

Transduction Efficiency of Zika Virus E Protein Pseudotyped HIV-1gfp and Its Oncolytic Activity Tested in Primary Glioblastoma Cell Cultures
 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Cell Lines
2.2. Primary GBM Cultures
2.3. Immunostaining
2.4. Production of ZIKV Envelope Pseudotyped LVs
2.5. HIV-1 p24-Antigen Assay
2.6. Transduction of Tumor Cells by Lentviral Vectors
3. Results
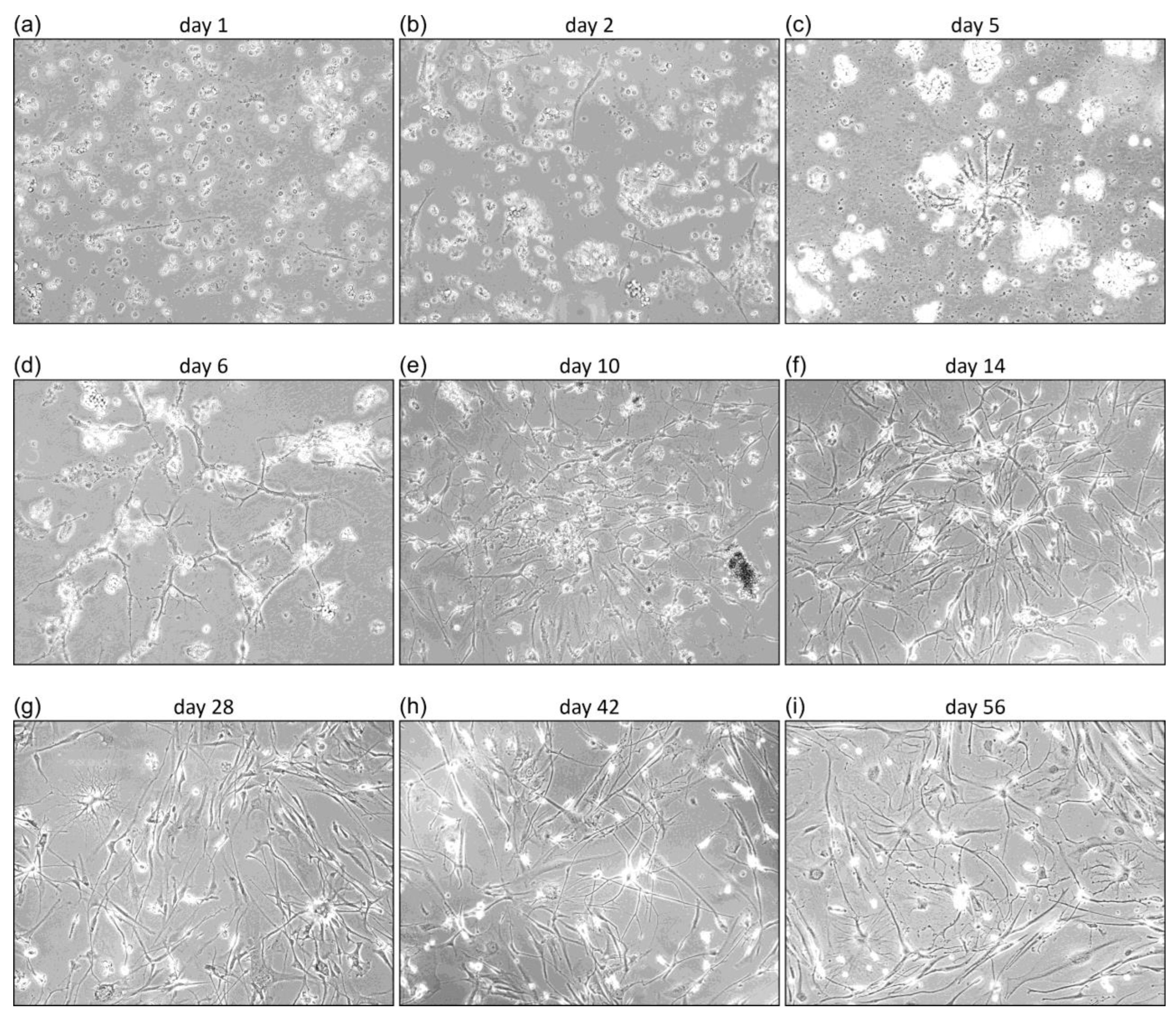
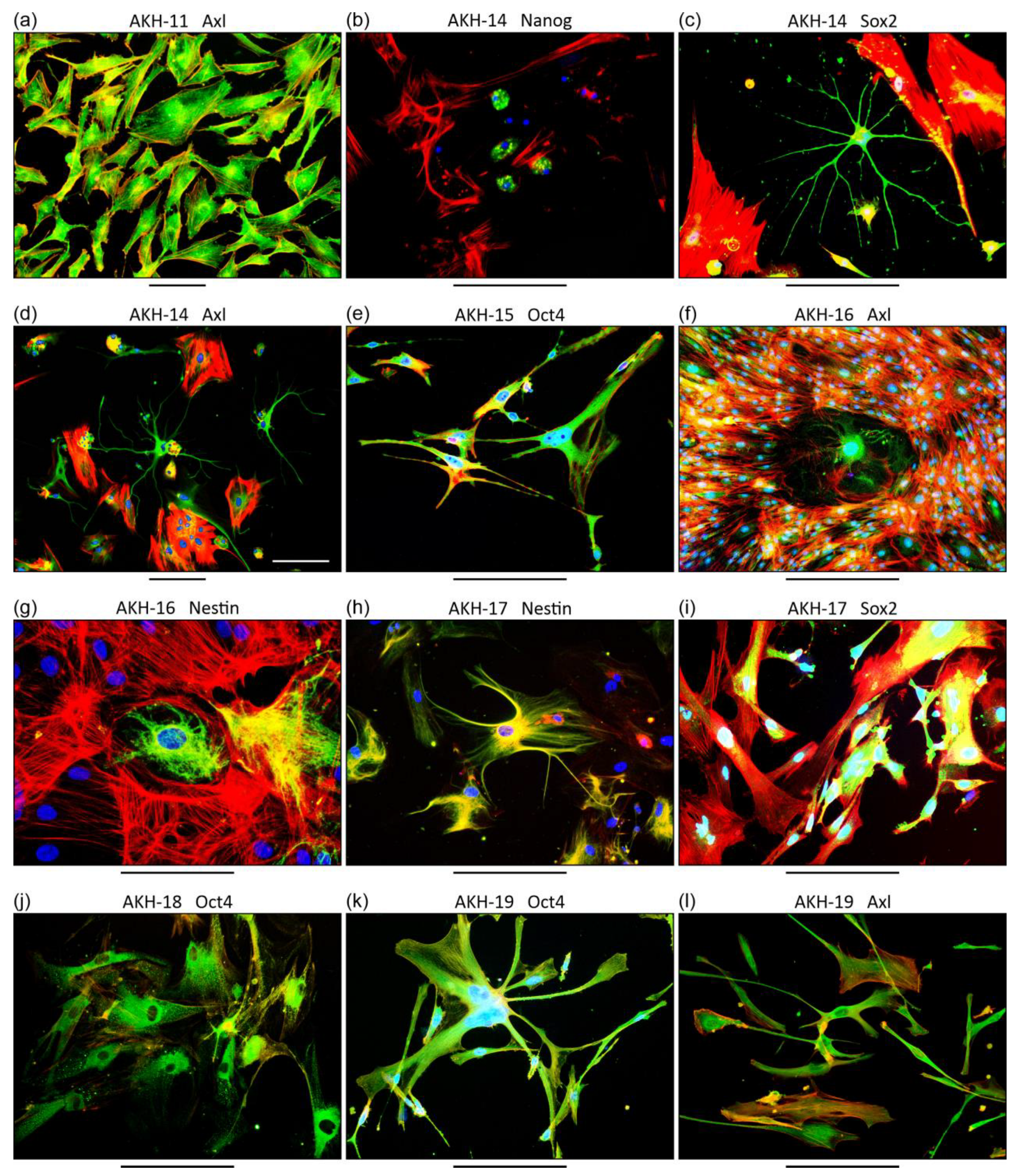
3.1. Characterization of Primary GBM Cell Cultures
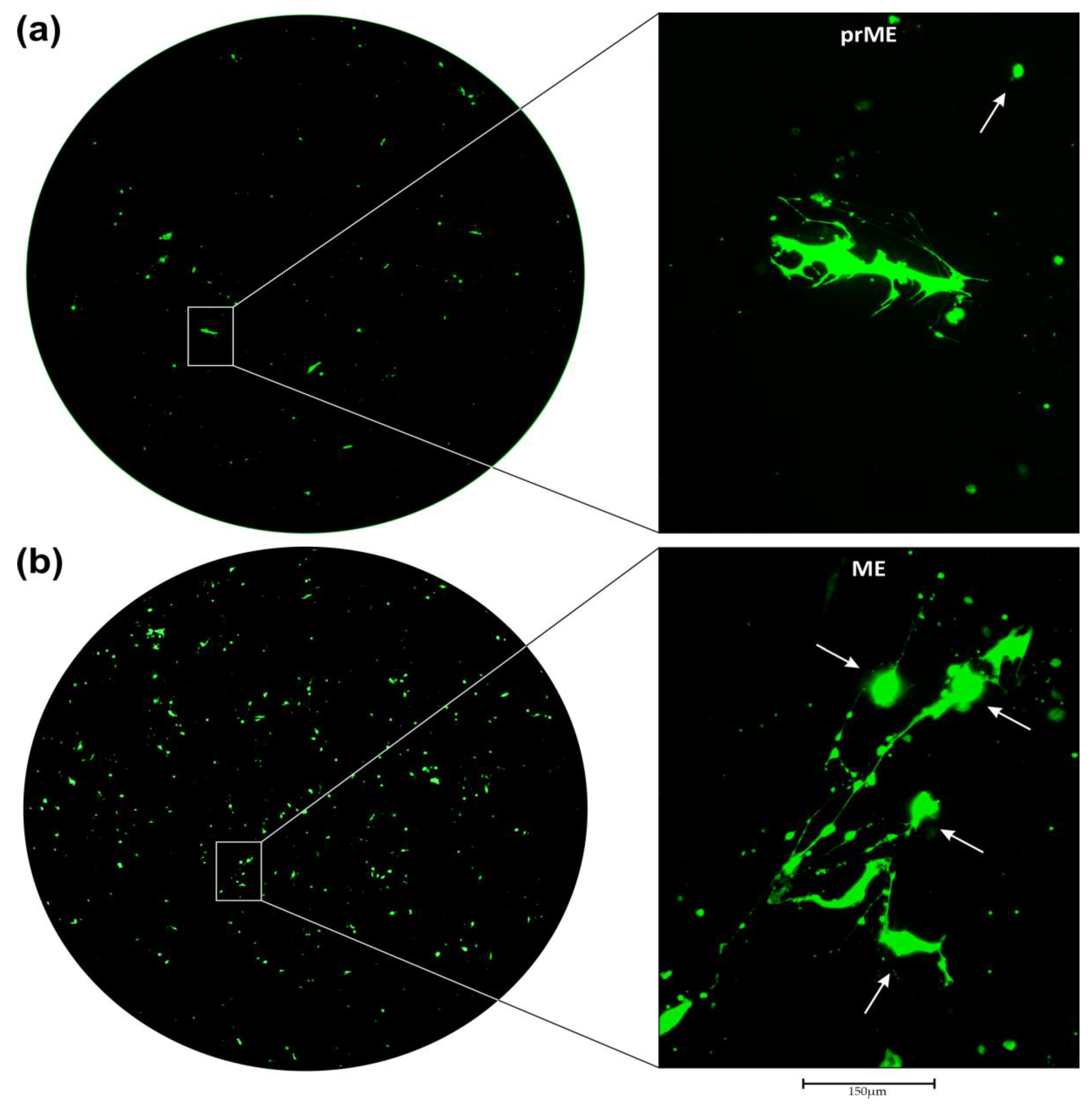
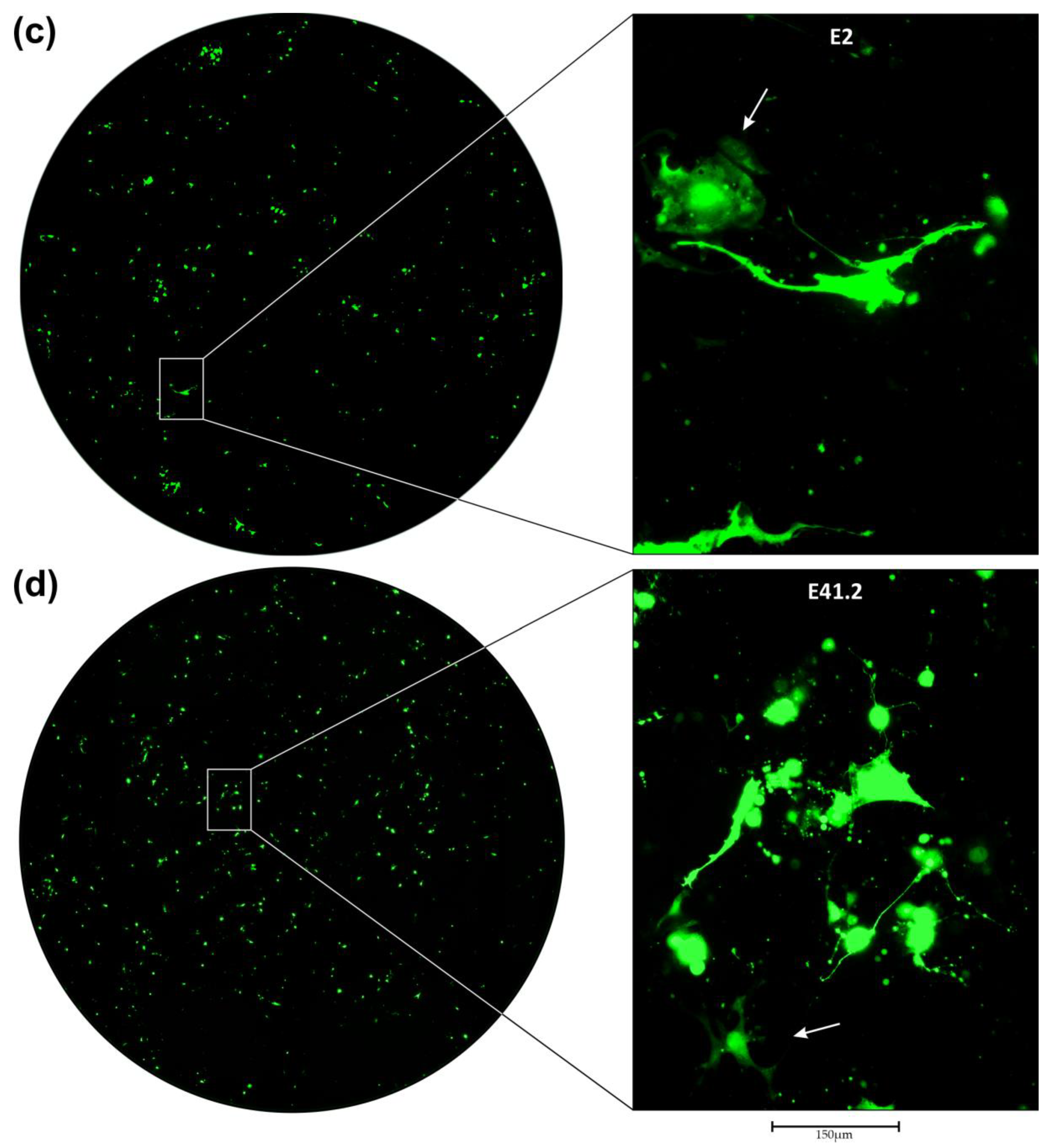
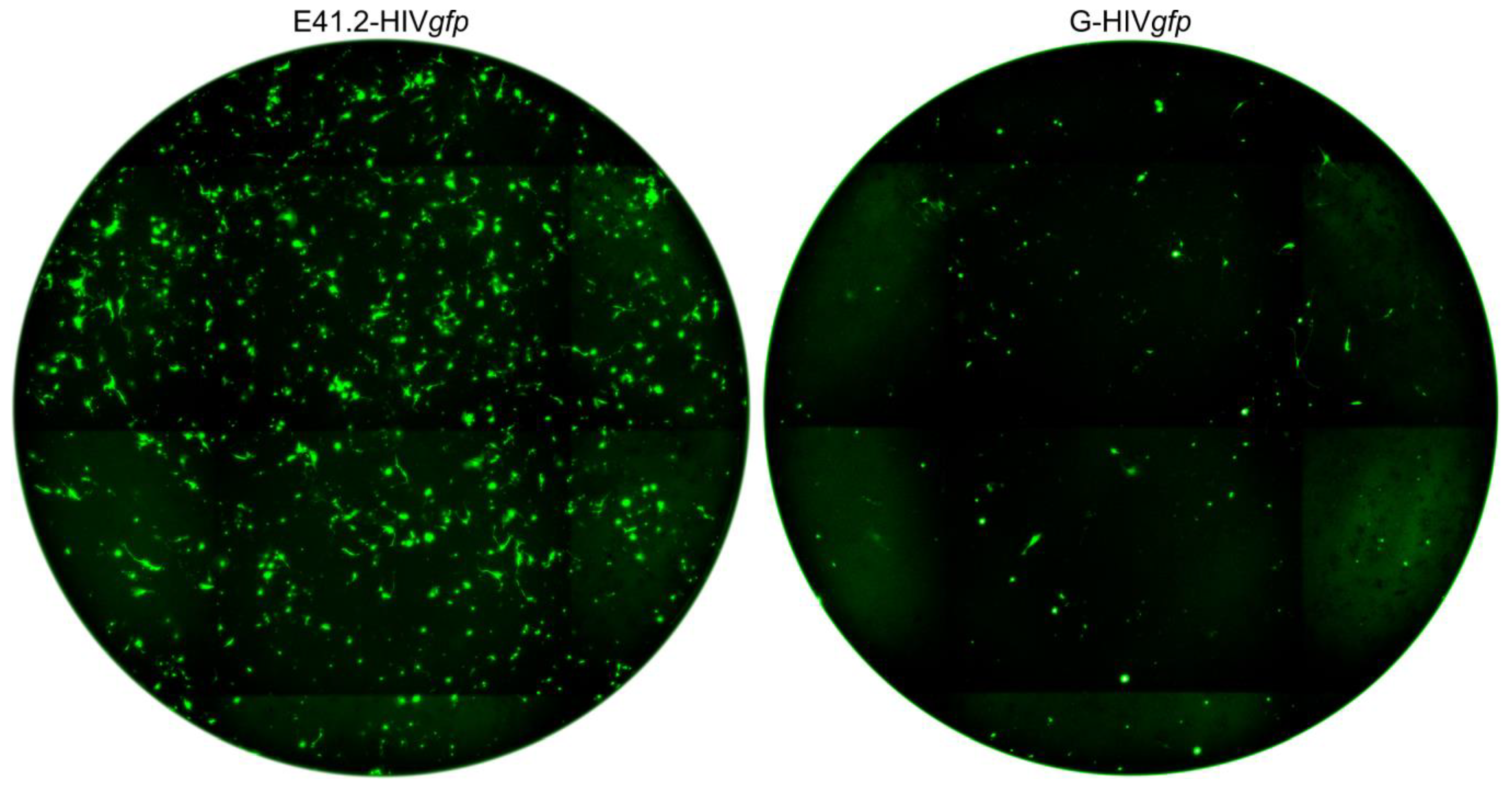
3.2. Transduction of Primary GBM Cells with Four Different LVs
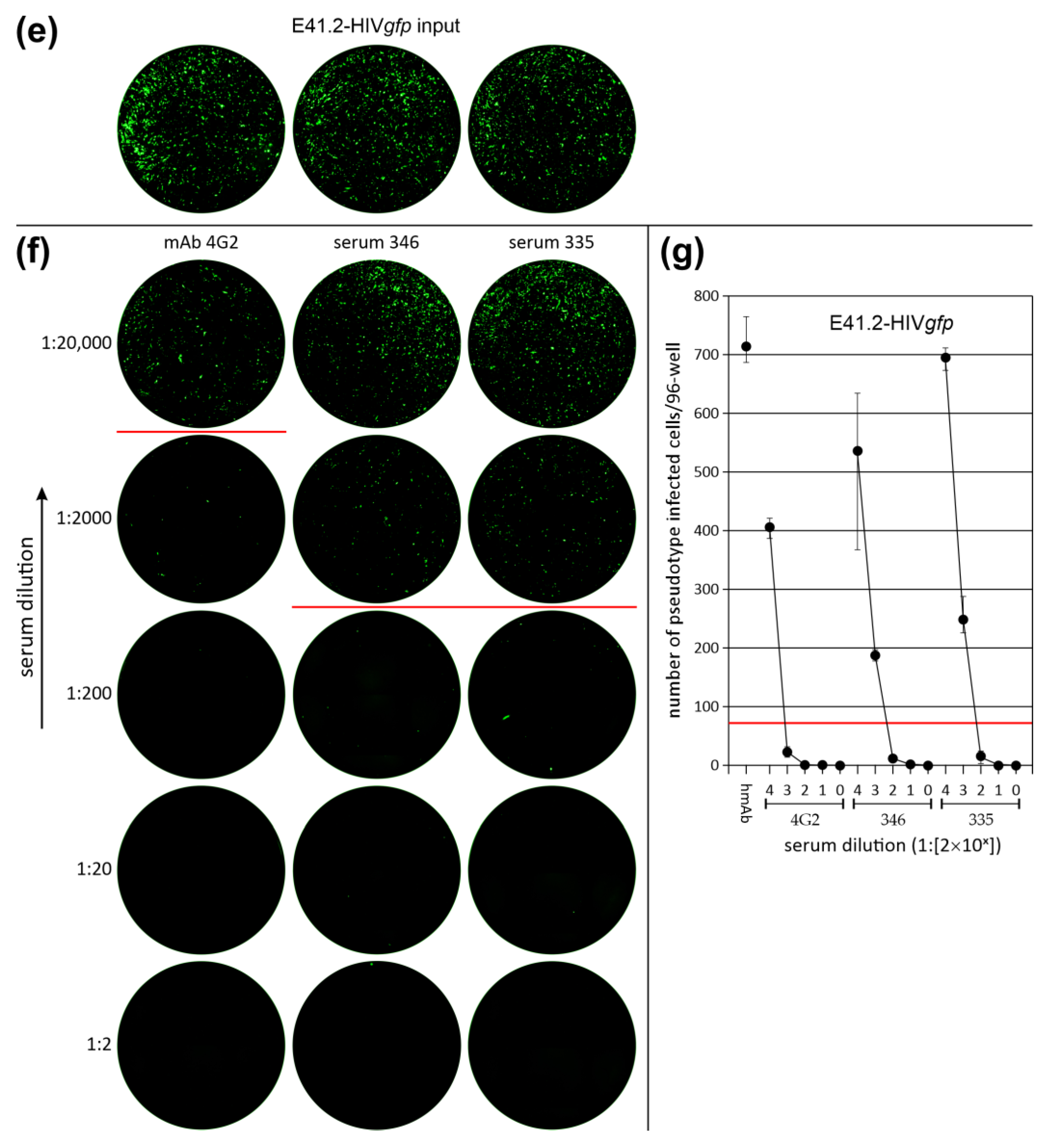
3.3. Neutralization of LV Entry into GBM Tumor Cells
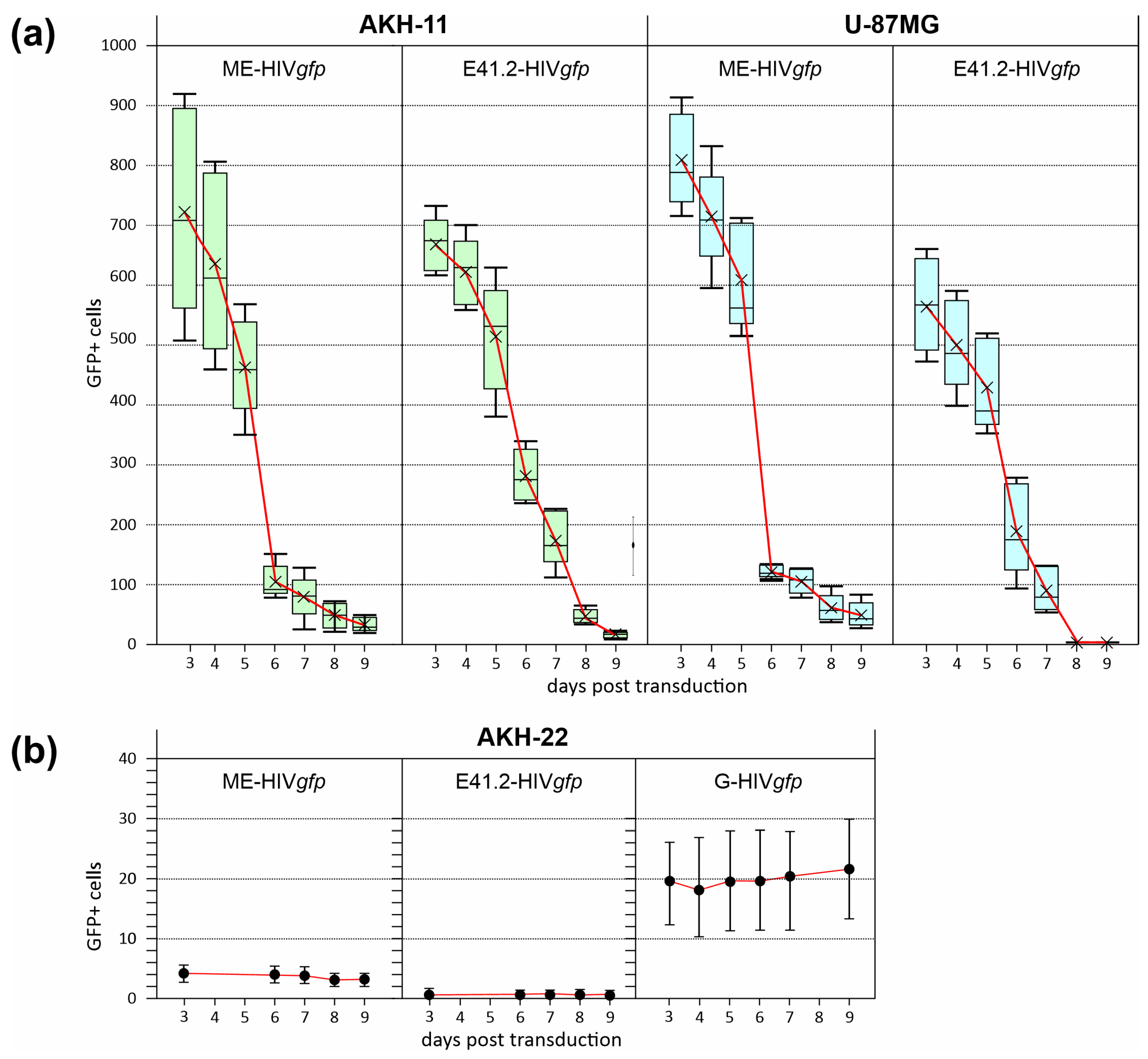
3.4. Efficiency of LV Entry in Primary GBM Cell Cultures
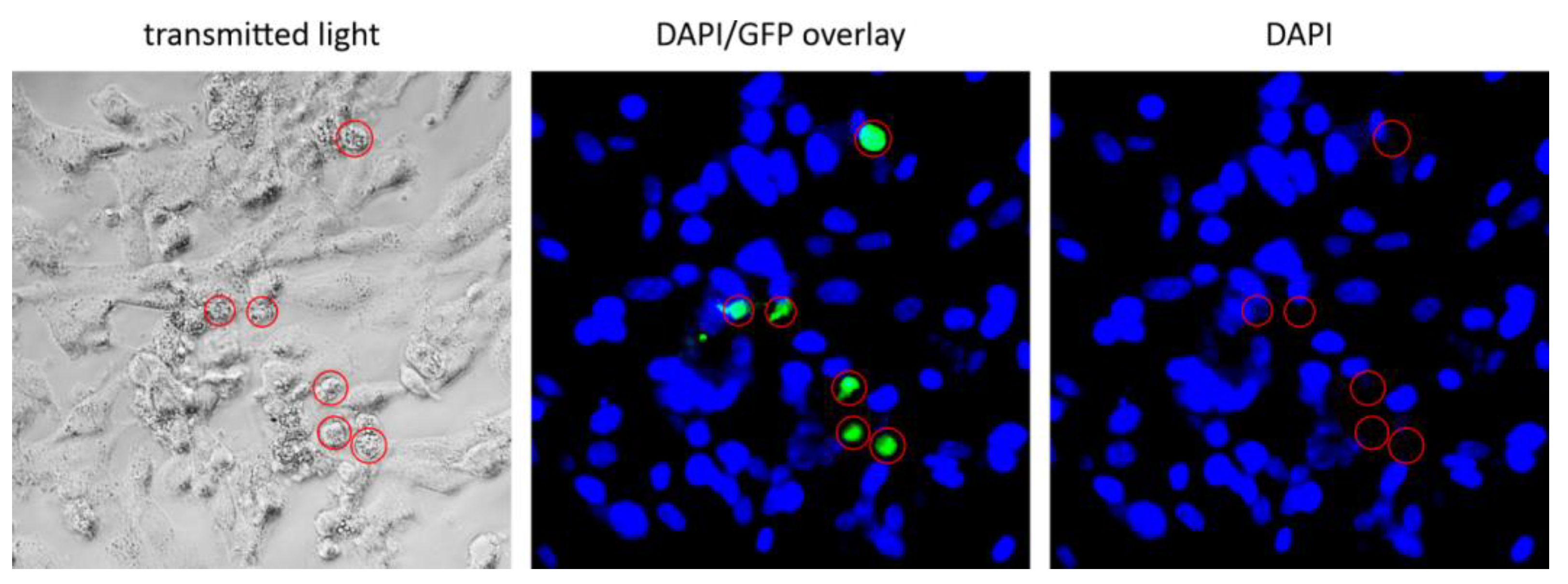
3.5. Oncolytic Activity of ZIKV/HIVgfp Lentiviral Vectors
4. Discussion
4.1. GBM Primary Cell Cultures
4.2. Virus Envelope for Pseudotyping Lentiviral Vectors
4.3. GFP as a Reporter for Transduction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes–Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro-Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Pająk, B. Looking for the Holy Grail—Drug Candidates for Glioblastoma Multiforme Chemotherapy. Biomedicines 2022, 10, 1001. [Google Scholar] [CrossRef] [PubMed]
- Rocha Pinheiro, S.L.; Lemos, F.F.B.; Marques, H.S.; Silva Luz, M.; de Oliveira Silva, L.G.; Faria Souza Mendes dos Santos, C.; da Costa Evangelista, K.; Calmon, M.S.; Sande Loureiro, M.; Freire de Melo, F. Immunotherapy in Glioblastoma Treatment: Current State and Future Prospects. World J. Clin. Oncol. 2023, 14, 138–159. [Google Scholar] [CrossRef]
- Mozhei, O.; Teschemacher, A.G.; Kasparov, S. Viral Vectors as Gene Therapy Agents for Treatment of Glioblastoma. Cancers 2020, 12, 3724. [Google Scholar] [CrossRef]
- Hamad, A.; Yusubalieva, G.M.; Baklaushev, V.P.; Chumakov, P.M.; Lipatova, A.V. Recent Developments in Glioblastoma Therapy: Oncolytic Viruses and Emerging Future Strategies. Viruses 2023, 15, 547. [Google Scholar] [CrossRef]
