
Intratumoral Delivery of Interleukin 9 via Oncolytic Vaccinia Virus Elicits Potent Antitumor Effects in Tumor Models

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Mice and Cell Lines
2.2. Virus Generation
2.3. Viral Replication and IL-9 Expression In Vitro
2.4. Cytotoxicity Assay In Vitro
2.5. Rodent Tumor Models
2.6. Flow Cytometry
2.7. RT-qPCR
2.8. Statistics
3. Results
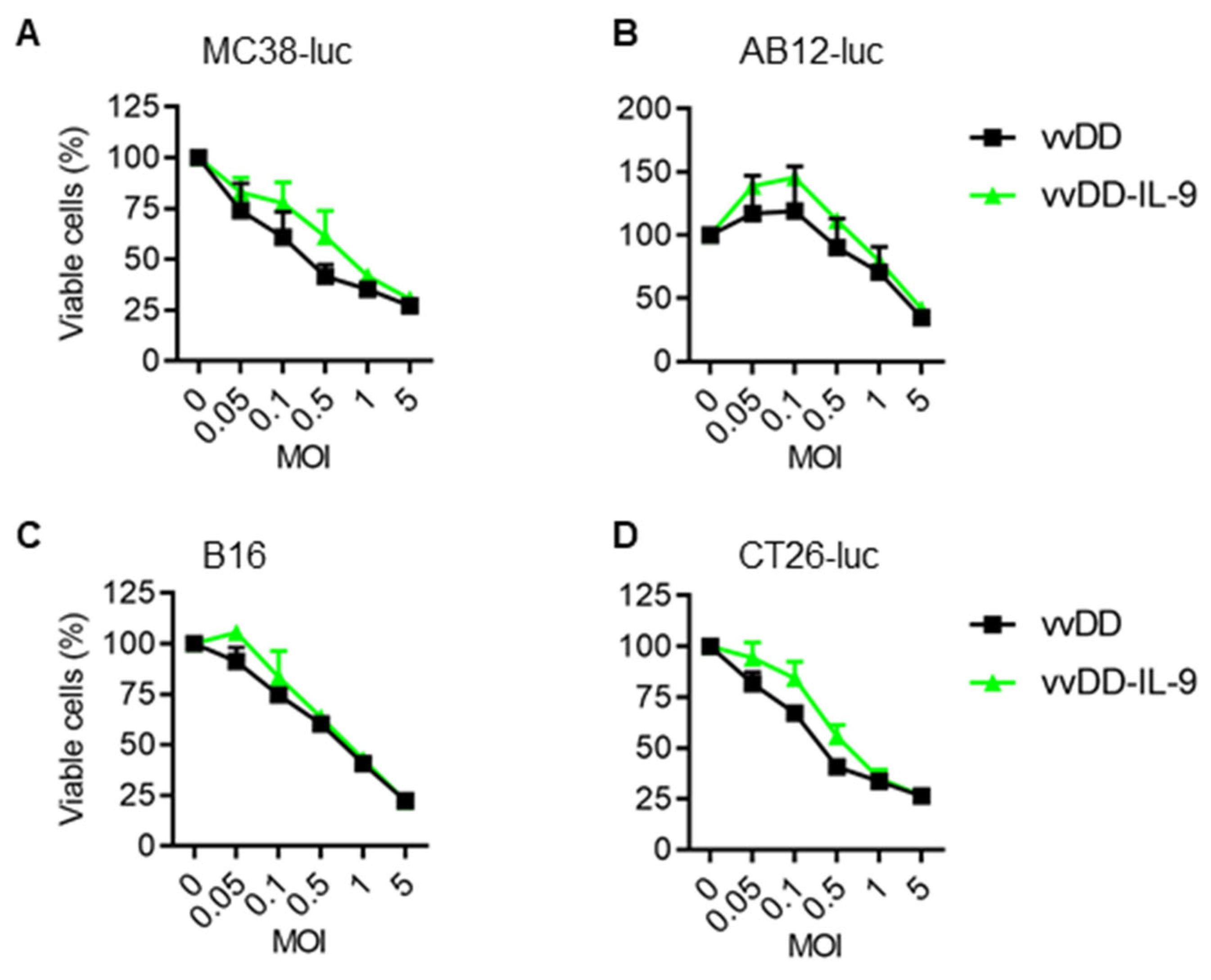
3.1. IL-9 Expression Does Not Impact Viral Replication and Cytotoxicity In Vitro
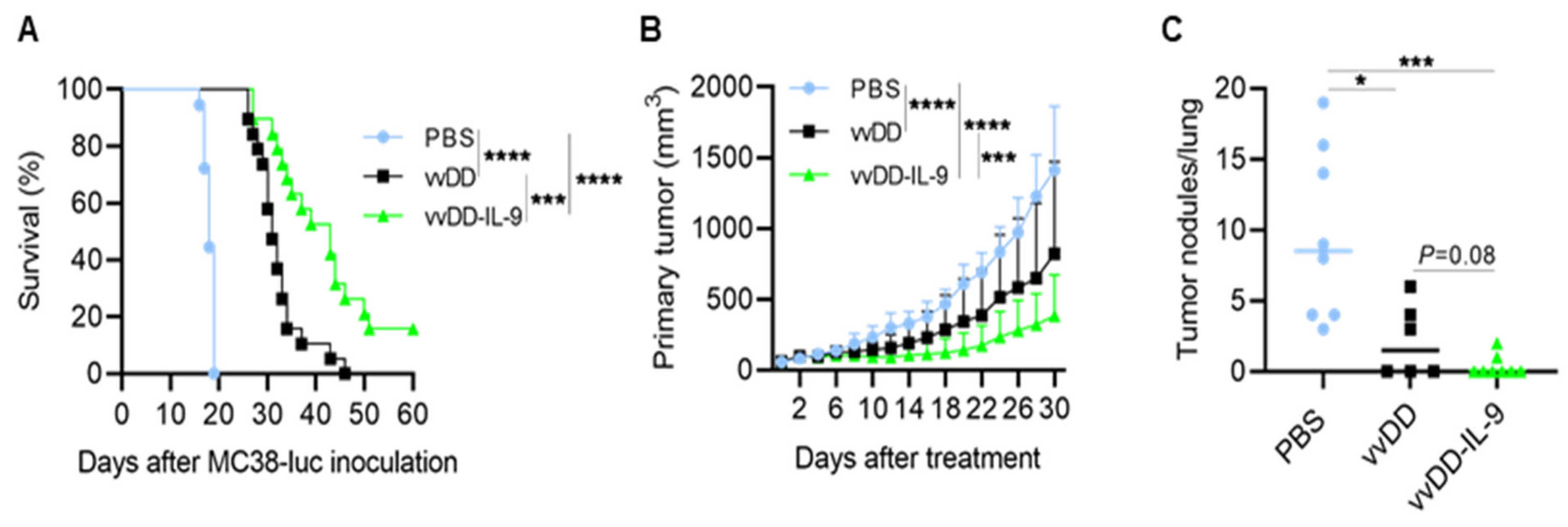
3.2. IL-9 Expressing oVV Elicits Antitumor Effects in Tumor Models
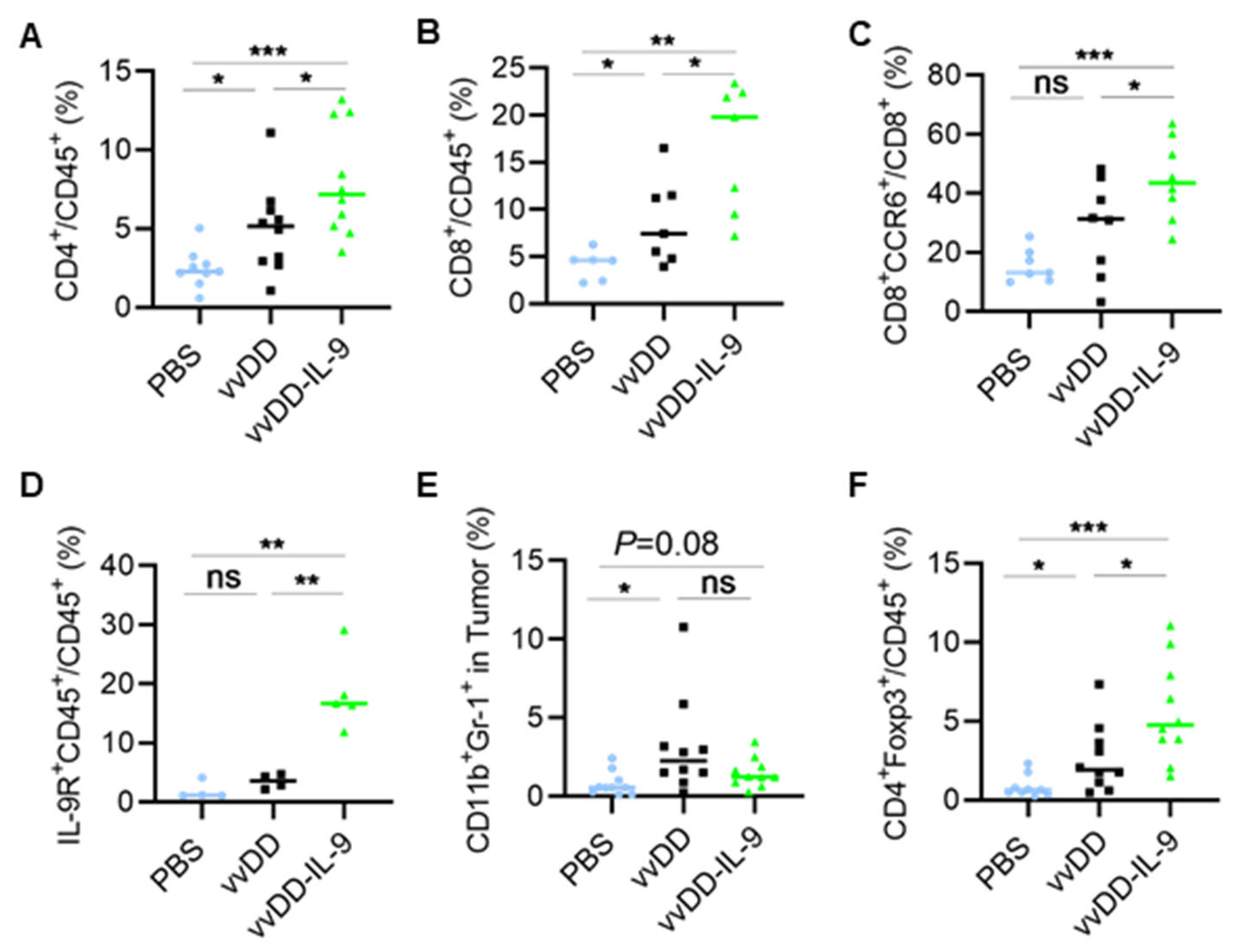
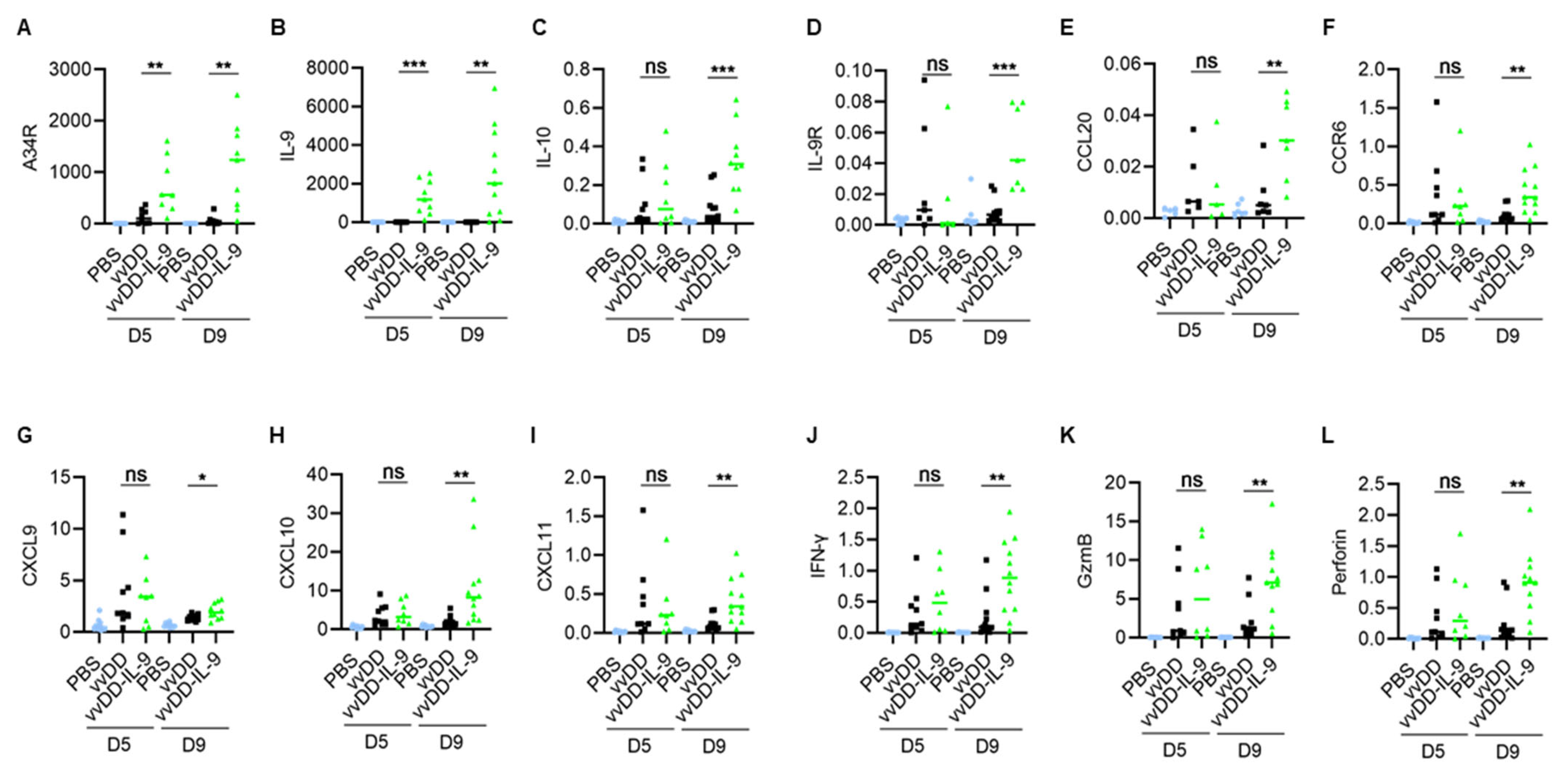
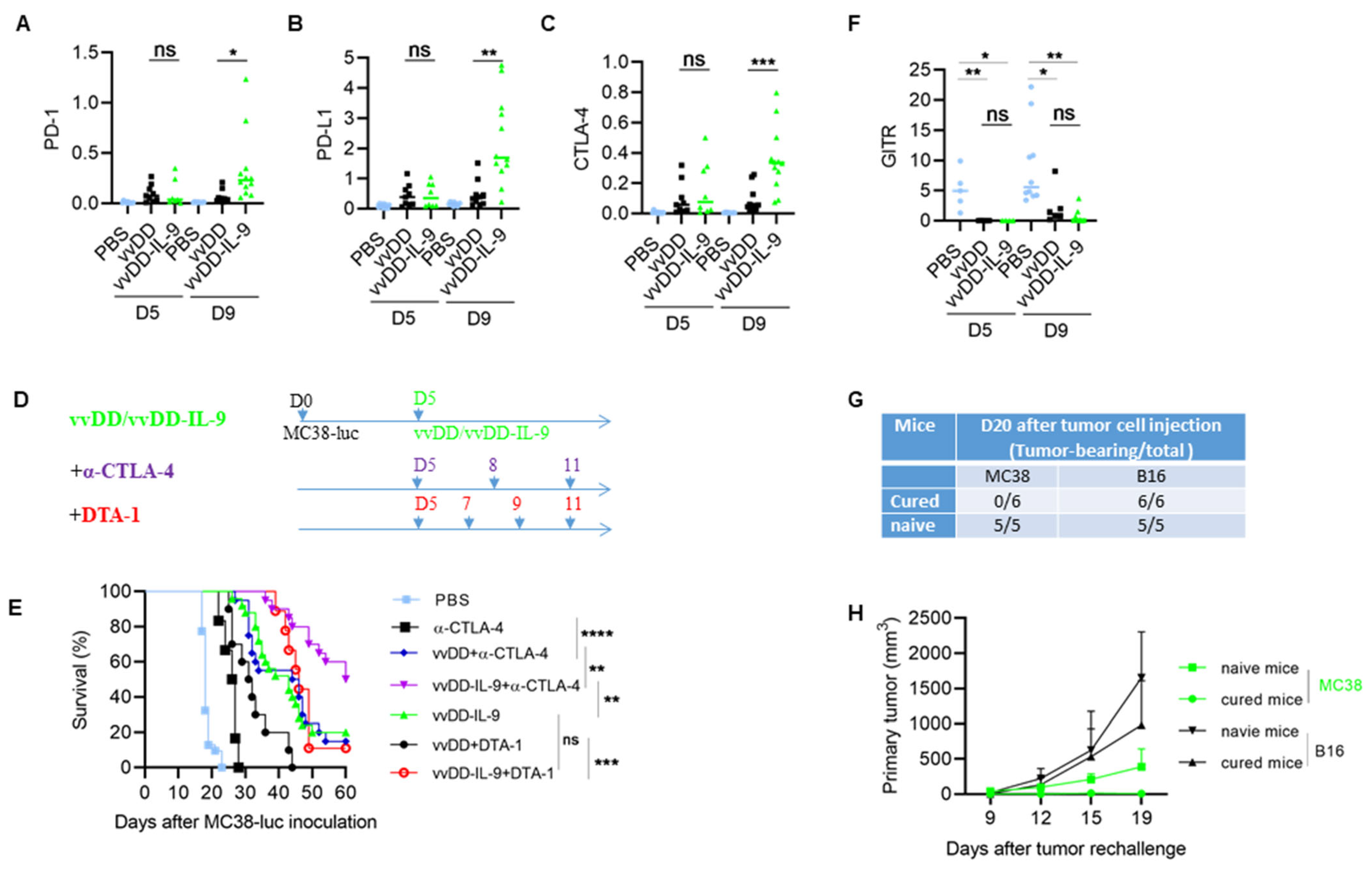
3.3. IL-9 Expressing oVV Modulates the Tumor Microenvironment
3.4. CTLA-4 Blockade Enhanced the Antitumor Effects Elicited by vvDD-IL-9
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Woude, L.L.; Gorris, M.A.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Bartlett, D.L.; Liu, Z.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.; He, Y.; Guo, Z.S. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 2013, 12, 103. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer 2019, 7, 6. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef]
- Liu, Z.; Ravindranathan, R.; Li, J.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. OncoImmunology 2016, 5, e1091554. [Google Scholar] [CrossRef]
- Guo, Z.S.; Liu, Z.; Kowalsky, S.; Feist, M.; Kalinski, P.; Lu, B.; Storkus, W.J.; Bartlett, D.L. Oncolytic Immunotherapy: Conceptual Evolution, Current Strategies, and Future Perspectives. Front. Immunol. 2017, 8, 555. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Pelin, A.; Boulton, S.; Tamming, L.A.; Bell, J.C.; Singaravelu, R. Engineering vaccinia virus as an immunotherapeutic battleship to overcome tumor heterogeneity. Expert. Opin. Biol. Ther. 2020, 20, 1083–1097. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Hemminki, O.; dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef]
