
Identification of Hypoxia Prognostic Signature in Glioblastoma Multiforme Based on Bulk and Single-Cell RNA-Seq

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
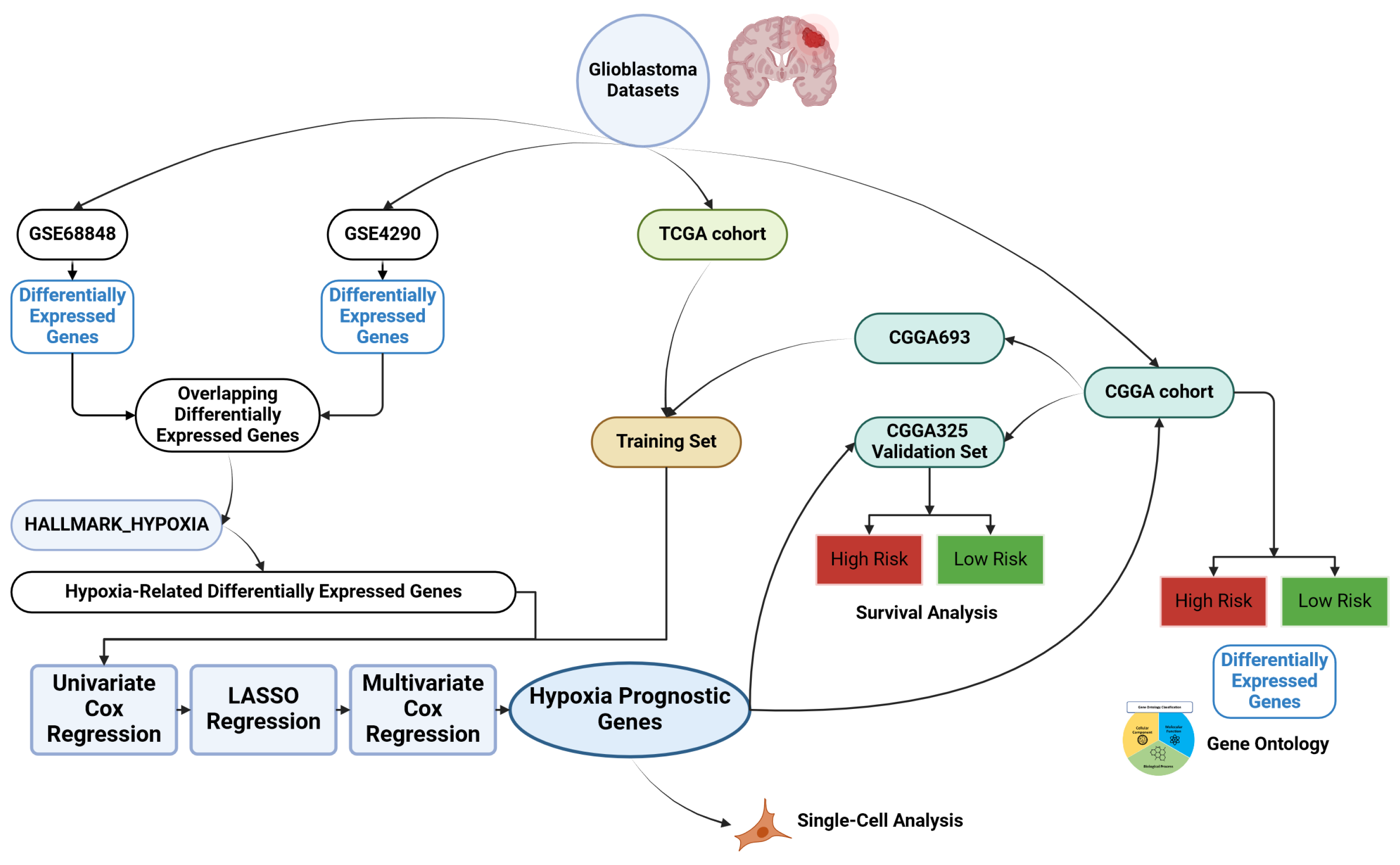
2. Materials and Methods
2.1. Data Sources
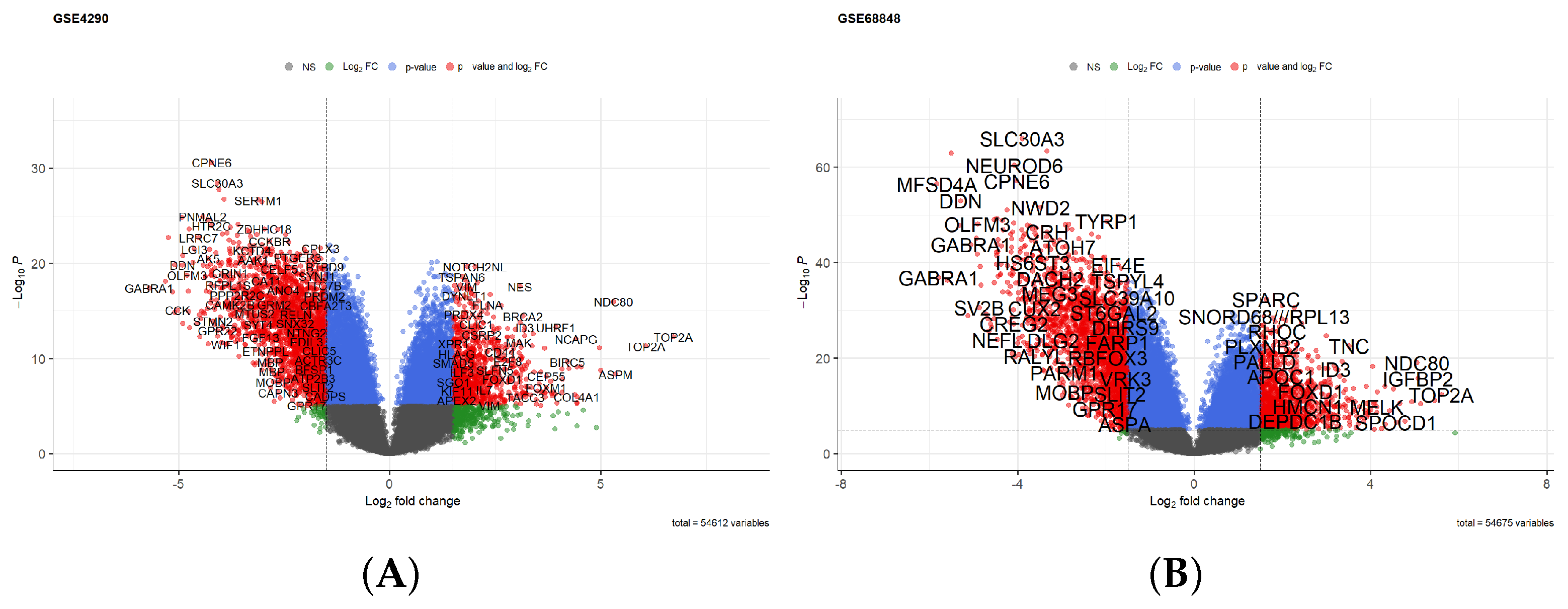
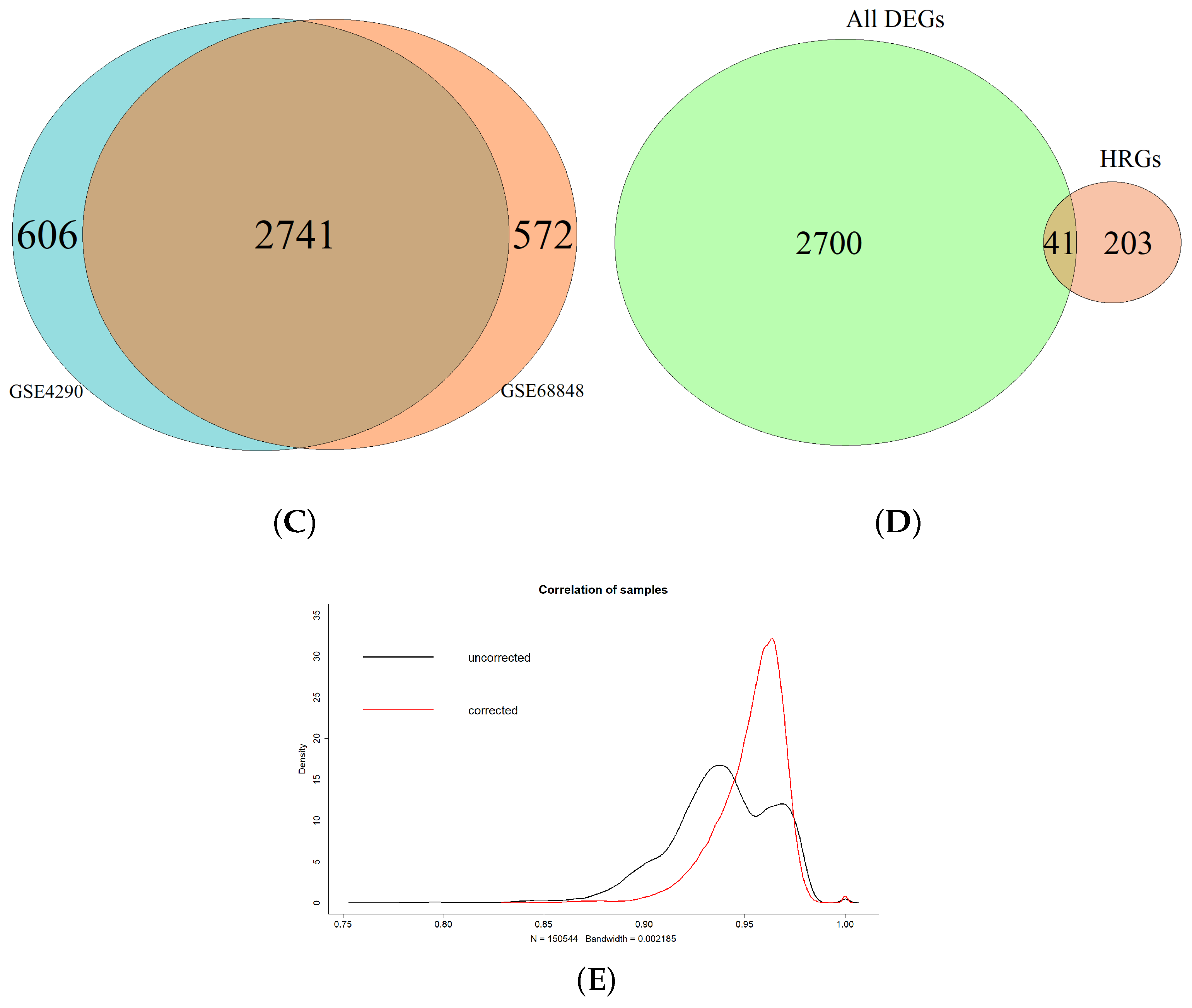
2.2. Identification of Hypoxia-Related Differentially Expressed Genes
2.3. Construction of Machine Learning Diagnostic Model
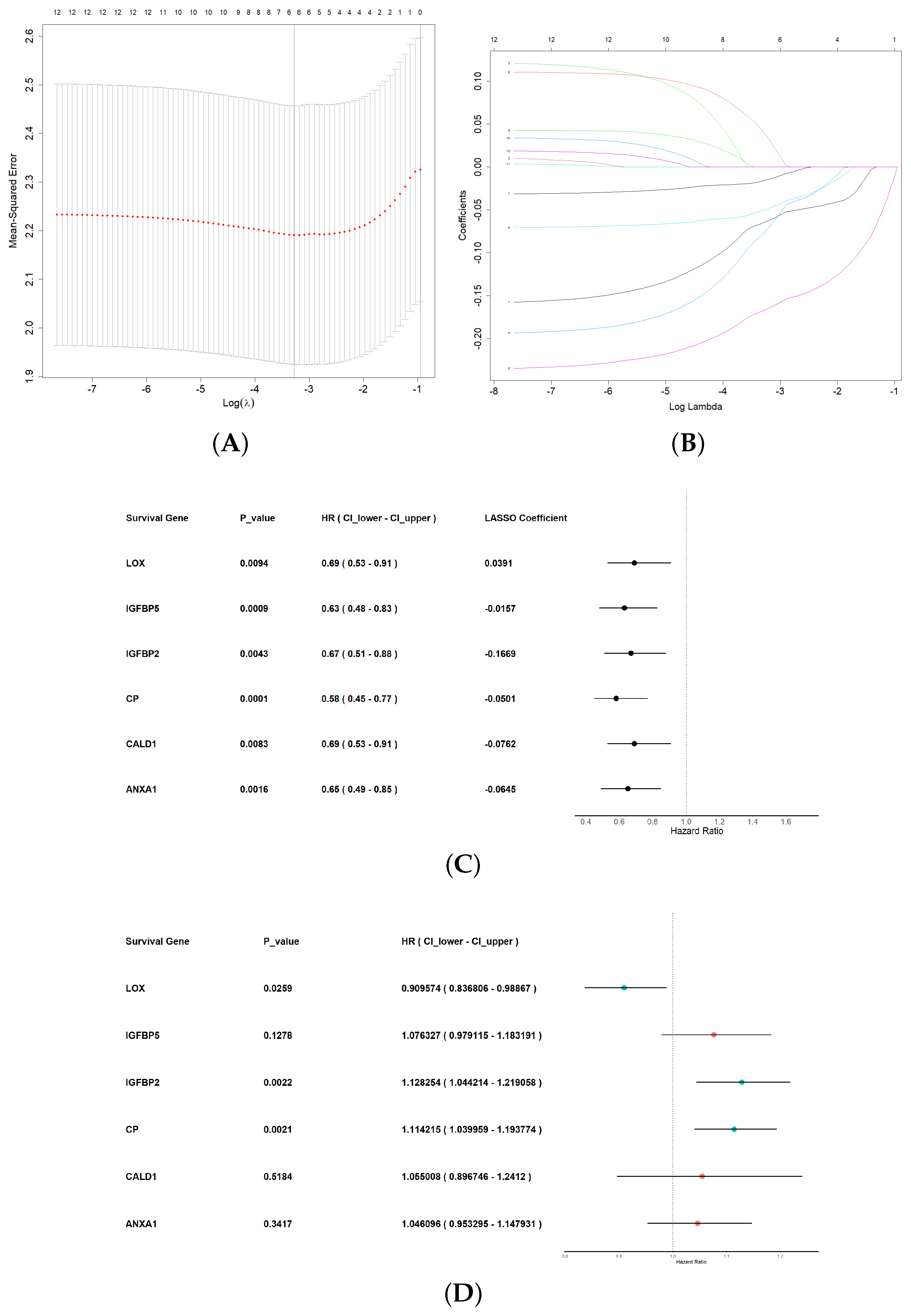
2.4. Construction of Hypoxia-Related Prognostic Signature
2.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analyses
2.6. Tumor Immune Microenvironment and Drug Sensitivity Analysis
2.7. Single-Cell Sequencing Analysis
2.8. Statistical Analysis and Data Manipulation
3. Results
3.1. Identification of Hypoxia-Related Differentially Expressed Genes
3.2. Allocating the Training and Validation Datasets
3.3. Construction of Machine Learning Diagnostic Model
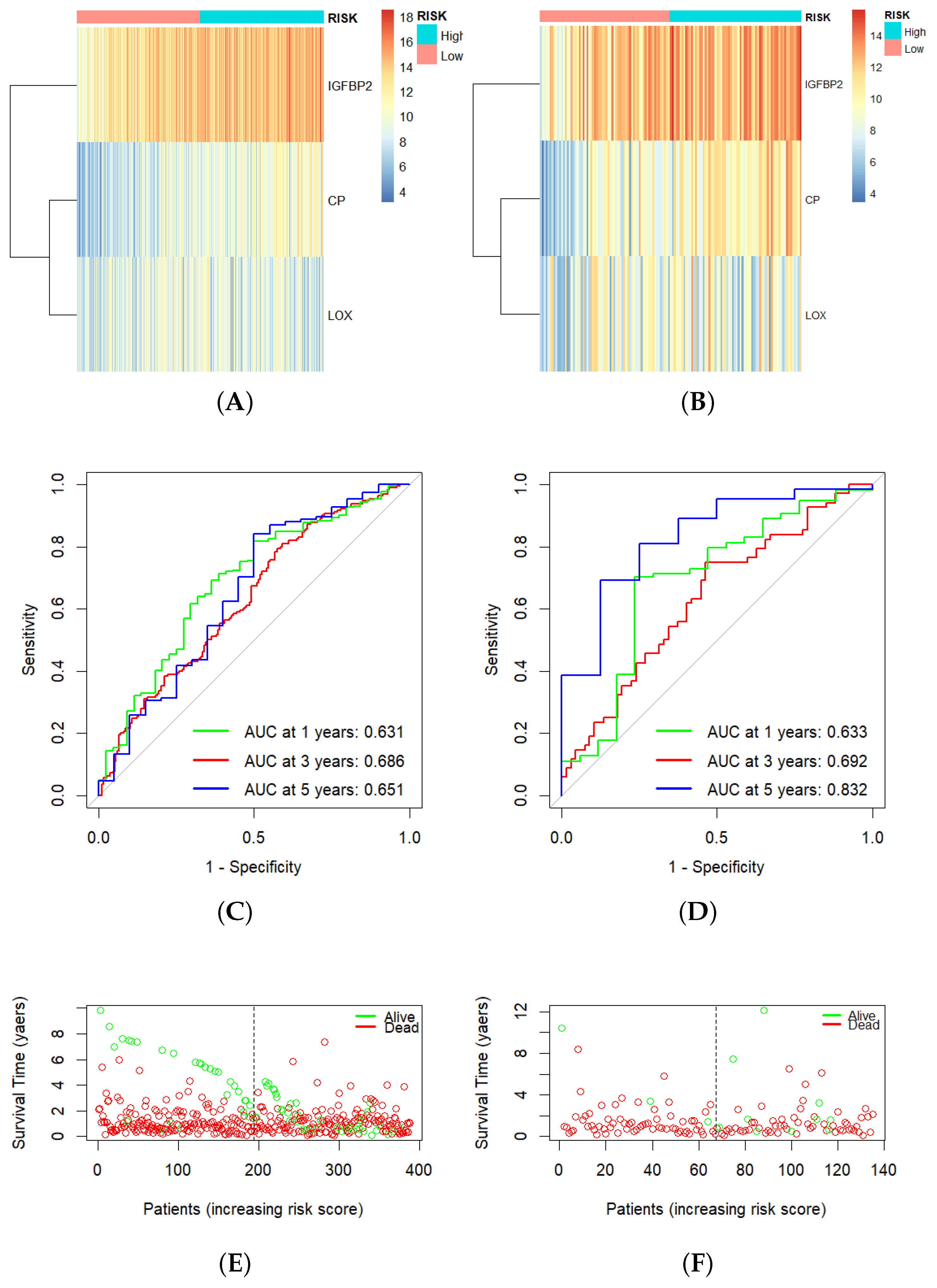
3.4. Construction of Hypoxia-Related Prognostic Signature
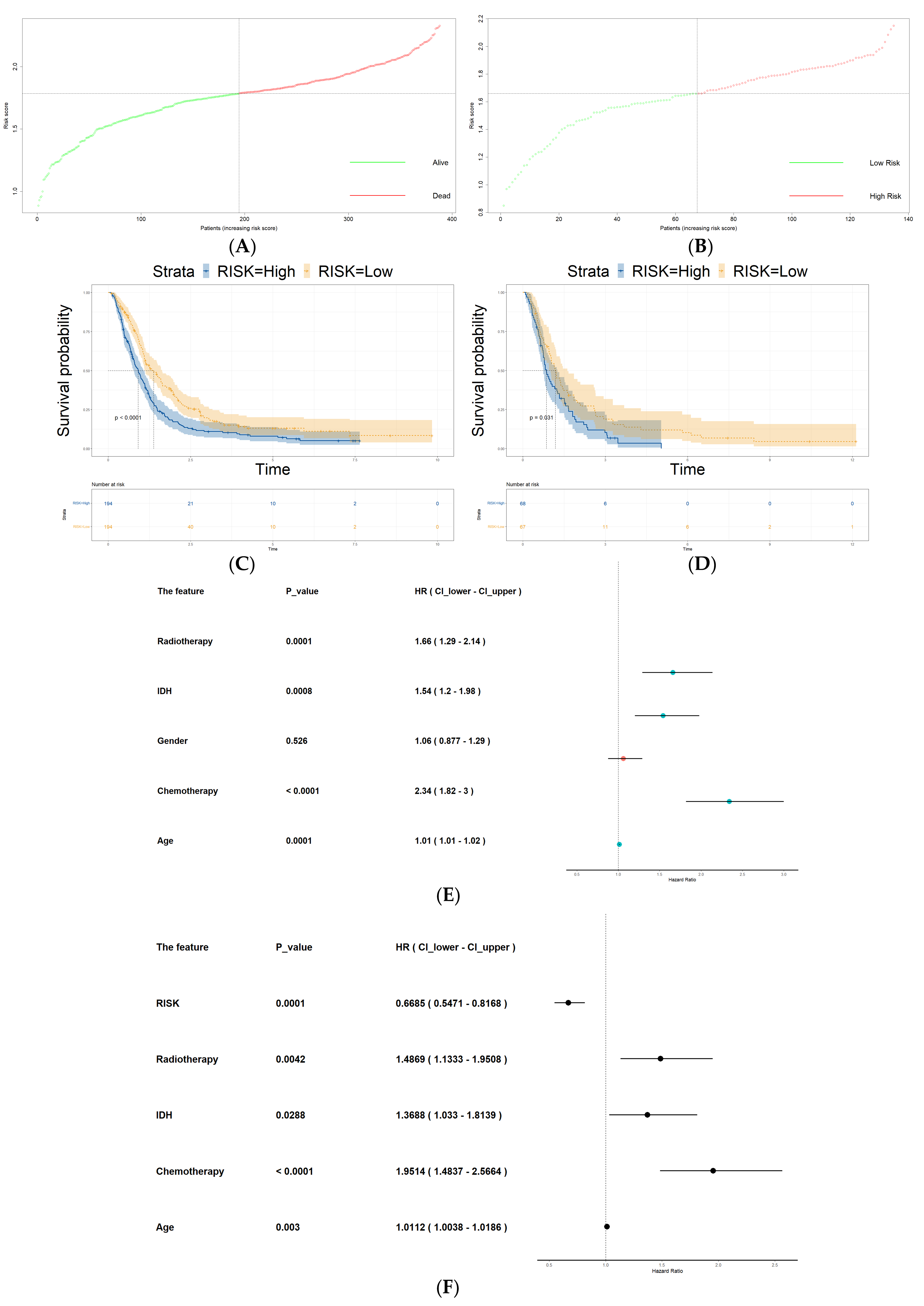
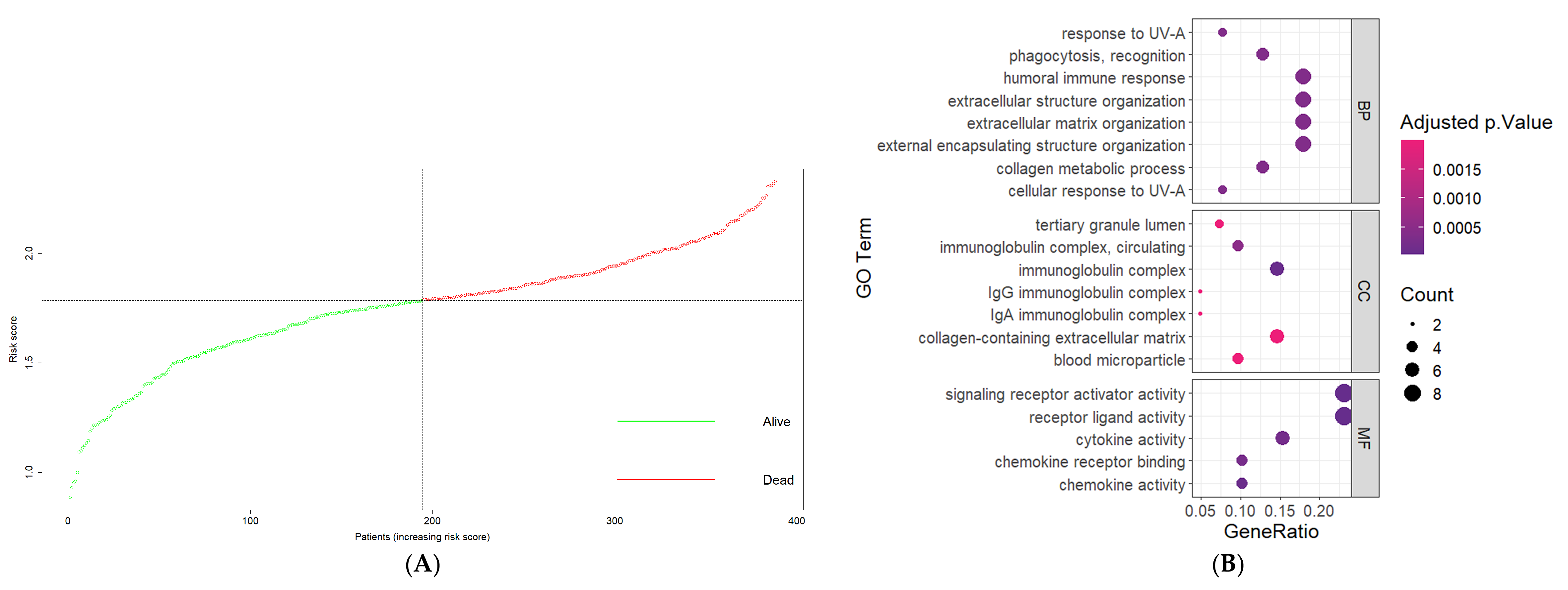
3.5. Risk Score as an Independent Prognostic Factor
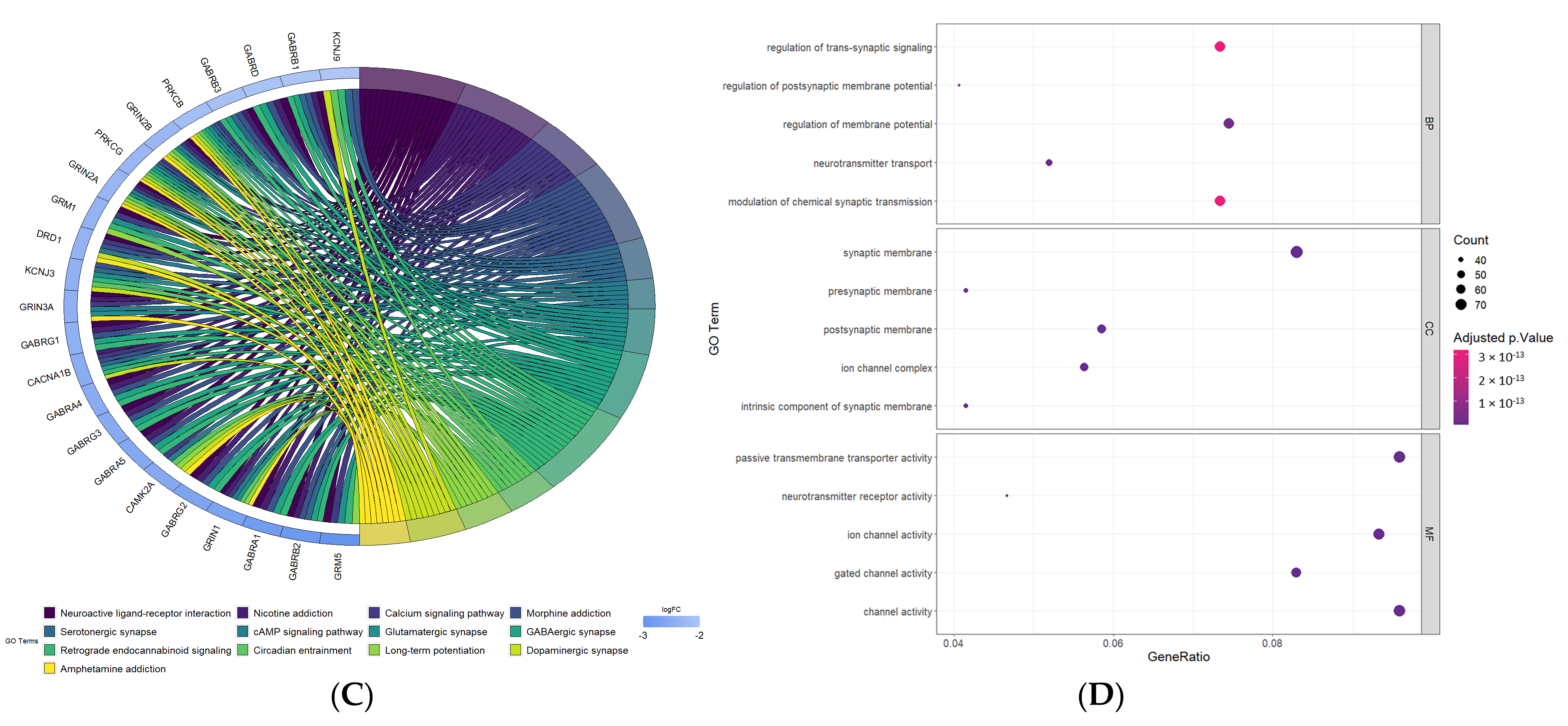
3.6. Functional Annotations of DEGs between High-Risk and Low-Risk Groups
3.7. Tumor Immune Microenvironment and Drug Sensitivity Analysis
3.8. Single-Cell Sequencing Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme—Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhang, G. Differential gene expression analysis in glioblastoma cells and normal human brain cells based on GEO database. Oncol. Lett. 2017, 14, 6040–6044. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Winter, S.C.; Buffa, F.M.; Silva, P.; Miller, C.; Valentine, H.R.; Turley, H.; Shah, K.A.; Cox, G.J.; Corbridge, R.J.; Homer, J.J.; et al. Relation of a Hypoxia Metagene Derived from Head and Neck Cancer to Prognosis of Multiple Cancers. Cancer Res. 2007, 67, 3441–3449. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Wang, X.; Chen, L.; Liu, W.; Zhang, Y.; Liu, D.; Zhou, C.; Shi, S.; Dong, J.; Lai, Z.; Zhao, B.; et al. TIMEDB: Tumor immune micro-environment cell composition database with automatic analysis and interactive visualization. Nucleic Acids Res. 2023, 51, D1417–D1424. [Google Scholar] [CrossRef] [PubMed]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Sceneay, J.; Gödde, N.; Kinwel, T.; Ham, S.; Thompson, E.W.; Humbert, P.O.; Möller, A. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene 2018, 37, 4214–4225. [Google Scholar] [CrossRef]
- Huang, Z.; Yang, M.; Li, Y.; Yang, F.; Feng, Y. Exosomes Derived from Hypoxic Colorectal Cancer Cells Transfer Wnt4 to Normoxic Cells to Elicit a Prometastatic Phenotype. Int. J. Biol. Sci. 2018, 14, 2094–2102. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhang, J.; Shi, Y.; Shen, H.; Li, Y.; Chen, Y.; Liang, L. ESM1/HIF-1α pathway modulates chronic intermittent hypoxia-induced non-small-cell lung cancer proliferation, stemness and epithelial-mesenchymal transition. Oncol. Rep. 2021, 45, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Xia, M.; Xu, Y.; Sun, Q.; Ye, L.; Yuan, F.; Wang, Y.; Cai, J.; Ye, Z.; Tian, D. Identification and validation of a novel prognostic signature based on mitochondria and oxidative stress related genes for glioblastoma. J. Transl. Med. 2023, 21, 136. [Google Scholar] [CrossRef]
- Zhang, B.; Xie, L.; Liu, J.; Liu, A.; He, M. Construction and validation of a cuproptosis-related prognostic model for glioblastoma. Front. Immunol. 2023, 14, 1082974. [Google Scholar] [CrossRef] [PubMed]
