
TRPA1 Contributes to FGFR2c Signaling and to Its Oncogenic Outcomes in Pancreatic Ductal Adenocarcinoma-Derived Cell Lines

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Treatments
2.2. Immunofluorescence
2.3. Western Blot Analysis
2.4. Invasion Assay
2.5. Primers
2.6. RNA Extraction and cDNA Synthesis
2.7. PCR Amplification and Real-Time Quantitation
2.8. Statistics
3. Results
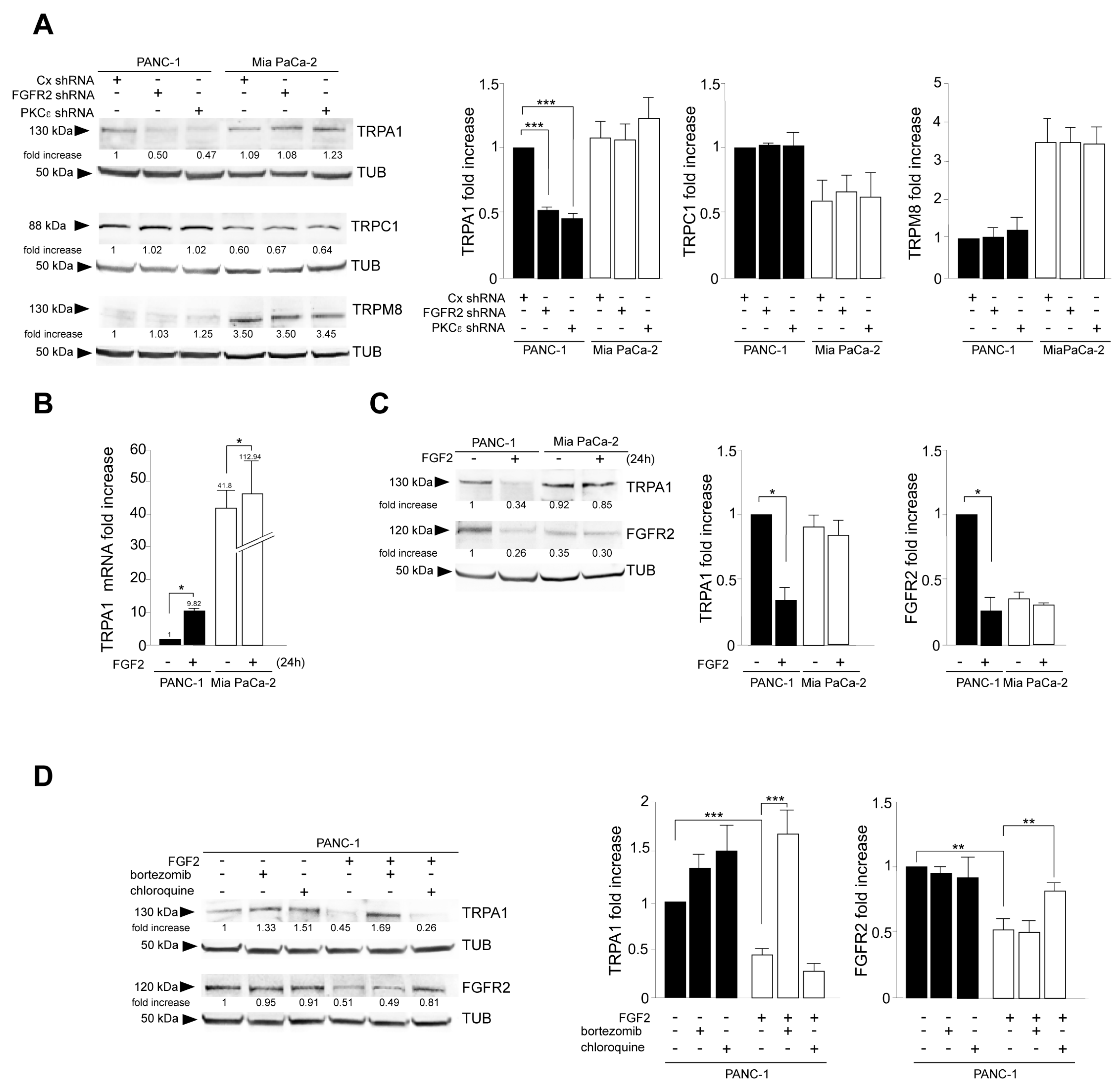
3.1. TRPA1 Is a Possible Candidate for FGFR2c Interplay
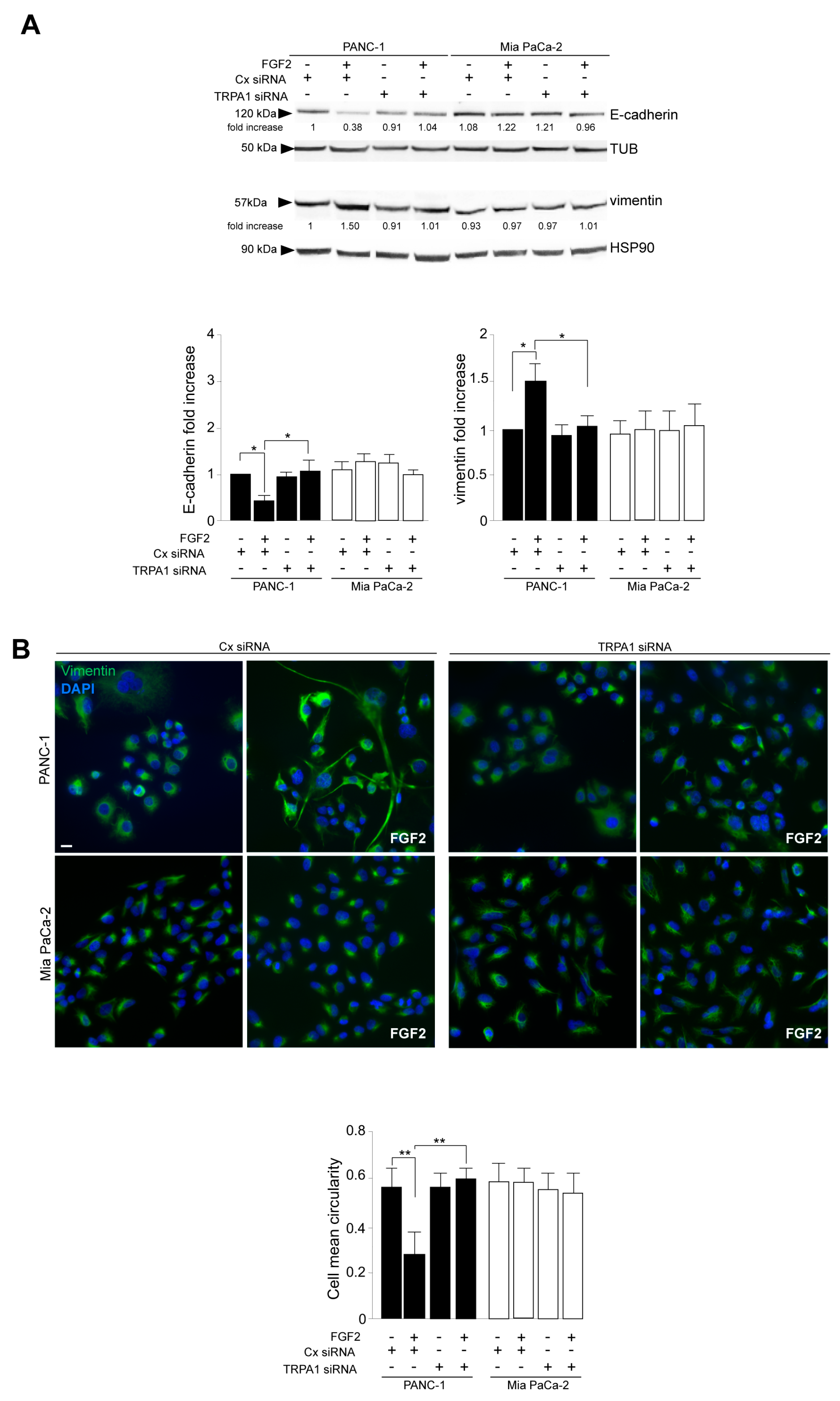
3.2. TRPA1 Contributes to FGFR2c-Established Aberrant Signaling and to the Consequent Enhancement of EMT and Invasive Traits
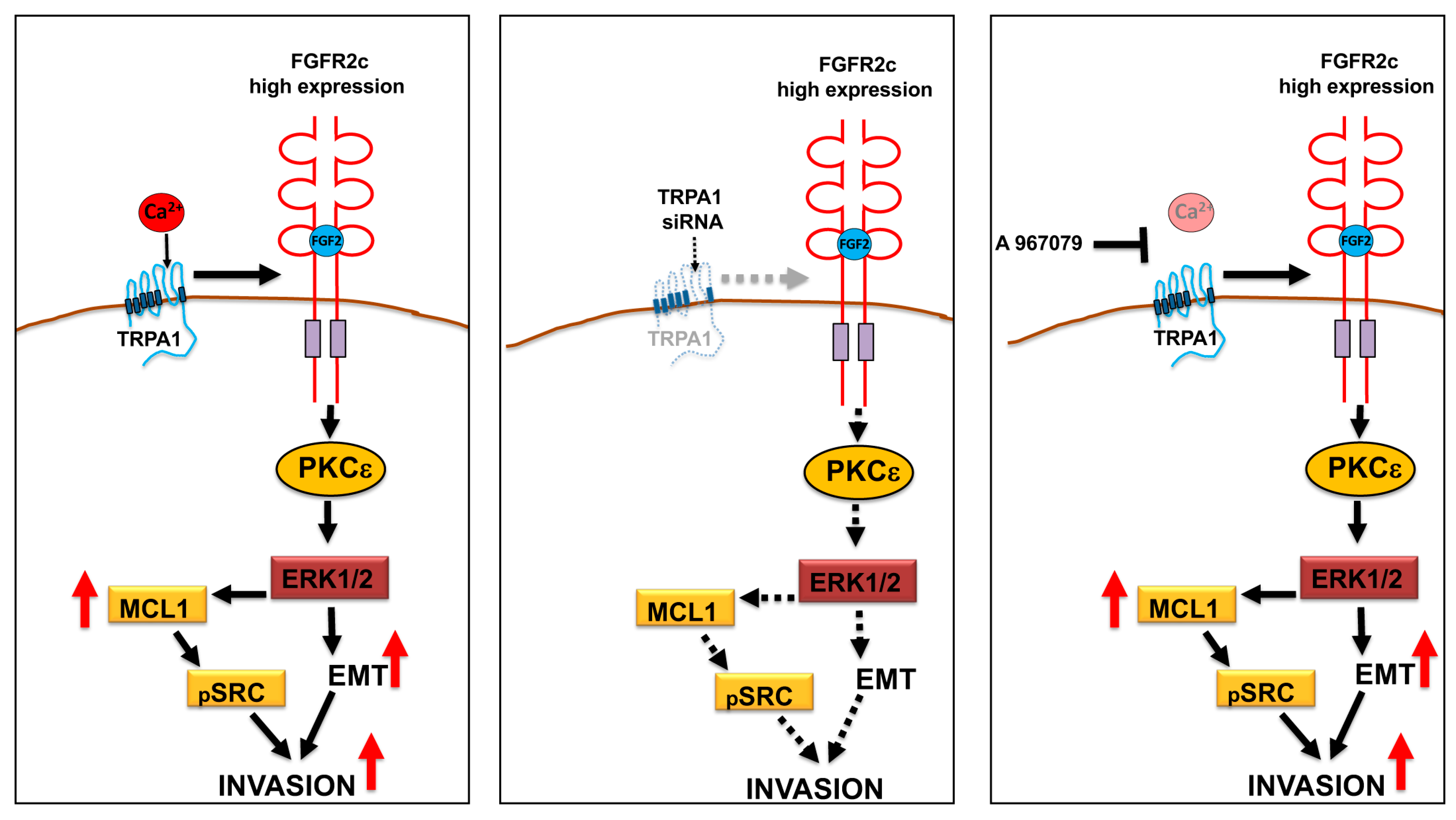
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Javadrashid, D.; Baghbanzadeh, D.; Derakhshani, A.; Leone, P.; Silvestris, N.; Racanelli, A.; Solimando, A.G.; Baradaran, B. Pancreatic Cancer Signaling Pathways, Genetic Alterations, and Tumor Microenvironment: The Barriers Affecting the Method of Treatment. Biomedicines 2021, 9, 373. [Google Scholar] [CrossRef]
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat. Cancer 2022, 3, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A.; Magliano, M.P.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef]
- Maneshi, P.; Mason, J.; Dongre, M.; Öhlund, D. Targeting Tumor-Stromal Interactions in Pancreatic Cancer: Impact of Collagens and Mechanical Traits. Front. Cell Dev. Biol. 2021, 9, 787485. [Google Scholar] [CrossRef] [PubMed]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma involvement in pancreatic ductal adenocarcinoma: An overview focusing on extracellular matrix proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hylander, B.L.; LeVea, C.; Repasky, E.A.; Straubinger, R.M.; Adjei, A.A.; Ma, W.W. Enhanced FGFR signalling predisposes pancreatic cancer to the effect of a potent FGFR inhibitor in preclinical models. Br. J. Cancer 2014, 110, 320–329. [Google Scholar] [CrossRef]