- Ralainirina, N.; Poli, A.; Michel, T.; Poos, L.; Andrès, E.; Hentges, F.; Zimmer, J. Control of NK cell functions by CD4+CD25+ regulatory T cells. J. Leukoc. Biol. 2007, 81, 144–153. [Google Scholar] [CrossRef]
- Lorvik, K.B.; Hammarström, C.; Fauskanger, M.; Haabeth, O.A.W.; Zangani, M.; Haraldsen, G.; Bogen, B.; Corthay, A. Adoptive Transfer of Tumor-Specific Th2 Cells Eradicates Tumors by Triggering an In Situ Inflammatory Immune Response. Cancer Res. 2016, 76, 6864–6876. [Google Scholar] [CrossRef]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef]
- Lu, Y.; Hong, S.; Li, H.; Park, J.; Hong, B.; Wang, L.; Zheng, Y.; Liu, Z.; Xu, J.; He, J.; et al. Th9 cells promote antitumor immune responses in vivo. J. Clin. Investig. 2012, 122, 4160–4171. [Google Scholar] [CrossRef]
- Sharma, S.; Stolina, M.; Lin, Y.; Gardner, B.; Miller, P.W.; Kronenberg, M.; Dubinett, S.M. T cell-derived IL-10 promotes lung cancer growth by suppressing both T cell and APC function. J. Immunol. 1999, 163, 5020–5028. [Google Scholar] [CrossRef]
- Duan, Z.; Miller, H.D.; Fu, X.; Ge, D.; Jin, B.; A Moustafa, A.; Lan, R.; Zhang, K.; Chen, Z.; You, Z. Th17 cells promote tumor growth in an immunocompetent orthotopic mouse model of prostate cancer. Am. J. Clin. Exp. Urol. 2019, 7, 249–261. [Google Scholar] [PubMed]
- Salazar, Y.; Zheng, X.; Brunn, D.; Raifer, H.; Picard, F.S.; Zhang, Y.; Winter, H.; Guenther, S.; Weigert, A.; Weigmann, B.; et al. Microenvironmental Th9 and Th17 lymphocytes induce metastatic spreading in lung cancer. J. Clin. Investig. 2020, 130, 3560–3575. [Google Scholar] [CrossRef]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.-C.; et al. IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3− effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef]