- Phillips, L.M.; Zhou, X.; Cogdell, D.E.; Chua, C.Y.; Huisinga, A.; Hess, K.R.; Fuller, G.N.; Zhang, W. Glioma progression is mediated by an addiction to aberrant IGFBP2 expression and can be blocked using anti-IGFBP2 strategies. J. Pathol. 2016, 239, 355–364. [Google Scholar] [CrossRef]
- Yao, X.; Sun, S.; Zhou, X.; Guo, W.; Zhang, L. IGF-binding protein 2 is a candidate target of therapeutic potential in cancer. Tumor Biol. 2016, 37, 1451–1459. [Google Scholar] [CrossRef]
- Liu, Y.; Song, C.; Shen, F.; Zhang, J.; Song, S.W. IGFBP2 promotes immunosuppression associated with its mesenchymal induction and FcγRIIB phosphorylation in glioblastoma. PLoS ONE 2019, 14, e0222999. [Google Scholar] [CrossRef]
- Yamini, B. NF-κB, Mesenchymal Differentiation and Glioblastoma. Cells 2018, 7, 125. [Google Scholar] [CrossRef]
- Cai, J.; Chen, Q.; Cui, Y.; Dong, J.; Chen, M.; Wu, P.; Jiang, C. Immune heterogeneity and clinicopathologic characterization of IGFBP2 in 2447 glioma samples. OncoImmunology 2018, 7, e1426516. [Google Scholar] [CrossRef]
- Yuan, Q.; Cai, H.Q.; Zhong, Y.; Zhang, M.J.; Cheng, Z.J.; Hao, J.J.; Wang, M.R.; Wan, J.H. Overexpression of IGFBP2 mRNA predicts poor survival in patients with glioblastoma. Biosci. Rep. 2019, 39, BSR20190045. [Google Scholar] [CrossRef] [PubMed]
- Elmlinger, M.W.; Deininger, M.H.; Schuett, B.S.; Meyermann, R.; Duffner, F.; Grote, E.H.; Ranke, M.B. In Vivo Expression of Insulin-Like Growth Factor-Binding Protein-2 in Human Gliomas Increases with the Tumor Grade. Endocrinology 2001, 142, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen Sensing, Homeostasis, and Disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.W.; Liao, A.; Qutub, A.A. Simulation Predicts IGFBP2-HIF1α Interaction Drives Glioblastoma Growth. PLoS Comput. Biol. 2015, 11, e1004169. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Weaver, V.M. Three-dimensional context regulation of metastasis. Clin. Exp. Metastasis 2009, 26, 35–49. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Kagan, H.M.; Trackman, P.C. Properties and function of lysyl oxidase. Am. J. Respir. Cell Mol. Biol. 1991, 5, 206–210. [Google Scholar] [CrossRef]
- Ji, F.; Wang, Y.; Qiu, L.; Li, S.; Zhu, J.; Liang, Z.; Wan, Y.; Di, W. Hypoxia inducible factor 1α -mediated LOX expression correlates with migration and invasion in epithelial ovarian cancer. Int. J. Oncol. 2013, 42, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Kii, I.; Saito, M.; Kudo, A. Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J. Biol. Chem. 2010, 285, 13294–13303. [Google Scholar] [CrossRef] [PubMed]
- De Donato, M.; Petrillo, M.; Martinelli, E.; Filippetti, F.; Zannoni, G.F.; Scambia, G.; Gallo, D. Uncovering the role of nuclear Lysyl oxidase (LOX) in advanced high grade serous ovarian cancer. Gynecol. Oncol. 2017, 146, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.d.; Uno, M.; Marie, S.K.N.; Oba-Shinjo, S.M. LOX Expression and Functional Analysis in Astrocytomas and Impact of IDH1 Mutation. PLoS ONE 2015, 10, e0119781. [Google Scholar] [CrossRef] [PubMed]
- Mukae, Y.; Ito, H.; Miyata, Y.; Araki, K.; Matsuda, T.; Aibara, N.; Nakamura, Y.; Matsuo, T.; Sakai, H.; Ohyama, K. Ceruloplasmin Levels in Cancer Tissues and Urine Are Significant Biomarkers of Pathological Features and Outcome in Bladder Cancer. Anticancer Res. 2021, 41, 3815–3823. [Google Scholar] [CrossRef] [PubMed]