- Kretschmer, M.; Kadlubowska, P.; Hoffmann, D.; Schwalbe, B.; Auerswald, H.; Schreiber, M. Zikavirus Pr ME Envelope Pseudotyped Human Immunodeficiency Virus Type-1 as a Novel Tool for Glioblastoma-Directed Virotherapy. Cancers 2020, 12, 1000. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Z.; Wu, X.; Li, T.; Wu, J.; Cai, M.; Nie, J.; Wang, W.; Cui, Z. Pseudotyped Viruses. Adv. Exp. Med. Biol. 2023, 1407, 1–27. [Google Scholar] [CrossRef]
- Deng, L.; Liang, P.; Cui, H. Pseudotyped Lentiviral Vectors: Ready for Translation into Targeted Cancer Gene Therapy? Genes Dis. 2023, 10, 1937–1955. [Google Scholar] [CrossRef]
- Miletic, H.; Fischer, Y.H.; Neumann, H.; Hans, V.; Stenzel, W.; Giroglou, T.; Hermann, M.; Deckert, M.; Von Laer, D. Selective Transduction of Malignant Glioma by Lentiviral Vectors Pseudotyped with Lymphocytic Choriomeningitis Virus Glycoproteins. Hum. Human. Gene Ther. 2004, 15, 1091–1100. [Google Scholar] [CrossRef]
- Huszthy, P.C.; Giroglou, T.; Tsinkalovsky, O.; Euskirchen, P.; Skaftnesmo, K.O.; Bjerkvig, R.; von Laer, D.; Miletic, H. Remission of Invasive, Cancer Stem-Like Glioblastoma Xenografts Using Lentiviral Vector-Mediated Suicide Gene Therapy. PLoS ONE 2009, 4, e6314. [Google Scholar] [CrossRef]
- Steffens, S.; Tebbets, J.; Kramm, C.M.; Lindemann, D.; Flake, A.; Sena-Esteves, M. Transduction of Human Glial and Neuronal Tumor Cells with Different Lentivirus Vector Pseudotypes. J. Neuro-Oncol. 2004, 70, 281–288. [Google Scholar] [CrossRef]
- Stachura, P.; Stencel, O.; Lu, Z.; Borkhardt, A.; Pandyra, A.A. Arenaviruses: Old Viruses Present New Solutions for Cancer Therapy. Front. Immunol. 2023, 14, 1110522. [Google Scholar] [CrossRef]
- Lee, H.J.; Min, K.-I.; Lee, J.; Kang, S.-H.; Jeon, W.; Nam, J.H.; Ju, Y.R.; Kim, Y.B. The prM-Independent Packaging of Pseudotyped Japanese Encephalitis Virus. Virol. J. 2009, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Choi, H.; Park, K.H.; Jang, Y.; Hong, Y.; Kim, Y.B. Retention of Neutralizing Antibodies to Japanese Encephalitis Vaccine in Age Groups above Fifteen Years in Korea. Int. J. Infect. Dis. 2020, 100, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika Virus Has Oncolytic Activity against Glioblastoma Stem Cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef] [PubMed]
- Komarasamy, T.V.; Adnan, N.A.A.; James, W.; Balasubramaniam, V.R.M.T. Zika Virus Neuropathogenesis: The Different Brain Cells, Host Factors and Mechanisms Involved. Front. Immunol. 2022, 13, 773191. [Google Scholar] [CrossRef] [PubMed]