- Veldhoen, M.; Uyttenhove, C.; van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol 2008, 9, 1341–1346. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Simpson, R.J.; Van Snick, J. Functional and structural characterization of P40, a mouse glycoprotein with T-cell growth factor activity. Proc. Natl. Acad. Sci. USA 1988, 85, 6934–6938. [Google Scholar] [CrossRef]
- Renauld, J.C.; Van Der Lugt, N.; Vink, A.; Van Roon, M.; Godfraind, C.; Warnier, G.; Merz, H.; Feller, A.; Berns, A.; Van Snick, J. Thymic lymphomas in interleukin 9 transgenic mice. Oncogene 1994, 9, 1327–1332. [Google Scholar]
- Nagato, T.; Kobayashi, H.; Kishibe, K.; Takahara, M.; Ogino, T.; Ishii, H.; Oikawa, K.; Aoki, N.; Sato, K.; Kimura, S.; et al. Expression of interleukin-9 in nasal natural killer/T-cell lymphoma cell lines and patients. Clin. Cancer Res. 2005, 11, 8250–8257. [Google Scholar] [CrossRef]
- Qiu, L.; Lai, R.; Lin, Q.; Lau, E.; Thomazy, D.M.; Calame, D.; Ford, R.J.; Kwak, L.W.; Kirken, R.A.; Amin, H.M. Autocrine release of interleukin-9 promotes Jak3-dependent survival of ALK+ anaplastic large-cell lymphoma cells. Blood 2006, 108, 2407–2415. [Google Scholar] [CrossRef]
- Feng, L.-L.; Gao, J.-M.; Li, P.-P.; Wang, X. IL-9 contributes to immunosuppression mediated by regulatory T cells and mast cells in B-cell non-hodgkin’s lymphoma. J. Clin. Immunol. 2011, 31, 1084–1094. [Google Scholar] [CrossRef]
- Purwar, R.; Schlapbach, C.; Xiao, S.; Kang, H.S.; Elyaman, W.; Jiang, X.; Jetten, A.M.; Khoury, S.J.; Fuhlbrigge, R.C.; Kuchroo, V.K.; et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat. Med. 2012, 18, 1248–1253. [Google Scholar] [CrossRef]
- Wang, J.; Sun, M.; Zhao, H.; Huang, Y.; Li, D.; Mao, D.; Zhang, Z.; Zhu, X.; Dong, X.; Zhao, X. IL-9 Exerts Antitumor Effects in Colon Cancer and Transforms the Tumor Microenvironment In Vivo. Technol. Cancer Res. Treat. 2019, 18, 1533033819857737. [Google Scholar] [CrossRef]
