V.; Ruiz-Garcia, E.; Kim, T.W.; Kuboki, Y.; et al. Sotorasib plus Panitumumab in Refractory Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 389, 2125–2139. [Google Scholar] [CrossRef]
- Yaeger, R.; Weiss, J.; Pelster, M.S.; Spira, A.I.; Barve, M.; Ou, S.I.; Leal, T.A.; Bekaii-Saab, T.S.; Paweletz, C.P.; Heavey, G.A.; et al. Adagrasib with or without Cetuximab in Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 388, 44–54. [Google Scholar] [CrossRef]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Van Schaeybroeck, S.; Kalimutho, M.; Dunne, P.D.; Carson, R.; Allen, W.; Jithesh, P.V.; Redmond, K.L.; Sasazuki, T.; Shirasawa, S.; Blayney, J.; et al. ADAM17-dependent c-MET-STAT3 signaling mediates resistance to MEK inhibitors in KRAS mutant colorectal cancer. Cell Rep. 2014, 7, 1940–1955. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Ghosh, S.; Powell, R.; Roszik, J.; Park, Y.; Sobieski, M.; Sorokin, A.; Stephan, C.; Kopetz, S.; Ellis, L.M.; et al. Combining MEK and SRC inhibitors for treatment of colorectal cancer demonstrate increased efficacy in vitro but not in vivo. PLoS ONE 2023, 18, e0281063. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Strickler, J.H.; Bekaii-Saab, T.S.; Yaeger, R. BRAF-Mutated Advanced Colorectal Cancer: A Rapidly Changing Therapeutic Landscape. J. Clin. Oncol. 2022, 40, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Cremolini, C.; Mazard, T.; Vidal, J.; Virchow, I.; Tougeron, D.; Cuyle, P.J.; Chibaudel, B.; Kim, S.; Ghanem, I.; et al. Real-world first-line treatment of patients with BRAF(V600E)-mutant metastatic colorectal cancer: The CAPSTAN CRC study. ESMO Open 2022, 7, 100603. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Wakatsuki, T.; Suenaga, M.; Ichimura, T.; Ogura, M.; Takahari, D.; Ooki, A.; Suzuki, T.; Ota, Y.; et al. Non-V600E BRAF mutations and EGFR signaling pathway in colorectal cancer. Int. J. Cancer 2019, 145, 2488–2495. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Antoniotti, C.; Stein, A.; Bendell, J.; Gruenberger, T.; Rossini, D.; Masi, G.; Ongaro, E.; Hurwitz, H.; Falcone, A.; et al. Individual Patient Data Meta-Analysis of FOLFOXIRI Plus Bevacizumab Versus Doublets Plus Bevacizumab as Initial Therapy of Unresectable Metastatic Colorectal Cancer. J. Clin. Oncol. 2020, 38, 3314–3324. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Kotani, D.; Mondaca, S.; Parikh, A.R.; Bando, H.; Van Seventer, E.E.; Taniguchi, H.; Zhao, H.; Thant, C.N.; de Stanchina, E.; et al. Response to Anti-EGFR Therapy in Patients with BRAF non-V600-Mutant Metastatic Colorectal Cancer. Clin. Cancer Res. 2019, 25, 7089–7097. [Google Scholar] [CrossRef]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients with Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition with Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef]
- Oddo, D.; Sennott, E.M.; Barault, L.; Valtorta, E.; Arena, S.; Cassingena, A.; Filiciotto, G.; Marzolla, G.; Elez, E.; van Geel, R.M.; et al. Molecular Landscape of Acquired Resistance to Targeted Therapy Combinations in BRAF-Mutant Colorectal Cancer. Cancer Res. 2016, 76, 4504–4515. [Google Scholar] [CrossRef]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273. [Google Scholar] [CrossRef]