- Scheinberg, I.H.; Gitlin, D. Deficiency of ceruloplasmin in patients with hepatolenticular degeneration (Wilson’s disease). Science 1952, 116, 484–485. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.H.; Venkatesh, R.; More, C.B. Determination of role of ceruloplasmin in oral potentially malignant disorders and oral malignancy—A cross-sectional study. Oral Dis. 2017, 23, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Balmaña, M.; Sarrats, A.; Llop, E.; Barrabés, S.; Saldova, R.; Ferri, M.J.; Figueras, J.; Fort, E.; de Llorens, R.; Rudd, P.M.; et al. Identification of potential pancreatic cancer serum markers: Increased sialyl-Lewis X on ceruloplasmin. Clin. Chim. Acta 2015, 442, 56–62. [Google Scholar] [CrossRef]
- Repetto, O.; Mussolin, L.; Elia, C.; Martina, L.; Bianchi, M.; Buffardi, S.; Sala, A.; Burnelli, R.; Mascarin, M.; De Re, V. Proteomic Identification of Plasma Biomarkers in Children and Adolescents with Recurrent Hodgkin Lymphoma. J. Cancer 2018, 9, 4650–4658. [Google Scholar] [CrossRef]
- Li, N.; Hu, H.; Wu, G.; Sun, B. Value of immune factors for monitoring risk of lung cancer in patients with interstitial lung disease. J. Int. Med Res. 2019, 47, 3344–3353. [Google Scholar] [CrossRef]
- Zhevak, T.; Shelekhova, T.; Chesnokova, N.; Tsareva, O.; Chanturidze, A.; Litvitsky, P.; Andriutsa, N.; Samburova, N.; Budnik, I. The relationship between oxidative stress and cytogenetic abnormalities in B-cell chronic lymphocytic leukemia. Exp. Mol. Pathol. 2020, 116, 104524. [Google Scholar] [CrossRef]
- Strickland, N.J.; Matsha, T.; Erasmus, R.T.; Zaahl, M.G. Molecular analysis of Ceruloplasmin in a South African cohort presenting with oesophageal cancer. Int. J. Cancer 2012, 131, 623–632. [Google Scholar] [CrossRef]