- Das, S.; Surve, V.; Marathe, S.; Wad, S.; Karulkar, A.; Srinivasan, S.; Dwivedi, A.; Barthel, S.R.; Purwar, R. IL-9 Abrogates the Metastatic Potential of Breast Cancer by Controlling Extracellular Matrix Remodeling and Cellular Contractility. J. Immunol. 2021, 206, 2740–2752. [Google Scholar] [CrossRef]
- Fortin, C.; Huang, X.; Yang, Y. NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J. Immunol. 2012, 189, 1843–1849. [Google Scholar] [CrossRef]
- Chard, L.S.; Maniati, E.; Wang, P.; Zhang, Z.; Gao, D.; Wang, J.; Cao, F.; Ahmed, J.; El Khouri, M.; Hughes, J.; et al. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin. Cancer Res. 2015, 21, 405–416. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Mahne, A.E.; Mauze, S.; Joyce-Shaikh, B.; Xia, J.; Bowman, E.P.; Beebe, A.M.; Cua, D.J.; Jain, R. Dual Roles for Regulatory T-cell Depletion and Costimulatory Signaling in Agonistic GITR Targeting for Tumor Immunotherapy. Cancer Res. 2017, 77, 1108–1118. [Google Scholar] [CrossRef]
- Gajewski, T.F. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non–T-Cell–Inflamed Tumor Microenvironment. Semin. Oncol. 2015, 42, 663–671. [Google Scholar] [CrossRef]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef]
- Chon, H.J.; Lee, W.S.; Yang, H.; Kong, S.J.; Lee, N.K.; Moon, E.S.; Choi, J.; Han, E.C.; Ahn, J.B.; Kim, J.H.; et al. Tumor Microenvironment Remodeling by Intratumoral Oncolytic Vaccinia Virus Enhances the Efficacy of Immune-Checkpoint Blockade. Clin. Cancer Res. 2019, 25, 1612–1623. [Google Scholar] [CrossRef]
- Smith, H.G.; Mansfield, D.; Roulstone, V.; Kyula-Currie, J.N.; McLaughlin, M.; Patel, R.R.; Bergerhoff, K.F.; Paget, J.T.; Dillon, M.T.; Khan, A.; et al. PD-1 Blockade Following Isolated Limb Perfusion with Vaccinia Virus Prevents Local and Distant Relapse of Soft-tissue Sarcoma. Clin. Cancer Res. 2019, 25, 3443–3454. [Google Scholar] [CrossRef]