- Carter, E.P.; Coetzee, A.S.; Bort, E.T.; Wang, Q.; Kocher, H.M.; Grose, R.P. Dissecting FGF Signalling to Target Cellular Crosstalk in Pancreatic Cancer. Cells 2021, 10, 847. [Google Scholar] [CrossRef]
- D’Agosto, S.; Pezzini, F.; Veghini, L.; Delfino, P.; Fiorini, C.; Temgue Tane, G.D.; Del Curatolo, A.; Vicentini, C.; Ferrari, G.; Pasini, D.; et al. Loss of FGFR4 promotes the malignant phenotype of PDAC. Oncogene 2022, 41, 4371–4384. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Oon, C.; Kothari, A.; Horton, W.; Link, J.; Sears, R.C.; Sherman, M.H. Acidic fibroblast growth factor underlies microenvironmental regulation of MYC in pancreatic cancer. J. Exp. Med. 2020, 217, e20191805. [Google Scholar] [CrossRef]
- Ranieri, D.; Guttieri, L.; Raffa, S.; Torrisi, M.R.; Belleudi, F. Role of FGFR2c and Its PKCε Downstream Signaling in the Control of EMT and Autophagy in Pancreatic Ductal Adenocarcinoma Cells. Cancers 2021, 13, 4993. [Google Scholar] [CrossRef]
- Ranieri, D.; French, D.; Persechino, F.; Guttieri, L.; Torrisi, M.R.; Belleudi, F. The FGFR2c/PKCε Axis Controls MCL-1-Mediated Invasion in Pancreatic Ductal Adenocarcinoma Cells: Perspectives for Innovative Target Therapies. Biomedicines 2022, 10, 1652. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, C.; Vriens, J.; De Clercq, K. Transient receptor potential channel regulation by growth factors. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118950. [Google Scholar] [CrossRef] [PubMed]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta 2015, 1848 Pt B, 2685–2702. [Google Scholar] [CrossRef] [PubMed]
