
The Role of Chemokines in Orchestrating the Immune Response to Pancreatic Ductal Adenocarcinoma



Abstract
Simple Summary
Abstract
1. Introduction
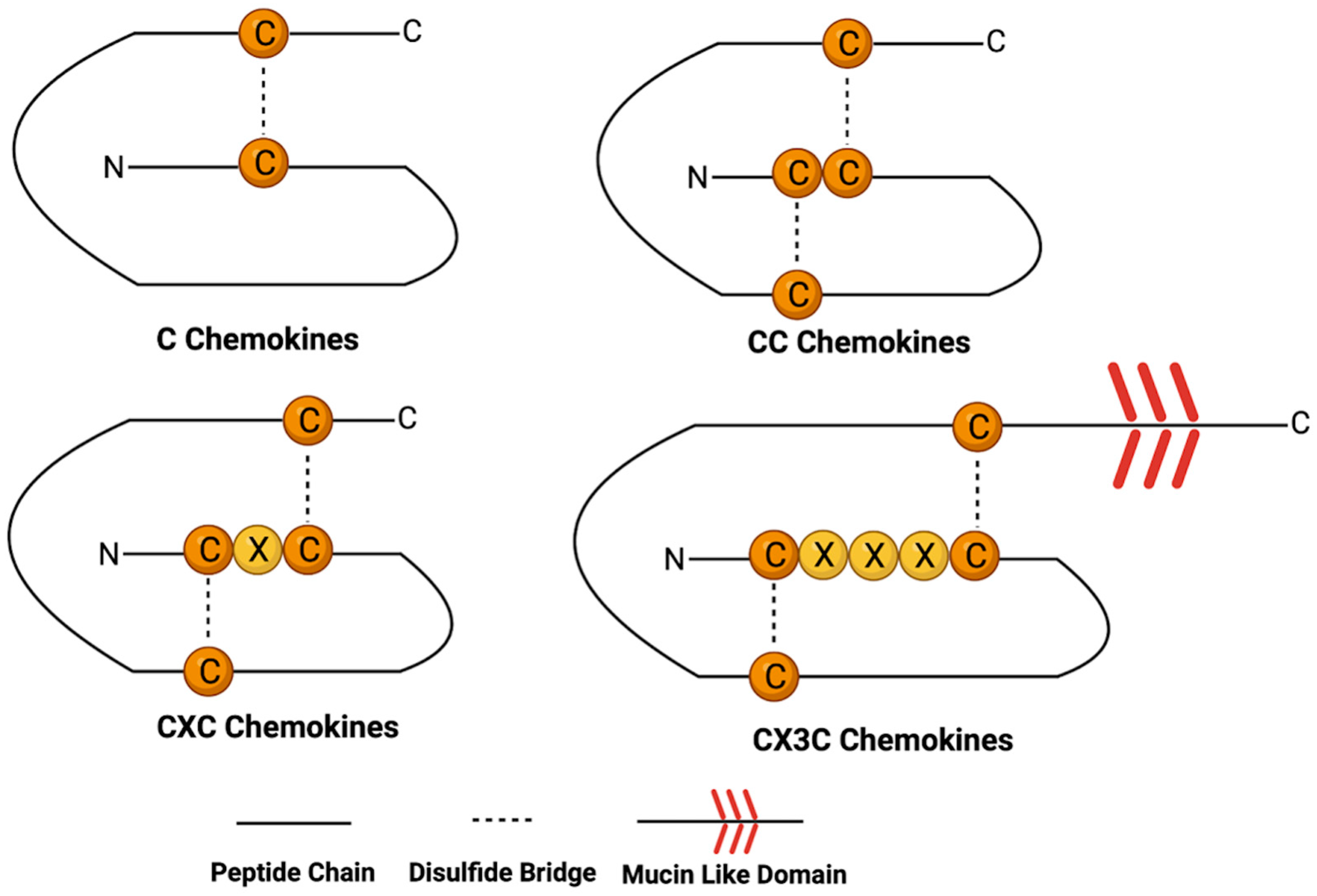
2. Chemokine Structure and Function
2.1. C Chemokines
2.2. CC Chemokines
2.3. CXC Chemokines
2.4. CX3C Chemokines
2.5. Chemokine Function
3. Chemokines Present in the PDAC Tumor Microenvironment
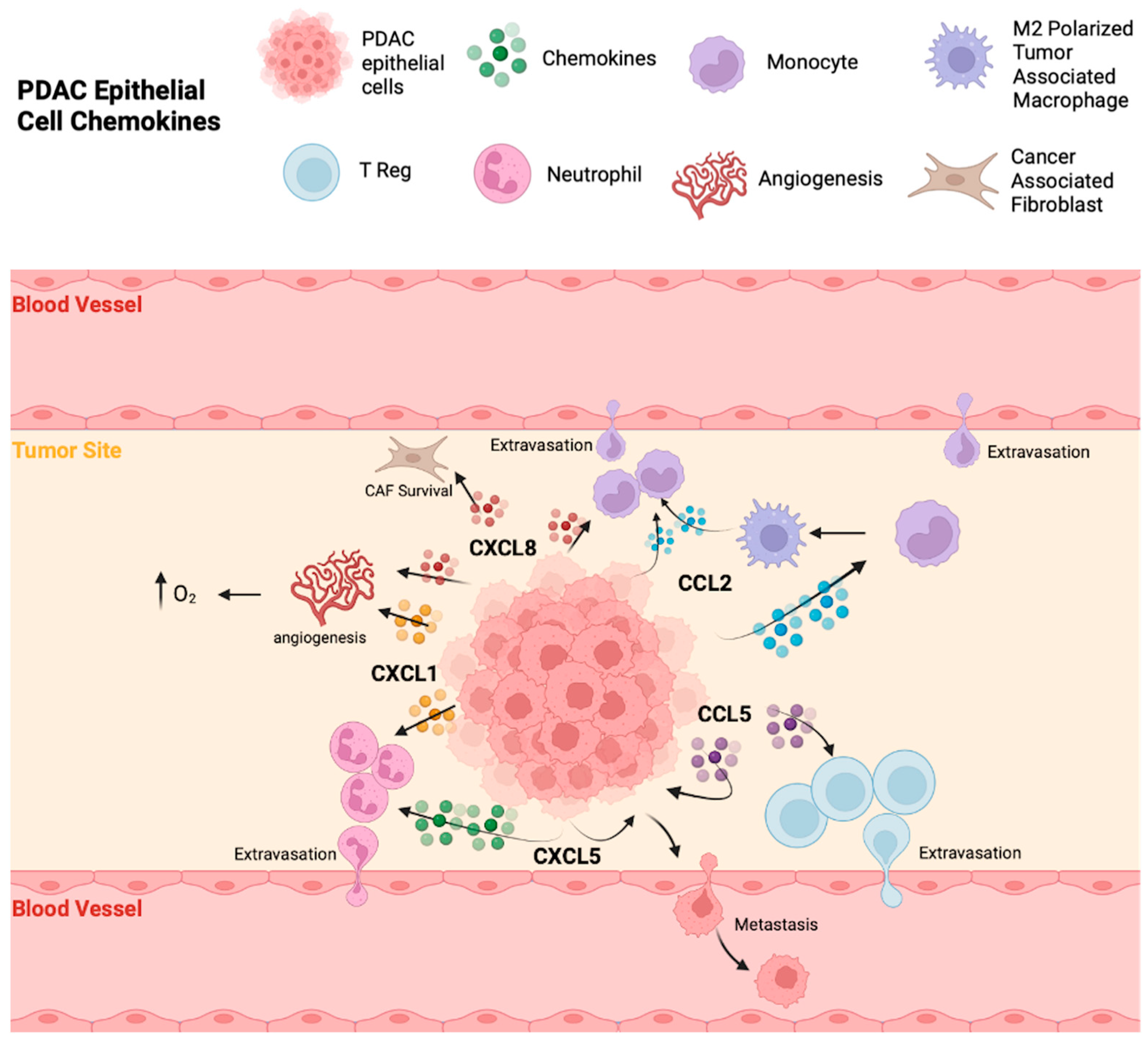
3.1. Chemokines Produced by Malignant Epithelial Cells
3.1.1. CCL2
3.1.2. CCL5
3.1.3. CXCL1
3.1.4. CXCL5
3.1.5. CXCL8
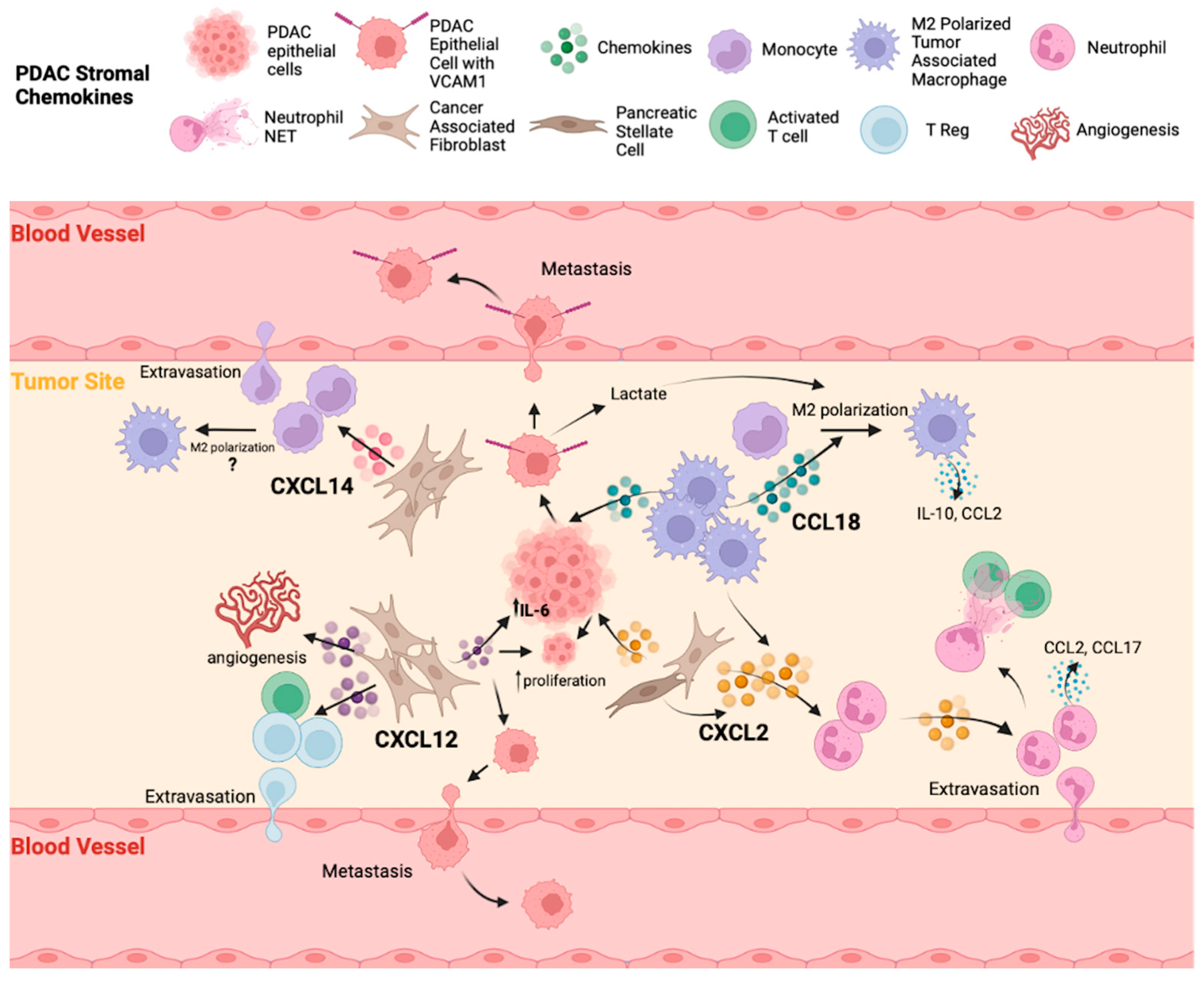
3.2. Chemokines Present in the PDAC Stroma
3.2.1. CCL18
3.2.2. CXCL2
3.2.3. CXCL12
3.2.4. CXCL14
4. Chemokines Influencing Immune Cell Accumulation in PDAC
4.1. Promoting Anti-Tumor Immune Responses
4.1.1. CD8+ T Cells
4.1.2. NK Cells
4.2. Promoting Pro-Tumor Immune Responses
4.2.1. Tumor-Associated Macrophages
4.2.2. Tumor-Associated Neutrophils
4.2.3. T Regulatory Cells
4.3. Pleotropic Chemokines
5. Chemokines and Patient Outcomes in PDAC
5.1. CC Chemokines
5.2. CXC Chemokines
5.3. CX3C Chemokines
6. Chemokines and Immunotherapy Approaches
6.1. Checkpoint Inhibitors
6.2. Cellular Therapies
6.3. Vaccines
6.4. Oncolytic Viruses
7. Targeting Chemokine Receptors and Ligands
7.1. CCR2/CCL2
7.2. CCR4
7.3. CCR5

{kind=link}
{kind=link}
{kind=link}
| Pathway Targeted | Clinical Trial Number | Study/Patient Info. | Chemokine-Targeted Therapy | Additional Treatment | Outcome | Reference |
|---|---|---|---|---|---|---|
| CCR2/CCL2 | NCT01413022 | Phase Ib, borderline resectable pancreatic cancer, n = 33 | CCR2 antagonist (PF-04136309) | FOLFIRINOX | Well-tolerated, 49% objective tumor response | [198] |
| NCT02732938 | Phase Ib/II, metastatic PDAC, n = 21 | CCR2 antagonist (PF-04136309) |
Gemcitabine Nab-paclitaxel | No additional benefit, high incidence of pulmonary toxicity (24%) | [199] | |