- Kowalsky, S.J.; Liu, Z.; Feist, M.; Berkey, S.E.; Ma, C.; Ravindranathan, R.; Dai, E.; Roy, E.J.; Guo, Z.S.; Bartlett, D.L. Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol. Ther. 2018, 26, 2476–2486. [Google Scholar] [CrossRef]
- Cervera-Carrascon, V.; Siurala, M.; Santos, J.M.; Havunen, R.; Tähtinen, S.; Karell, P.; Sorsa, S.; Kanerva, A.; Hemminki, A. TNFa and IL-2 armed adenoviruses enable complete responses by anti-PD-1 checkpoint blockade. OncoImmunology 2018, 7, e1412902. [Google Scholar] [CrossRef]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e5. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef]
- Liu, Z.; Ge, Y.; Wang, H.; Ma, C.; Feist, M.; Ju, S.; Guo, Z.S.; Bartlett, D.L. Modifying the cancer-immune set point using vaccinia virus expressing re-designed interleukin-2. Nat. Commun. 2018, 9, 4682. [Google Scholar] [CrossRef]
- Bourgeois-Daigneault, M.-C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef]
- Liu, W.; Dai, E.; Liu, Z.; Ma, C.; Guo, Z.S.; Bartlett, D.L. In Situ Therapeutic Cancer Vaccination with an Oncolytic Virus Expressing Membrane-Tethered IL-2. Mol. Ther. Oncolytics 2020, 17, 350–360. [Google Scholar] [CrossRef]
- Ge, Y.; Wang, H.; Ren, J.; Liu, W.; Chen, L.; Chen, H.; Ye, J.; Dai, E.; Ma, C.; Ju, S.; et al. Oncolytic vaccinia virus delivering tethered IL-12 enhances antitumor effects with improved safety. J. Immunother. Cancer 2020, 8, e000710. [Google Scholar] [CrossRef]
- Chen, T.; Ding, X.; Liao, Q.; Gao, N.; Chen, Y.; Zhao, C.; Zhang, X.; Xu, J. IL-21 arming potentiates the anti-tumor activity of an oncolytic vaccinia virus in monotherapy and combination therapy. J. Immunother. Cancer 2021, 9, e001647. [Google Scholar] [CrossRef]
- Marelli, G.; Dunmall, L.S.C.; Yuan, M.; Di Gioia, C.; Miao, J.; Cheng, Z.; Zhang, Z.; Liu, P.; Ahmed, J.; Gangeswaran, R.; et al. A systemically deliverable Vaccinia virus with increased capacity for intertumoral and intratumoral spread effectively treats pancreatic cancer. J. Immunother. Cancer 2021, 9, e001624. [Google Scholar] [CrossRef]