- Marbán-Castro, E.; Goncé, A.; Fumadó, V.; Romero-Acevedo, L.; Bardají, A. Zika Virus Infection in Pregnant Women and Their Children: A Review. Eur. J. Obs. Obstet. Gynecol. Reprod. Biol. 2021, 265, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Ko, A.I.; Baud, D. Zika Virus Infection—After the Pandemic. N. Engl. J. Med. 2019, 381, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.I.; Gonzalez-Orozco, M.; Rajsbaum, R. Pathogenesis of Zika Virus Infection. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 181–203. [Google Scholar] [CrossRef] [PubMed]
- Hastings, A.K.; Yockey, L.J.; Jagger, B.W.; Hwang, J.; Uraki, R.; Gaitsch, H.F.; Parnell, L.A.; Cao, B.; Mysorekar, I.U.; Rothlin, C.V.; et al. TAM Receptors Are Not Required for Zika Virus Infection in Mice. Cell Rep. 2017, 19, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Su, W.; Jin, G.; Xu, F.; Hao, S.; Guan, F.; Jia, W.; Liu, F. Glioma Stem Cells Targeted by Oncolytic Virus Carrying Endostatin–Angiostatin Fusion Gene and the Expression of Its Exogenous Gene in Vitro. Brain Res. 2011, 1390, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zwernik, S.D.; Adams, B.H.; Raymond, D.A.; Warner, C.M.; Kassam, A.B.; Rovin, R.A.; Akhtar, P. AXL Receptor Is Required for Zika Virus Strain MR-766 Infection in Human Glioblastoma Cell Lines. Mol. Ther. Oncolytics 2021, 23, 447–457. [Google Scholar] [CrossRef]
- Villinger, F.; V Silveira, E.L.; Yan Su, K.; M T Balasubramaniam, V.R. Zika Virus as Oncolytic Therapy for Brain Cancer: Myth or Reality? Front. Microbiol. 2019, 10, 2715. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Acker, T. Isolation and Culture of Primary Glioblastoma Cells from Human Tumor Specimens. Methods Mol. Biol. 2015, 1235, 263–275. [Google Scholar] [CrossRef]
- Pöhlking, C.; Beier, S.; Formanski, J.P.; Friese, M.; Schreiber, M.; Schwalbe, B. Isolation of Cells from Glioblastoma Multiforme Grade 4 Tumors for Infection with Zika Virus prME and ME Pseudotyped HIV-1. Int. J. Mol. Sci. 2023, 24, 4467. [Google Scholar] [CrossRef]
- Ledur, P.F.; Onzi, G.R.; Zong, H.; Lenz, G. Culture Conditions Defining Glioblastoma Cells Behavior: What Is the Impact for Novel Discoveries? Oncotarget 2017, 8, 69185–69197. [Google Scholar] [CrossRef]
- Wilson, C.B.; Barker, M. Cerebrospinal Fluid as a Culture Medium for Human Brain Tumors. Neurology 1966, 16, 1064. [Google Scholar] [CrossRef]