| NCT02345408 | Phase Ib, locally advanced or metastatic, non-resectable PDAC, n = 50 | CCR2 antagonist (CCX872-B) | FOLFIRINOX | Well-tolerated, overall survival of 29% | [200] | |
| NCT03778879 | Phase I/II, PDAC | CCR2 antagonist (CCX872-B) | Stereotactic body radiation therapy (25 Gy in 5 fractions) | Withdrawn due to lack of CCX872-B availability | CT.gov | |
| NCT03851237 | Phase I, PDAC | CCR2 imaging/therapeutic agent (64Cu-DOTA-ECLIi) | Correlating CCR2 expression with response to standard of care chemotherapy/CCR2-targeted therapy | Patient recruitment underway | CT.gov | |
| - | Phase I, advanced solid tumors, n = 44 (1 PDAC) | Anti-CCL2 antibody (Carlumab, CNTO 888) | - | Well-tolerated, 0% objective anti-tumor response | [201] | |
| CCR4 | NCT02476123 | Phase I, advanced solid tumors, n = 96 (15 pancreatic adenocarcinoma) | Anti-CCR4 antibody (Mogamulizumab) | Nivolumab (anti-PD1) | Well-tolerated, 1 confirmed objective response in PDAC | [202] |
| NCT02301130 | Phase I, advanced solid tumors, n = 64 (27 pancreatic cancer) | Anti-CCR4 antibody (Mogamulizumab) | Durvalumab (anti-PD1) or Tremelimumab (anti-CTLA4) | Well-tolerated, 0% ORR in pancreatic cancer | [203] | |
| NCT02705105 | Phase I/II, locally advanced or metastatic solid tumors, n = 114 (18 pancreatic cancer) | Anti-CCR4 antibody (Mogamulizumab) | Nivolumab | Acceptable tolerability, no enhanced anti-tumor effects, 0% ORR in pancreatic cancer | [204] | |
| CCR5 | NCT03496662 | Phase I/II, PDAC, n = 40 | CCR2/CCR5 dual antagonist (BMS-813160) | Nivolumab Gemcitabine Nab-paclitaxel | Results not yet reported | CT.gov |
| NCT03184870 | Phase I/II, colorectal cancer and PDAC, n = 332 | CCR2/CCR5 dual antagonist (BMS-813160) | Nivolumab or FOLFIRI or Gemcitabine + Nab-paclitaxel | Results not yet reported | CT.gov | |
| NCT03767582 | Phase I/II, PDAC | CCR2/CCR5 dual antagonist (BMS-813160) | Nivolumab with or without GVAX | Safe dosage identified, proceeding to Phase II portion | [206] | |