- Liu, W.; Chen, H.; Zhu, Z.; Liu, Z.; Ma, C.; Lee, Y.J.; Bartlett, D.L.; Guo, Z.-S. Ferroptosis Inducer Improves the Efficacy of Oncolytic Virus-Mediated Cancer Immunotherapy. Biomedicines 2022, 10, 1425. [Google Scholar] [CrossRef]
- Van Vloten, J.P.; Matuszewska, K.; A A Minow, M.; A Minott, J.; A Santry, L.; Pereira, M.; A Stegelmeier, A.; McAusland, T.M.; Klafuric, E.M.; Karimi, K.; et al. Oncolytic Orf virus licenses NK cells via cDC1 to activate innate and adaptive antitumor mechanisms and extends survival in a murine model of late-stage ovarian cancer. J. Immunother. Cancer 2022, 10, e004335. [Google Scholar] [CrossRef]
- Jung, B.-K.; Ko, H.Y.; Kang, H.; Hong, J.; Ahn, H.M.; Na, Y.; Kim, H.; Kim, J.S.; Yun, C.-O. Relaxin-expressing oncolytic adenovirus induces remodeling of physical and immunological aspects of cold tumor to potentiate PD-1 blockade. J. Immunother. Cancer 2020, 8, e000763. [Google Scholar] [CrossRef]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar]
- Zeh, H.J.; Downs-Canner, S.; McCart, J.A.; Guo, Z.S.; Rao, U.N.M.; Ramalingam, L.; Thorne, S.H.; Jones, H.L.; Kalinski, P.; Wieckowski, E.; et al. First-in-man study of western reserve strain oncolytic vaccinia virus: Safety, systemic spread, and antitumor activity. Mol. Ther. 2015, 23, 202–214. [Google Scholar] [CrossRef]
- Downs-Canner, S.; Guo, Z.S.; Ravindranathan, R.; Breitbach, C.J.; O’Malley, M.E.; Jones, H.L.; Moon, A.; McCart, J.A.; Shuai, Y.; Zeh, H.J.; et al. Phase 1 Study of Intravenous Oncolytic Poxvirus (vvDD) in Patients with Advanced Solid Cancers. Mol. Ther. 2016, 24, 1492–1501. [Google Scholar] [CrossRef]
- Chen, L.; Chen, H.; Ye, J.; Ge, Y.; Wang, H.; Dai, E.; Ren, J.; Liu, W.; Ma, C.; Ju, S.; et al. Intratumoral expression of interleukin 23 variants using oncolytic vaccinia virus elicit potent antitumor effects on multiple tumor models via tumor microenvironment modulation. Theranostics 2021, 11, 6668–6681. [Google Scholar] [CrossRef]