- Vrenken, K.S.; Jalink, K.; Van Leeuwen, F.N.; Middelbeek, J. Beyond ion-conduction: Channel-dependent and-independent roles of TRP channels during development and tissue homeostasis. Biochim. Biophys. Acta 2016, 1863, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Berrout, J.; Kyriakopoulou, E.; Moparthi, L.; Hogea, A.S.; Berrout, L.; Ivan, C.; Lorger, M.; Boyle, J.; Peers, C.; Muench, S.; et al. TRPA1–FGFR2 binding event is a regulatory oncogenic driver modulated by miRNA-142-3p. Nat. Commun. 2017, 8, 947. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, H.; Du, Q.; Gu, J.; Wu, J.; Liu, Q.; Li, Z.; Zhang, T.; Xu, J.; Xie, R. Research Progress on TRPA1 in Diseases. J. Membr. Biol. 2023, 256, 301–316. [Google Scholar] [CrossRef]
- Cojocaru, F.; Şelescu, T.; Domocoş, D.; Măruţescu, L.; Chiritoiu, G.; Chelaru, N.R.; Dima, S.; Mihăilescu, D.; Babes, A.; Cucu, D. Functional expression of the transient receptor potential ankyrin type 1 channel in pancreatic adenocarcinoma cells. Sci. Rep. 2021, 11, 2018. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Nurmagambetova, A.; Mustyatsa, V.; Saidova, A.; Vorobjev, I. Morphological and cytoskeleton changes in cells after EMT. Sci. Rep. 2023, 13, 22164. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Guttieri, L.; Raffa, S.; Salerno, G.; Bigi, R.; Persechino, F.; Visco, V.; Torrisi, M.R.; Ranieri, D.; Belleudi, F. FGFR2c Upregulation Contributes to Cancer-Associated Fibroblast Program Activation and to Enhanced Autophagy in Actinic Keratosis-Derived Dermal Fibroblasts: A Possible Role in Precancerous Cell/Stromal Cell Crosstalk. Biology 2023, 12, 463. [Google Scholar] [CrossRef]