- Smalley, K.S.; Lioni, M.; Dalla Palma, M.; Xiao, M.; Desai, B.; Egyhazi, S.; Hansson, J.; Wu, H.; King, A.J.; Van Belle, P.; et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol. Cancer Ther. 2008, 7, 2876–2883. [Google Scholar] [CrossRef]
- Houles, T.; Lavoie, G.; Nourreddine, S.; Cheung, W.; Vaillancourt-Jean, E.; Guerin, C.M.; Bouttier, M.; Grondin, B.; Lin, S.; Saba-El-Leil, M.K.; et al. CDK12 is hyperactivated and a synthetic-lethal target in BRAF-mutated melanoma. Nat. Commun. 2022, 13, 6457. [Google Scholar] [CrossRef] [PubMed]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat. Commun. 2018, 9, 4782. [Google Scholar] [CrossRef]
- Papadatos-Pastos, D.; Rabbie, R.; Ross, P.; Sarker, D. The role of the PI3K pathway in colorectal cancer. Crit. Rev. Oncol. Hematol. 2015, 94, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shi, Y.L.; Zhou, K.; Wang, L.L.; Yan, Z.X.; Liu, Y.L.; Xu, L.L.; Zhao, S.W.; Chu, H.L.; Shi, T.T.; et al. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 2018, 9, 739. [Google Scholar] [CrossRef]
- Coleman, N.; Moyers, J.T.; Harbery, A.; Vivanco, I.; Yap, T.A. Clinical Development of AKT Inhibitors and Associated Predictive Biomarkers to Guide Patient Treatment in Cancer Medicine. Pharmgenomics Pers. Med. 2021, 14, 1517–1535. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.D.; Der, C.J. Differential involvement of RalA and RalB in colorectal cancer. Small GTPases 2012, 3, 126–130. [Google Scholar] [CrossRef]
- Moghadam, A.R.; Patrad, E.; Tafsiri, E.; Peng, W.; Fangman, B.; Pluard, T.J.; Accurso, A.; Salacz, M.; Shah, K.; Ricke, B.; et al. Ral signaling pathway in health and cancer. Cancer Med. 2017, 6, 2998–3013. [Google Scholar] [CrossRef]
- Naszai, M.; Bellec, K.; Yu, Y.; Roman-Fernandez, A.; Sandilands, E.; Johansson, J.; Campbell, A.D.; Norman, J.C.; Sansom, O.J.; Bryant, D.M.; et al. RAL GTPases mediate EGFR-driven intestinal stem cell proliferation and tumourigenesis. Elife 2021, 10, e63807. [Google Scholar] [CrossRef]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Uno, Y.; Ohbayashi, N.; Ohata, H.; Mimata, A.; Kukimoto-Niino, M.; Moriyama, H.; Kashimoto, S.; Inoue, T.; Goto, N.; et al. TNIK inhibition abrogates colorectal cancer stemness. Nat. Commun. 2016, 7, 12586. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Oh, J.; Peterson, H.M.; Carlson, J.C.T.; Pittet, M.J.; Weissleder, R. TNIK Inhibition Has Dual Synergistic Effects on Tumor and Associated Immune Cells. Adv. Biol. 2022, 6, e2200030. [Google Scholar] [CrossRef] [PubMed]
- Martins-Neves, S.R.; Paiva-Oliveira, D.I.; Fontes-Ribeiro, C.; Bovee, J.; Cleton-Jansen, A.M.; Gomes, C.M.F. IWR-1, a tankyrase inhibitor, attenuates Wnt/beta-catenin signaling in cancer stem-like cells and inhibits in vivo the growth of a subcutaneous human osteosarcoma xenograft. Cancer Lett. 2018, 414, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, L.; Pollock, K.; Guettler, S. Regulation of Wnt/beta-catenin signalling by tankyrase-dependent poly(ADP-ribosyl)ation and scaffolding. Br. J. Pharmacol. 2017, 174, 4611–4636. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A.; et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef] [PubMed]