- Grunwald, V.; Ngo, H.D.; Formanski, J.P.; Jonas, J.S.; Pöhlking, C.; Schwalbe, B.; Schreiber, M. Development of Zika Virus E Variants for Pseudotyping Retroviral Vectors Targeting Glioblastoma Cells. Int. J. Mol. Sci. 2023, 24, 14487. [Google Scholar] [CrossRef] [PubMed]
- Polzer, S.; van Yperen, M.; Kirst, M.; Schwalbe, B.; Schaal, H.; Schreiber, M. Neutralization of X4- and R5-Tropic HIV-1 NL4-3 Variants by HOCl-Modified Serum Albumins. BMC Res. Notes 2010, 3, 155. [Google Scholar] [CrossRef]
- Pugach, P.; Marozsan, A.J.; Ketas, T.J.; Landes, E.L.; Moore, J.P.; Kuhmann, S.E. HIV-1 Clones Resistant to a Small Molecule CCR5 Inhibitor Use the Inhibitor-Bound Form of CCR5 for Entry. Virology 2007, 361, 212. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.D.; Formanski, J.P.; Grunwald, V.; Schwalbe, B.; Schreiber, M. Generation of Viral Particles with Brain Cell-Specific Tropism by Pseudotyping HIV-1 with the Zika Virus E Protein. Methods Protoc. 2024, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Auerswald, H.; Klepsch, L.; Schreiber, S.; Hülsemann, J.; Franzke, K.; Kann, S.; Bunthin, Y.; Duong, V.; Buchy, P.; Schreiber, M.; et al. The Dengue ED3 Dot Assay, a Novel Serological Test for the Detection of Denguevirus Type-Specific Antibodies and Its Application in a Retrospective Seroprevalence Study. Viruses 2019, 11, 304. [Google Scholar] [CrossRef] [PubMed]
- Grube, S.; Freitag, D.; Kalff, R.; Ewald, C.; Walter, J. Characterization of Adherent Primary Cell Lines from Fresh Human Glioblastoma Tissue, Defining Glial Fibrillary Acidic Protein as a Reliable Marker in Establishment of Glioblastoma Cell Culture. Cancer Rep. 2021, 4, e1324. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.M.; Diez-Valle, R.; Manterola, L.; Rubio, A.; Liu, D.; Cortes-Santiago, N.; Urquiza, L.; Jauregi, P.; Lopez de Munain, A.; Sampron, N.; et al. Genetic and Epigenetic Modifications of Sox2 Contribute to the Invasive Phenotype of Malignant Gliomas. PLoS ONE 2011, 6, e26740. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Jia, D.; Liu, S.; Wang, F.; Li, G.; Zhang, Y.; Cao, X.; Ling, E.-A.; Hao, A. Oct4 Is Expressed in Human Gliomas and Promotes Colony Formation in Glioma Cells. Glia 2009, 57, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Neradil, J.; Veselska, R. Nestin as a Marker of Cancer Stem Cells. Cancer Sci. 2015, 106, 803. [Google Scholar] [CrossRef]