- Kondo, T.; Takata, H.; Takiguchi, M. Functional expression of chemokine receptor CCR6 on human effector memory CD8+ T cells. Eur. J. Immunol. 2007, 37, 54–65. [Google Scholar] [CrossRef]
- Kearley, J.; Erjefalt, J.S.; Andersson, C.; Benjamin, E.; Jones, C.P.; Robichaud, A.; Pegorier, S.; Brewah, Y.; Burwell, T.J.; Bjermer, L.; et al. IL-9 governs allergen-induced mast cell numbers in the lung and chronic remodeling of the airways. Am. J. Respir. Crit. Care Med. 2011, 183, 865–875. [Google Scholar] [CrossRef]
- Elyaman, W.; Bradshaw, E.M.; Uyttenhove, C.; Dardalhon, V.; Awasthi, A.; Imitola, J.; Bettelli, E.; Oukka, M.; van Snick, J.; Renauld, J.-C.; et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12885–12890. [Google Scholar] [CrossRef]
- Wan, J.; Wu, Y.; Ji, X.; Huang, L.; Cai, W.; Su, Z.; Wang, S.; Xu, H. IL-9 and IL-9-producing cells in tumor immunity. Cell Commun. Signal. 2020, 18, 50. [Google Scholar] [CrossRef]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef]
- Ferguson, M.S.; Dunmall, L.S.C.; Gangeswaran, R.; Marelli, G.; Tysome, J.R.; Burns, E.; Whitehead, M.A.; Aksoy, E.; Alusi, G.; Hiley, C.; et al. Transient Inhibition of PI3Kdelta Enhances the Therapeutic Effect of Intravenous Delivery of Oncolytic Vaccinia Virus. Mol. Ther. 2020, 28, 1263–1275. [Google Scholar] [CrossRef]
- Giehl, E.; Kosaka, H.; Liu, Z.; Feist, M.; Kammula, U.S.; Lotze, M.T.; Ma, C.; Guo, Z.S.; Bartlett, D.L. In Vivo Priming of Peritoneal Tumor-Reactive Lymphocytes with a Potent Oncolytic Virus for Adoptive Cell Therapy. Front. Immunol. 2021, 12, 610042. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, M.E.; Jang, W.S.; Kim, J.; Park, S.M.; Oh, K.; Lee, N.; Ham, W.S. Systemic Injection of Oncolytic Vaccinia Virus Suppresses Primary Tumor Growth and Lung Metastasis in Metastatic Renal Cell Carcinoma by Remodeling Tumor Microenvironment. Biomedicines 2022, 10, 173. [Google Scholar] [CrossRef]