- Burock, S.; Daum, S.; Keilholz, U.; Neumann, K.; Walther, W.; Stein, U. Phase II trial to investigate the safety and efficacy of orally applied niclosamide in patients with metachronous or sychronous metastases of a colorectal cancer progressing after therapy: The NIKOLO trial. Bmc Cancer 2018, 18, 297. [Google Scholar] [CrossRef] [PubMed]
- Handeli, S.; Simon, J.A. A small-molecule inhibitor of Tcf/beta-catenin signaling down-regulates PPARgamma and PPARdelta activities. Mol. Cancer Ther. 2008, 7, 521–529. [Google Scholar] [CrossRef]
- Fang, L.; Zhu, Q.; Neuenschwander, M.; Specker, E.; Wulf-Goldenberg, A.; Weis, W.I.; von Kries, J.P.; Birchmeier, W. A Small-Molecule Antagonist of the beta-Catenin/TCF4 Interaction Blocks the Self-Renewal of Cancer Stem Cells and Suppresses Tumorigenesis. Cancer Res. 2016, 76, 891–901. [Google Scholar] [CrossRef]
- Kortum, B.; Radhakrishnan, H.; Zincke, F.; Sachse, C.; Burock, S.; Keilholz, U.; Dahlmann, M.; Walther, W.; Dittmar, G.; Kobelt, D.; et al. Combinatorial treatment with statins and niclosamide prevents CRC dissemination by unhinging the MACC1-beta-catenin-S100A4 axis of metastasis. Oncogene 2022, 41, 4446–4458. [Google Scholar] [CrossRef]
- Park, W.J.; Kim, M.J. A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities. Cells 2023, 12, 1110. [Google Scholar] [CrossRef]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Giantonio, B.J.; Catalano, P.J.; Meropol, N.J.; O’Dwyer, P.J.; Mitchell, E.P.; Alberts, S.R.; Schwartz, M.A.; Benson, A.B., 3rd; Eastern Cooperative Oncology Group Study, E. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 2007, 25, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Van der Veldt, A.A.; Lubberink, M.; Bahce, I.; Walraven, M.; de Boer, M.P.; Greuter, H.N.; Hendrikse, N.H.; Eriksson, J.; Windhorst, A.D.; Postmus, P.E.; et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: Implications for scheduling of anti-angiogenic drugs. Cancer Cell 2012, 21, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Hopirtean, C.; Nagy, V. Optimizing the use of anti VEGF targeted therapies in patients with metastatic colorectal cancer: Review of literature. Clujul Med. 2018, 91, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, T.; Ullah, S.; Piantadosi, C.; Karapetis, C.S.; Townsend, A.R.; Roy, A.; Geerinckx, B.; Maddern, G.; Roder, D.; Padbury, R.; et al. First-line anti-EGFR or anti-VEGF therapy, tumour sidedness, and survival: Results from the South Australian (SA) Metastatic Colorectal Cancer (mCRC) Registry. J. Clin. Oncol. 2023, 41, 42. [Google Scholar] [CrossRef]
- Ulivi, P.; Scarpi, E.; Chiadini, E.; Marisi, G.; Valgiusti, M.; Capelli, L.; Casadei Gardini, A.; Monti, M.; Ruscelli, S.; Frassineti, G.L.; et al. Right- vs. Left-Sided Metastatic Colorectal Cancer: Differences in Tumor Biology and Bevacizumab Efficacy. Int. J. Mol. Sci. 2017, 18, 1240. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, X.; Li, W.; Ye, Y.; Du, X.; Sun, S.; Liu, L.; Zhang, H. Combination of Anti-EGFR and Anti-VEGF Drugs for the Treatment of Previously Treated Metastatic Colorectal Cancer: A Case Report and Literature Review. Front. Oncol. 2021, 11, 684309. [Google Scholar] [CrossRef]
- Weng, J.; Li, S.; Zhu, Z.; Liu, Q.; Zhang, R.; Yang, Y.; Li, X. Exploring immunotherapy in colorectal cancer. J. Hematol. Oncol. 2022, 15, 95. [Google Scholar] [CrossRef]