- Niu, C.-S.; Li, D.-X.; Liu, Y.-H.; Fu, X.-M.; Tang, S.-F.; Li, J. Expression of NANOG in Human Gliomas and Its Relationship with Undifferentiated Glioma Cells. Oncol. Rep. 2011, 26, 593–601. [Google Scholar] [CrossRef]
- Annovazzi, L.; Mellai, M.; Caldera, V.; Valente, G.; Schiffer, D. SOX2 Expression and Amplification in Gliomas and Glioma Cell Lines. Cancer Genom. Proteom. 2011, 8, 139–147. [Google Scholar]
- Zhu, Z.; Mesci, P.; Bernatchez, J.A.; Gimple, R.C.; Wang, X.; Schafer, S.T.; Wettersten, H.I.; Beck, S.; Clark, A.E.; Wu, Q.; et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin Avβ5 Axis. Cell Stem Cell 2020, 26, 187–204.e10. [Google Scholar] [CrossRef]
- Miyan, J.A.; Zendah, M.; Mashayekhi, F.; Owen-Lynch, P.J. Cerebrospinal Fluid Supports Viability and Proliferation of Cortical Cells in Vitro, Mirroring in Vivo Development. Cerebrospinal Fluid. Res. 2006, 3, 2. [Google Scholar] [CrossRef]
- Pontes Soares, C.; Midlej, V.; de Oliveira, M.E.W.; Benchimol, M.; Costa, M.L.; Mermelstein, C. 2D and 3D-Organized Cardiac Cells Shows Differences in Cellular Morphology, Adhesion Junctions, Presence of Myofibrils and Protein Expression. PLoS ONE 2012, 7, e38147. [Google Scholar] [CrossRef]
- Cumberworth, S.L.; Barrie, J.A.; Cunningham, M.E.; de Figueiredo, D.P.G.; Schultz, V.; Wilder-Smith, A.J.; Brennan, B.; Pena, L.J.; Freitas de Oliveira França, R.; Linington, C.; et al. Zika Virus Tropism and Interactions in Myelinating Neural Cell Cultures: CNS Cells and Myelin Are Preferentially Affected. Acta Neuropathol. Commun. 2017, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.; Chakravarty, N.; Paiola, S.; Urena, E.; Gyani, P.; Tse, C.; French, S.W.; Danielpour, M.; Breunig, J.J.; Nathanson, D.A.; et al. Differential Susceptibility of Ex Vivo Primary Glioblastoma Tumors to Oncolytic Effect of Modified Zika Virus. Cells 2023, 12, 2384. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Regla-Nava, J.A.; Chai, G.; Sheets, N.; Tang, W.; Terskikh, A.V.; Shresta, S.; Gleeson, J.G. Zika Virus Infects Neural Progenitors in the Adult Mouse Brain and Alters Proliferation. Cell Stem Cell 2016, 19, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Rusu, P.; Shao, C.; Neuerburg, A.; Acikgöz, A.A.; Wu, Y.; Zou, P.; Phapale, P.; Shankar, T.S.; Döring, K.; Dettling, S.; et al. GPD1 Specifically Marks Dormant Glioma Stem Cells with a Distinct Metabolic Profile. Cell Stem Cell 2019, 25, 241–257.e8. [Google Scholar] [CrossRef] [PubMed]