- Delaunay, T.; Nader, J.; Grard, M.; Farine, I.; Hedwig, V.; Foloppe, J.; Blondy, T.; Violland, M.; Pouliquen, D.; Grégoire, M.; et al. High Oncolytic Activity of a Double-Deleted Vaccinia Virus Copenhagen Strain against Malignant Pleural Mesothelioma. Mol. Ther. Oncolytics 2020, 18, 573–578. [Google Scholar] [CrossRef]
- Lauer, U.M.; Schell, M.; Beil, J.; Berchtold, S.; Koppenhöfer, U.; Glatzle, J.; Königsrainer, A.; Möhle, R.; Nann, D.; Fend, F.; et al. Phase I Study of Oncolytic Vaccinia Virus GL-ONC1 in Patients with Peritoneal Carcinomatosis. Clin. Cancer Res. 2018, 24, 4388–4398. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, J.; Chen, L.; Waltermire, J.; Zhao, J.; Ren, J.; Guo, Z.; Bartlett, D.L.; Liu, Z. Intratumoral Delivery of Interleukin 9 via Oncolytic Vaccinia Virus Elicits Potent Antitumor Effects in Tumor Models. Cancers 2024, 16, 1021. https://doi.org/10.3390/cancers16051021
Ye J, Chen L, Waltermire J, Zhao J, Ren J, Guo Z, Bartlett DL, Liu Z. Intratumoral Delivery of Interleukin 9 via Oncolytic Vaccinia Virus Elicits Potent Antitumor Effects in Tumor Models. Cancers. 2024; 16(5):1021. https://doi.org/10.3390/cancers16051021
Chicago/Turabian StyleYe, Junjie, Lingjuan Chen, Julia Waltermire, Jinshun Zhao, Jinghua Ren, Zongsheng Guo, David L. Bartlett, and Zuqiang Liu. 2024. "Intratumoral Delivery of Interleukin 9 via Oncolytic Vaccinia Virus Elicits Potent Antitumor Effects in Tumor Models" Cancers 16, no. 5: 1021. https://doi.org/10.3390/cancers16051021
APA StyleYe, J., Chen, L., Waltermire, J., Zhao, J., Ren, J., Guo, Z., Bartlett, D. L., & Liu, Z. (2024). Intratumoral Delivery of Interleukin 9 via Oncolytic Vaccinia Virus Elicits Potent Antitumor Effects in Tumor Models. Cancers, 16(5), 1021. https://doi.org/10.3390/cancers16051021