- Kullmann, F.; Strissel, P.L.; Strick, R.; Stoehr, R.; Eckstein, M.; Bertz, S.; Wullich, B.; Sikic, D.; Wach, S.; Taubert, H.; et al. Frequency of microsatellite instability (MSI) in upper tract urothelial carcinoma: Comparison of the Bethesda panel and the Idylla MSI assay in a consecutively collected, multi-institutional cohort. J. Clin. Pathol. 2023, 76, 126–132. [Google Scholar] [CrossRef]
- Gmeiner, W.H. A narrative review of genetic factors affecting fluoropyrimidine toxicity. Precis. Cancer Med. 2021, 4, 38. [Google Scholar] [CrossRef]
- Baxter, N.N.; Kennedy, E.B.; Bergsland, E.; Berlin, J.; George, T.J.; Gill, S.; Gold, P.J.; Hantel, A.; Jones, L.; Lieu, C.; et al. Adjuvant Therapy for Stage II Colon Cancer: ASCO Guideline Update. J. Clin. Oncol. 2022, 40, 892–910. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Kim, S.Y.; Lee, J.; Yun, S.H.; Kim, H.C.; Lee, W.Y.; Kim, T.W.; Hong, Y.S.; Lim, S.B.; Baek, J.Y.; et al. Oxaliplatin (3 months v 6 months) with 6 Months of Fluoropyrimidine as Adjuvant Therapy in Patients with Stage II/III Colon Cancer: KCSG CO09-07. J. Clin. Oncol. 2022, 40, 3868–3877. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Lo, S.N.; Gibbs, P. Circulating Tumor DNA Guiding Adjuvant Therapy in Colon Cancer. Reply. N. Engl. J. Med. 2022, 387, 760. [Google Scholar] [CrossRef]
- Slater, S.; Bryant, A.; Chen, H.C.; Begum, R.; Rana, I.; Aresu, M.; Peckitt, C.; Zhitkov, O.; Lazaro-Alcausi, R.; Borja, V.; et al. ctDNA guided adjuvant chemotherapy versus standard of care adjuvant chemotherapy after curative surgery in patients with high risk stage II or stage III colorectal cancer: A multi-centre, prospective, randomised control trial (TRACC Part C). BMC Cancer 2023, 23, 257. [Google Scholar] [CrossRef] [PubMed]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef]
- Arbuck, S.G. Overview of clinical trials using 5-fluorouracil and leucovorin for the treatment of colorectal cancer. Cancer 1989, 63, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.; Saltz, L.; Twelves, C.; Van Cutsem, E.; Hoff, P.; Kang, Y.; Saini, J.P.; Gilberg, F.; Cunningham, D. Efficacy of capecitabine versus 5-fluorouracil in colorectal and gastric cancers: A meta-analysis of individual data from 6171 patients. Ann. Oncol. 2011, 22, 2604–2609. [Google Scholar] [CrossRef]
- de Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Cunningham, D.; Roth, A.D.; Navarro, M.; James, R.D.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: A multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef]
- Tournigand, C.; Andre, T.; Achille, E.; Lledo, G.; Flesh, M.; Mery-Mignard, D.; Quinaux, E.; Couteau, C.; Buyse, M.; Ganem, G.; et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: A randomized GERCOR study. J. Clin. Oncol. 2004, 22, 229–237. [Google Scholar] [CrossRef]