- Han, I.W.; Jang, J.Y.; Kwon, W.; Park, T.; Kim, Y.; Lee, K.B.; Kim, S.W. Ceruloplasmin as a prognostic marker in patients with bile duct cancer. Oncotarget 2017, 8, 29028–29037. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhi, Q.; Xie, Q.; Wu, X.; Gao, Y.; Chen, X.; Shi, L. Reduced expression of ferroportin1 and ceruloplasmin predicts poor prognosis in adrenocortical carcinoma. J. Trace Elem. Med. Biol. 2019, 56, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Bleu, M.; Gaulis, S.; Lopes, R.; Sprouffske, K.; Apfel, V.; Holwerda, S.; Pregnolato, M.; Yildiz, U.; Cordo’, V.; Dost, A.F.M.; et al. PAX8 activates metabolic genes via enhancer elements in Renal Cell Carcinoma. Nat. Commun. 2019, 10, 3739. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, R.; Shiba-Ishii, A.; Nakano, N.; Togayachi, A.; Sakashita, S.; Sato, Y.; Minami, Y.; Noguchi, M. Heterotopic production of ceruloplasmin by lung adenocarcinoma is significantly correlated with prognosis. Lung Cancer 2018, 118, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Han, B.; Meng, Y.; Han, Y.; Liu, B.; Zhang, B.; Chang, Y.; Cao, P.; Fan, Y.; Tan, K. Ceruloplasmin correlates with immune infiltration and serves as a prognostic biomarker in breast cancer. Aging 2021, 13, 20438–20467. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.; Avril, S.; Legendre, C.; Lelièvre, B.; Vellenriter, H.; Boni, S.; Cayon, J.; Guillet, C.; Guilloux, Y.; Chérel, M.; et al. A role for ceruloplasmin in the control of human glioblastoma cell responses to radiation. BMC Cancer 2022, 22, 843. [Google Scholar] [CrossRef] [PubMed]
- Osaki, S.; Johnson, D.A.; Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem. 1966, 241, 2746–2751. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Ward, D.M.; di Patti, M.C.B.; Jeong, S.Y.; David, S.; Musci, G.; Kaplan, J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007, 26, 2823–2831. [Google Scholar] [CrossRef]