- Hoogstrate, Y.; Ghisai, S.A.; de Wit, M.; de Heer, I.; Draaisma, K.; van Riet, J.; van de Werken, H.J.G.; Bours, V.; Buter, J.; Vanden Bempt, I.; et al. The EGFRvIII Transcriptome in Glioblastoma: A Meta-Omics Analysis. Neuro-Oncol. 2022, 24, 429–441. [Google Scholar] [CrossRef]
- Morris, K.V.; Rossi, J.J. Lentiviral-Mediated Delivery of siRNAs for Antiviral Therapy. Gene Ther. 2006, 13, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Kantor, B. Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses 2021, 13, 1288. [Google Scholar] [CrossRef]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef]
- Poletti, V.; Mavilio, F. Designing Lentiviral Vectors for Gene Therapy of Genetic Diseases. Viruses 2021, 13, 1526. [Google Scholar] [CrossRef]
- Joglekar, A.V.; Sandoval, S. Pseudotyped Lentiviral Vectors: One Vector, Many Guises. Hum. Human. Gene Ther. Methods 2017, 28, 291–301. [Google Scholar] [CrossRef]
- Liu, J.; Mao, Y.; Li, Q.; Qiu, Z.; Li, J.; Li, X.; Liang, W.; Xu, M.; Li, A.; Cai, X.; et al. Efficient Gene Transfer to Kidney Using a Lentiviral Vector Pseudotyped with Zika Virus Envelope Glycoprotein. Hum. Gene Ther. 2022, 33, 1269–1278. [Google Scholar] [CrossRef]
- Votteler, J.; Sundquist, W.I. Virus Budding and the ESCRT Pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Teis, D. The ESCRT Machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. Role of ESCRT-I in Retroviral Budding. J. Virol. 2003, 77, 4794–4804. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.M.; Ahmed, A.K.; Matsangos, A.E.; Lay, F.; Born, L.J.; Marti, G.; Harmon, J.W.; Sun, Z. Cellular GFP Toxicity and Immunogenicity: Potential Confounders in in Vivo Cell Tracking Experiments. Stem Cell Rev. Rep. 2016, 12, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.S.; Jan, M.S.; Chou, C.K.; Chen, P.H.; Ke, N.J. Is Green Fluorescent Protein Toxic to the Living Cells? Biochem. Biophys. Res. Commun. 1999, 260, 712–717. [Google Scholar] [CrossRef]
- Taghizadeh, R.R.; Sherley, J.L. CFP and YFP, but Not GFP, Provide Stable Fluorescent Marking of Rat Hepatic Adult Stem Cells. J. Biomed. Biotechnol. 2008, 2008, 453590. [Google Scholar] [CrossRef] [PubMed]