| NCT05940844 | Phase I, treatment-refractory metastatic solid tumors | CCR5 antagonist (OB-002) | - | Trial has not yet started | CT.gov | |
| NCT04721301 | Phase I, advanced metastatic colorectal and pancreatic cancer | CCR5 antagonist (Maraviroc) | Nivolumab Ipilimumab (anti-CTLA4) | Results not yet reported | CT.gov | |
| CXCR1/2 | NCT04477343 | Phase I, metastatic PDAC | CXCR1/2 antagonist (SX-682) | Nivolumab | Patient recruitment underway | [207] |
| NCT05604560 | Phase II, resectable pancreatic cancer | CXCR1/2 antagonist (SX-682) | Tislelizumab (anti-PD1) | Patient recruitment underway | CT.gov | |
| NCT00851955 | PDAC, n = 150 | Observational study to correlate CXCR2 receptor/ligand expression and patient outcomes | - | Results not yet reported | CT.gov | |
| CXCR4/CXCL12 | NCT02179970 | Phase I, advanced pancreatic, ovarian, and colorectal cancers, n = 26 (10 PDAC) | CXCR4 antagonist (AMD3100) | - | Well-tolerated, increased T-cell and NK-cell activation and infiltration | [208] |
| NCT04177810 | Phase II, metastatic PDAC, n = 25 | CXCR4 antagonist (AMD3100) | Cemiplimab (anti-PD1) | Results not yet reported | CT.gov | |
| NCT02826486 | Phase IIa, metastatic PDAC, n = 37 (chemotherapy resistant cohort) | CXCR4 antagonist (BL-8040) | Pembrolizumab (anti-PD1) | Well-tolerated, 34.5% disease control rate with 1 partial response, increased CD8+ T-cell infiltration | [209] | |
| NCT02907099 | Phase II, metastatic pancreatic cancer n = 20 (15 evaluable for response) | CXCR4 antagonist (BL-8040) | Pembrolizumab | 1 partial response, increased cytotoxic CD8+ T-cell infiltration post-therapy compared to baseline | [210] | |
| NCT04543071 | Phase II, metastatic treatment-naive pancreatic adenocarcinoma | CXCR4 antagonist (BL-8040) | Cemiplimab Gemcitabine Nab-paclitaxel | Patient recruitment underway | CT.gov | |