- Shi, T.; Zhu, J.; Zhang, X.; Mao, X. The Role of Hypoxia and Cancer Stem Cells in Development of Glioblastoma. Cancers 2023, 15, 2613. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Varn, F.S.; Park, S.H.; Yoon, B.W.; Park, H.R.; Lee, C.; Verhaak, R.G.W.; Paek, S.H. Perspective of mesenchymal transformation in glioblastoma. Acta Neuropathol. Commun. 2021, 9, 50. [Google Scholar] [CrossRef]
- Verma, B.K.; Kondaiah, P. Regulation of β-catenin by IGFBP2 and its cytoplasmic actions in glioma. J. Neuro-Oncol. 2020, 149, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef]
- Coelho, B.P.; Fernandes, C.F.d.L.; Boccacino, J.M.; Souza, M.C.d.S.; Melo-Escobar, M.I.; Alves, R.N.; Prado, M.B.; Iglesia, R.P.; Cangiano, G.; Mazzaro, G.L.R.; et al. Multifaceted WNT Signaling at the Crossroads between Epithelial-Mesenchymal Transition and Autophagy in Glioblastoma. Front. Oncol. 2020, 10, 597743. [Google Scholar] [CrossRef]
- Ang, H.L.; Mohan, C.D.; Shanmugam, M.K.; Leong, H.C.; Makvandi, P.; Rangappa, K.S.; Bishayee, A.; Kumar, A.P.; Sethi, G. Mechanism of epithelial-mesenchymal transition in cancer and its regulation by natural compounds. Med. Res. Rev. 2023, 43, 1141–1200. [Google Scholar] [CrossRef]
- Kim, S.M.; Lim, E.J.; Yoo, K.C.; Zhao, Y.; Kang, J.H.; Lim, E.J.; Shin, I.; Kang, S.G.; Lim, H.W.; Lee, S.J. Glioblastoma-educated mesenchymal stem-like cells promote glioblastoma infiltration via extracellular matrix remodelling in the tumour microenvironment. Clin. Transl. Med. 2022, 12, e997. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef]
- Ishii, A.; Kimura, T.; Sadahiro, H.; Kawano, H.; Takubo, K.; Suzuki, M.; Ikeda, E. Histological Characterization of the Tumorigenic “Peri-Necrotic Niche” Harboring Quiescent Stem-like Tumor Cells in Glioblastoma. PLoS ONE 2016, 11, e0147366. [Google Scholar] [CrossRef]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ Necrosis in Glioblastoma: A Familiar Morphologic Feature