- Detrait, E.R.; Bowers, W.J.; Halterman, M.W.; Giuliano, R.E.; Bennice, L.; Federoff, H.J.; Richfield, E.K. Reporter Gene Transfer Induces Apoptosis in Primary Cortical Neurons. Mol. Ther. 2002, 5, 723–730. [Google Scholar] [CrossRef]
- Krestel, H.E.; Mihaljevic, A.L.A.; Hoffman, D.A.; Schneider, A. Neuronal Co-Expression of EGFP and Beta-Galactosidase in Mice Causes Neuropathology and Premature Death. Neurobiol. Dis. 2004, 17, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Ganini, D.; Leinisch, F.; Kumar, A.; Jiang, J.; Tokar, E.J.; Malone, C.C.; Petrovich, R.M.; Mason, R.P. Fluorescent Proteins Such as eGFP Lead to Catalytic Oxidative Stress in Cells. Redox Biol. 2017, 12, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Yang, B.; Petersen, D.; Pepper, K.A.; Alfaro, P.A.; Kohn, D.B.; Reynolds, C.P. Transduction of Green Fluorescent Protein Increased Oxidative Stress and Enhanced Sensitivity to Cytotoxic Drugs in Neuroblastoma Cell Lines. Mol. Cancer Ther. 2003, 2, 911–917. [Google Scholar] [PubMed]
- Shemiakina, I.I.; Ermakova, G.V.; Cranfill, P.J.; Baird, M.A.; Evans, R.A.; Souslova, E.A.; Staroverov, D.B.; Gorokhovatsky, A.Y.; Putintseva, E.V.; Gorodnicheva, T.V.; et al. A Monomeric Red Fluorescent Protein with Low Cytotoxicity. Nat. Commun. 2012, 3, 1204. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Geng, Y.; Lovett-Barron, M.; Niu, X.; Deng, M.; Wang, L.; Ataie, N.; Sens, A.; Ng, H.-L.; Chen, S.; et al. A Bright, Nontoxic, and Non-Aggregating Red Fluorescent Protein for Long-Term Labeling of Fine Structures in Neurons. Front. Cell Dev. Biol. 2022, 10, 893468. [Google Scholar] [CrossRef] [PubMed]
- Agbulut, O.; Huet, A.; Niederländer, N.; Puceat, M.; Menasché, P.; Coirault, C. Green Fluorescent Protein Impairs Actin-Myosin Interactions by Binding to the Actin-Binding Site of Myosin. J. Biol. Chem. 2007, 282, 10465–10471. [Google Scholar] [CrossRef]
- Agbulut, O.; Coirault, C.; Niederländer, N.; Huet, A.; Vicart, P.; Hagège, A.; Puceat, M.; Menasché, P. GFP Expression in Muscle Cells Impairs Actin-Myosin Interactions: Implications for Cell Therapy. Nat. Methods 2006, 3, 331. [Google Scholar] [CrossRef]