- Falcone, A.; Ricci, S.; Brunetti, I.; Pfanner, E.; Allegrini, G.; Barbara, C.; Crino, L.; Benedetti, G.; Evangelista, W.; Fanchini, L.; et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: The Gruppo Oncologico Nord Ovest. J. Clin. Oncol. 2007, 25, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Atreya, C.E.; Yaeger, R.; Chu, E. Systemic Therapy for Metastatic Colorectal Cancer: From Current Standards to Future Molecular Targeted Approaches. Am. Soc. Clin. Oncol. Educ. Book. 2017, 37, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Taieb, J.; Kuboki, Y.; Pfeiffer, P.; Kumar, A.; Hochster, H.S. Trifluridine/tipiracil with or without bevacizumab in metastatic colorectal cancer: Results of a systematic review and meta-analysis. Ther. Adv. Med. Oncol. 2023, 15, 17588359221146137. [Google Scholar] [CrossRef] [PubMed]
- Gmeiner, W.H.; Dominijanni, A.; Haber, A.O.; Ghiraldeli, L.P.; Caudell, D.L.; D’agostino, R.; Pasche, B.C.; Smith, T.L.; Deng, Z.; Kiren, S.; et al. Improved anti-Tumor Activity of the Fluoropyrimidine Polymer CF10 in pre-Clinical Colorectal Cancer Models thru Distinct Mechanistic and Pharmacological Properties. Mol. Cancer Ther. 2020, 20, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wang, C.; Lee, P.P.; Chu, P.; Fakih, M. Response to PD-1 Blockade in Microsatellite Stable Metastatic Colorectal Cancer Harboring a POLE Mutation. J. Natl. Compr. Cancer Netw. 2017, 15, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. High tumor mutation burden (TMB) in microsatellite stable (MSS) colorectal cancers: Diverse molecular associations point to variable pathophysiology. Cancer Treat. Res. Commun. 2023, 36, 100746. [Google Scholar] [CrossRef] [PubMed]
- Gandini, A.; Puglisi, S.; Pirrone, C.; Martelli, V.; Catalano, F.; Nardin, S.; Seeber, A.; Puccini, A.; Sciallero, S. The role of immunotherapy in microsatellites stable metastatic colorectal cancer: State of the art and future perspectives. Front. Oncol. 2023, 13, 1161048. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, P.; Yilmaz, M.; Moller, S.; Zitnjak, D.; Krogh, M.; Petersen, L.N.; Poulsen, L.O.; Winther, S.B.; Thomsen, K.G.; Qvortrup, C. TAS-102 with or without bevacizumab in patients with chemorefractory metastatic colorectal cancer: An investigator-initiated, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 412–420. [Google Scholar] [CrossRef]
- Gmeiner, W.H. Recent Advances in Our Knowledge of mCRC Tumor Biology and Genetics: A Focus on Targeted Therapy Development. Onco Targets Ther. 2021, 14, 2121–2130. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Drug | Brand Name | Company | Mutation | Status |
|---|---|---|---|---|
| Sotorasib | Lumakras | AMGEN | G12C | Approved |
| Adagrasib | KRAZATI | Mirati | G12C | Approved |
| JDQ443 | Novartis | G12C | Phase 2 | |
| MRTX1133 | Mirati | G12D | Phase 1 | |
| RMC-6236 | Revolution | Multiple | Phase 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gmeiner, W.H. Recent Advances in Therapeutic Strategies to Improve Colorectal Cancer Treatment. Cancers 2024, 16, 1029. https://doi.org/10.3390/cancers16051029
Gmeiner WH. Recent Advances in Therapeutic Strategies to Improve Colorectal Cancer Treatment. Cancers. 2024; 16(5):1029. https://doi.org/10.3390/cancers16051029
Chicago/Turabian StyleGmeiner, William H. 2024. "Recent Advances in Therapeutic Strategies to Improve Colorectal Cancer Treatment" Cancers 16, no. 5: 1029. https://doi.org/10.3390/cancers16051029
APA StyleGmeiner, W. H. (2024). Recent Advances in Therapeutic Strategies to Improve Colorectal Cancer Treatment. Cancers, 16(5), 1029. https://doi.org/10.3390/cancers16051029