- Ishiwata, T.; Matsuda, Y.; Yamamoto, T.; Uchida, E.; Korc, M.; Naito, Z. Enhanced expression of fibroblast growth factor receptor 2 IIIc promotes human pancreatic cancer cell proliferation. Am. J. Pathol. 2012, 180, 1928–1941. [Google Scholar] [CrossRef]
- Ueda, J.; Matsuda, Y.; Yamahatsu, K.; Uchida, E.; Naito, Z.; Korc, M.; Ishiwata, T. Epithelial splicing regulatory protein 1 is a favorable prognostic factor in pancreatic cancer that attenuates pancreatic metastases. Oncogene 2014, 33, 4485–4495. [Google Scholar] [CrossRef]
- Schnipper, J.; Kouba, S.; Hague, F.; Girault, A.; Rybarczyk, P.; Telliez, M.S.; Guénin, S.; Tebbakha, R.; Sevestre, H.; Ahidouch, A.; et al. The TRPC1 Channel Forms a PI3K/CaM Complex and Regulates Pancreatic Ductal Adenocarcinoma Cell Proliferation in a Ca2+-Independent Manner. Int. J. Mol. Sci. 2022, 23, 7923. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, G.; Prevarskaya, N.; Schwab, A.; Lehen’kyi, V.Y. Role of the TRP channels in pancreatic ductal adenocarcinoma development and progression. Cells 2021, 10, 1021. [Google Scholar] [CrossRef] [PubMed]
- Hofschröer, V.; Najder, K.; Rugi, M.; Bouazzi, R.; Cozzolino, M.; Arcangeli, A.; Panyi, G.; Schwab, A. Ion channels orchestrate pancreatic ductal adenocarcinoma progression and therapy. Front. Pharmacol. 2021, 11, 586599. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Oh, M.S.; Lee, D.W.; Kuh, H.J. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J. Exp. Clin. Cancer Res. 2019, 38, 258. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.D.; Corsa, C.A.S.; Biswas, H.; Aft, R.L.; Longmore, G.D. Temporal and spatial cooperation of Snail1 and Twist1 during epithelial–mesenchymal transition predicts for human breast cancer recurrence. Mol. Cancer Res. 2011, 9, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- Young, A.I.; Law, A.M.; Castillo, L.; Chong, S.; Cullen, H.D.; Koehler, M.; Herzog, S.; Brummer, T.; Lee, E.F.; Fairlie, W.D.; et al. MCL-1 inhibition provides a new way to suppress breast cancer metastasis and increase sensitivity to dasatinib. Breast Cancer Res. 2016, 18, 258. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Will, M.; Qin, A.C.; Toy, W.; Yao, Z.; Rodrik-Outmezguine, V.; Schneider, C.; Huang, X.; Monian, P.; Jiang, X.; De Stanchina, E.; et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov. 2014, 4, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, A.; Jalava, N.; Bratty, R.; Pertovaara, A. TRPA1 antagonists for pain relief. Pharmaceuticals 2018, 11, 117. [Google Scholar] [CrossRef]
- Hennig, A.; Baenke, F.; Klimova, A.; Drukewitz, S.; Jahnke, B.; Brückmann, S.; Secci, R.; Winter, C.; Schmäche, T.; Seidlitz, T.; et al. Detecting drug resistance in pancreatic cancer organoids guides optimized chemotherapy treatment. J. Pathol. 2022, 257, 607–619. [Google Scholar] [CrossRef]
- Espiau-Romera, P.; Courtois, S.; Parejo-Alonso, B.; Sancho, P. Molecular and metabolic subtypes correspondence for pancreatic ductal adenocarcinoma classification. J. Clin. Med. 2020, 9, 4128. [Google Scholar] [CrossRef]
- Espinet, E.; Klein, L.; Puré, E.; Singh, S.K. Mechanisms of PDAC subtype heterogeneity and therapy response. Trends Cancer. 2022, 8, 1060–1071. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the impact of immunotherapy to pancreatic cancer: Challenges and opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Liudahl, S.M.; Betts, C.B.; Sivagnanam, S.; Morales-Oyarvide, V.; Da Silva, A.; Yuan, C.; Hwang, S.; Grossblatt-Wait, A.; Leis, K.R.; Larson, W.; et al. Leukocyte heterogeneity in pancreatic ductal adenocarcinoma: Phenotypic and spatial features associated with clinical outcome. Cancer Discov. 