- Perelroizen, R.; Philosof, B.; Budick-Harmelin, N.; Chernobylsky, T.; Ron, A.; Katzir, R.; Shimon, D.; Tessler, A.; Adir, O.; Gaoni-Yogev, A.; et al. Astrocyte Immunometabolic Regulation of the Tumour Microenvironment Drives Glioblastoma Pathogenicity. Brain 2022, 145, 3288–3307. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AKH Cells | DF | CSF | CSF-DF |
|---|---|---|---|
| 01 | ++ | − | +++ |
| 02 | ++ | + | ++ |
| 05 | ++ | + | +++ |
| 09 | +++ | + | +++ |
| 10 | ++ | + | +++ |
| 11 | ++ | − | +++ |
| 12 | +++ | + | +++ |
| 13 | ++ | + | n.d. |
| 14 | ++ | + | +++ |
| 15 | + | − | ++ |
| 16 | ++ | − | +++ |
| 17 | ++ | − | +++ |
| 18 | n.d. | n.d. | +++ |
| 19 | n.d. | n.d. | +++ |
| Cell Cultures | E2-HIVgfp | E41.2-HIVgfp | G-HIVgfp |
|---|---|---|---|
| AKH-13 | 3.6 ± 0.2 | 3.2 ± 0.3 | 0.5 ± 0.1 |
| AKH-14 | 6.3 ± 1.1 | 11.7 ± 0.7 | 5.4 ± 0.5 |
| AKH-16 | 8.2 ± 0.6 | 24.0 ± 1.3 | 5.9 ± 1.6 |
| AKH-17 | 10.0 ± 3.7 | n.d. | 6.8 ± 1.1 |
| AKH-18 | 5.7 ± 0.9 | n.d. | 6.5 ± 1.2 |
| U-87MG | 1.9 ± 0.2 | 7.8 ± 1.0 | 1.7 ± 0.4 |
| U-138MG | 2.4 ± 0.5 | 9.8 ± 2.4 | 1.6 ± 0.5 |
| U-343MG | 2.7 ± 1.3 | n.d. | 0.8± 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Formanski, J.P.; Ngo, H.D.; Grunwald, V.; Pöhlking, C.; Jonas, J.S.; Wohlers, D.; Schwalbe, B.; Schreiber, M. Transduction Efficiency of Zika Virus E Protein Pseudotyped HIV-1gfp and Its Oncolytic Activity Tested in Primary Glioblastoma Cell Cultures. Cancers 2024, 16, 814. https://doi.org/10.3390/cancers16040814
Formanski JP, Ngo HD, Grunwald V, Pöhlking C, Jonas JS, Wohlers D, Schwalbe B, Schreiber M. Transduction Efficiency of Zika Virus E Protein Pseudotyped HIV-1gfp and Its Oncolytic Activity Tested in Primary Glioblastoma Cell Cultures. Cancers. 2024; 16(4):814. https://doi.org/10.3390/cancers16040814
Chicago/Turabian StyleFormanski, Jan Patrick, Hai Dang Ngo, Vivien Grunwald, Celine Pöhlking, Jana Sue Jonas, Dominik Wohlers, Birco Schwalbe, and Michael Schreiber. 2024. "Transduction Efficiency of Zika Virus E Protein Pseudotyped HIV-1gfp and Its Oncolytic Activity Tested in Primary Glioblastoma Cell Cultures" Cancers 16, no. 4: 814. https://doi.org/10.3390/cancers16040814
APA StyleFormanski, J. P., Ngo, H. D., Grunwald, V., Pöhlking, C., Jonas, J. S., Wohlers, D., Schwalbe, B., & Schreiber, M. (2024). Transduction Efficiency of Zika Virus E Protein Pseudotyped HIV-1gfp and Its Oncolytic Activity Tested in Primary Glioblastoma Cell Cultures. Cancers, 16(4), 814. https://doi.org/10.3390/cancers16040814