That Links Vascular Pathology, Hypoxia, and Angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Giambra, M.; Di Cristofori, A.; Valtorta, S.; Manfrellotti, R.; Bigiogera, V.; Basso, G.; Moresco, R.M.; Giussani, C.; Bentivegna, A. The peritumoral brain zone in glioblastoma: Where we are and where we are going. J. Neurosci. Res. 2023, 101, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; Van Meir, E.G. Pseudopalisades in Glioblastoma Are Hypoxic, Express Extracellular Matrix Proteases, and Are Formed by an Actively Migrating Cell Population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, Y.B.; Ababneh, O.E.; Al-Khalili, A.A.; Serhan, A.; Hatamleh, Z.; Ghammaz, O.; Alkhaldi, M.; Alomari, S. Identification of Hypoxia Prognostic Signature in Glioblastoma Multiforme Based on Bulk and Single-Cell RNA-Seq. Cancers 2024, 16, 633. https://doi.org/10.3390/cancers16030633
Ahmed YB, Ababneh OE, Al-Khalili AA, Serhan A, Hatamleh Z, Ghammaz O, Alkhaldi M, Alomari S. Identification of Hypoxia Prognostic Signature in Glioblastoma Multiforme Based on Bulk and Single-Cell RNA-Seq. Cancers. 2024; 16(3):633. https://doi.org/10.3390/cancers16030633
Chicago/Turabian StyleAhmed, Yaman B., Obada E. Ababneh, Anas A. Al-Khalili, Abdullah Serhan, Zaid Hatamleh, Owais Ghammaz, Mohammad Alkhaldi, and Safwan Alomari. 2024. "Identification of Hypoxia Prognostic Signature in Glioblastoma Multiforme Based on Bulk and Single-Cell RNA-Seq" Cancers 16, no. 3: 633. https://doi.org/10.3390/cancers16030633
APA StyleAhmed, Y. B., Ababneh, O. E., Al-Khalili, A. A., Serhan, A., Hatamleh, Z., Ghammaz, O., Alkhaldi, M., & Alomari, S. (2024). Identification of Hypoxia Prognostic Signature in Glioblastoma Multiforme Based on Bulk and Single-Cell RNA-Seq. Cancers, 16(3), 633. https://doi.org/10.3390/cancers16030633