| NCT03168139 | Phase I/II, microsatellite stable metastatic colon and pancreatic cancer, n = 20 (9 PDAC) | CXCL12 inhibitor (NOX-A12) | Pembrolizumab | Well-tolerated, 22% disease stabilization in PDAC patients | [211] | |
| NCT04901741 | Phase II, metastatic pancreatic cancer | CXCL12 inhibitor (NOX-A12) | Pembrolizumab and Nanoliposomal Irinotecan or Gemcitabine/Nab-paclitaxel | Trial has not yet started | CT.gov |
7.4. CXCR1/2
7.5. CXCR4/CXCL12
8. Future Directions
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Edwards, J.; Wilmott, J.S.; Madore, J.; Gide, T.N.; Quek, C.; Tasker, A.; Ferguson, A.; Chen, J.; Hewavisenti, R.; Hersey, P.; et al. CD103(+) Tumor-Resident CD8(+) T Cells Are Associated with Improved Survival in Immunotherapy-Naive Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Clin. Cancer Res. 2018, 24, 3036–3045. [Google Scholar] [CrossRef]
- Lee, J.S.; Ruppin, E. Multiomics Prediction of Response Rates to Therapies to Inhibit Programmed Cell Death 1 and Programmed Cell Death 1 Ligand 1. JAMA Oncol. 2019, 5, 1614–1618. [Google Scholar] [CrossRef]
- Ullman, N.A.; Burchard, P.R.; Dunne, R.F.; Linehan, D.C. Immunologic Strategies in Pancreatic Cancer: Making Cold Tumors Hot. J. Clin. Oncol. 2022, 40, 2789–2805. [Google Scholar] [CrossRef]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Malchiodi, Z.X.; Weiner, L.M. Understanding and Targeting Natural Killer Cell-Cancer-Associated Fibroblast Interactions in Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 405. [Google Scholar] [CrossRef] [PubMed]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, A.A.; Wang, S.; Agarwal, V.; Marcisak, E.F.; Zuo, A.; Jablonski, S.A.; Loth, M.; Fertig, E.J.; MacDougall, J.; Zhukovsky, E.; et al. DPP inhibition alters the CXCR3 axis and enhances NK and CD8+ T cell infiltration to improve anti-PD1 efficacy in murine models of pancreatic ductal adenocarcinoma. J. Immunother. Cancer 2021, 9, e002837. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.J.; Lolis, E. Structure, Function, and Inhibition of Chemokines. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 469–499. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Matsushima, K.; Tanaka, S.; Robinson, E.A.; Appella, E.; Oppenheim, J.J.; Leonard, E.J. Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc. Natl. Acad. Sci. USA 1987, 84, 9233–9237. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.K.; Basu, S.; Aithal, A.; Dwivedi, N.V.; Gulati, M.; Jain, M. Regulation of pancreatic cancer therapy resistance by chemokines. Semin. Cancer Biol. 2022, 86, 69–80. [Google Scholar] [CrossRef]
- Zhang, S.; Ding, J.; Zhang, Y.; Liu, S.; Yang, J.; Yin, T. Regulation and Function of Chemokines at the Maternal-Fetal Interface. Front. Cell Dev. Biol. 2022, 10, 826053. [Google Scholar] [CrossRef]
- Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The Role of Chemokines in Wound Healing. Int. J. Mol. Sci. 2018, 19, 3217. [Google Scholar] [CrossRef]
- Sarvaiya, P.J.; Guo, D.; Ulasov, I.; Gabikian, P.; Lesniak, M.S. Chemokines in tumor progression and metastasis. Oncotarget 2013, 4, 2171. [Google Scholar] [CrossRef]
- Stein, J.V.; Nombela-Arrieta, C. Chemokine control of lymphocyte trafficking: A general overview. Immunology 2005, 116, 1–12. [Google Scholar] [CrossRef]
- Gouwy, M.; Struyf, S.; Proost, P.; Van Damme, J. Synergy in cytokine and chemokine networks amplifies the inflammatory response. Cytokine Growth Factor. Rev. 2005, 16, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, S.; Vafaei, S.; Zhong, X. Tumor-Infiltrating Immune Cell Signature Predicts the Prognosis and Chemosensitivity of Patients With Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2020, 10, 557638. [Google Scholar] [CrossRef]