2021, 11, 2014–2031. [Google Scholar] [CrossRef]
- Väyrynen, S.A.; Zhang, J.; Yuan, C.; Väyrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer cells co-opt the neuronal redox-sensing channel TRPA1 to promote oxidative-stress tolerance. Cancer Cell 2018, 33, 985–1003. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancini, V.; Raffa, S.; Fiorio Pla, A.; French, D.; Torrisi, M.R.; Ranieri, D.; Belleudi, F. TRPA1 Contributes to FGFR2c Signaling and to Its Oncogenic Outcomes in Pancreatic Ductal Adenocarcinoma-Derived Cell Lines. Cancers 2024, 16, 609. https://doi.org/10.3390/cancers16030609
Mancini V, Raffa S, Fiorio Pla A, French D, Torrisi MR, Ranieri D, Belleudi F. TRPA1 Contributes to FGFR2c Signaling and to Its Oncogenic Outcomes in Pancreatic Ductal Adenocarcinoma-Derived Cell Lines. Cancers. 2024; 16(3):609. https://doi.org/10.3390/cancers16030609
Chicago/Turabian StyleMancini, Vanessa, Salvatore Raffa, Alessandra Fiorio Pla, Deborah French, Maria Rosaria Torrisi, Danilo Ranieri, and Francesca Belleudi. 2024. "TRPA1 Contributes to FGFR2c Signaling and to Its Oncogenic Outcomes in Pancreatic Ductal Adenocarcinoma-Derived Cell Lines" Cancers 16, no. 3: 609. https://doi.org/10.3390/cancers16030609
APA StyleMancini, V., Raffa, S., Fiorio Pla, A., French, D., Torrisi, M. R., Ranieri, D., & Belleudi, F. (2024). TRPA1 Contributes to FGFR2c Signaling and to Its Oncogenic Outcomes in Pancreatic Ductal Adenocarcinoma-Derived Cell Lines. Cancers, 16(3), 609. https://doi.org/10.3390/cancers16030609