- Kufareva, I.; Salanga, C.L.; Handel, T.M. Chemokine and chemokine receptor structure and interactions: Implications for therapeutic strategies. Immunol. Cell Biol. 2015, 93, 372–383. [Google Scholar] [CrossRef]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D. International Union of Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Müller, S.; Dorner, B.; Korthäuer, U.; Mages, H.W.; D’Apuzzo, M.; Senger, G.; Kroczek, R.A. Cloning of ATAC, an activation-induced, chemokine-related molecule exclusively expressed in CD8+ T lymphocytes. Eur. J. Immunol. 1995, 25, 1744–1748. [Google Scholar] [CrossRef] [PubMed]
- Giancarlo, B.; Silvano, S.; Albert, Z.; Alberto, M.; Paola, A. Migratory response of human natural killer cells to lymphotactin. Eur. J. Immunol. 1996, 26, 3238–3241. [Google Scholar] [CrossRef]
- Crozat, K.; Tamoutounour, S.; Vu Manh, T.-P.; Fossum, E.; Luche, H.; Ardouin, L.; Guilliams, M.; Azukizawa, H.; Bogen, B.; Malissen, B. Cutting edge: Expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8α+ type. J. Immunol. 2011, 187, 4411–4415. [Google Scholar] [CrossRef] [PubMed]
- Burrack, A.L.; Schmiechen, Z.C.; Patterson, M.T.; Miller, E.A.; Spartz, E.J.; Rollins, M.R.; Raynor, J.F.; Mitchell, J.S.; Kaisho, T.; Fife, B.T.; et al. Distinct myeloid antigen-presenting cells dictate differential fates of tumor-specific CD8+ T cells in pancreatic cancer. JCI Insight 2022, 7, e151593. [Google Scholar] [CrossRef] [PubMed]
- Beauvillain, C.; Cunin, P.; Doni, A.; Scotet, M.; Jaillon, S.; Loiry, M.L.; Magistrelli, G.; Masternak, K.; Chevailler, A.; Delneste, Y.; et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood 2011, 117, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Lanca, T.; Costa, M.F.; Goncalves-Sousa, N.; Rei, M.; Grosso, A.R.; Penido, C.; Silva-Santos, B. Protective role of the inflammatory CCR2/CCL2 chemokine pathway through recruitment of type 1 cytotoxic gammadelta T lymphocytes to tumor beds. J. Immunol. 2013, 190, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Mou, T.; Xie, F.; Zhong, P.; Hua, H.; Lai, L.; Yang, Q.; Wang, J. MiR-345-5p functions as a tumor suppressor in pancreatic cancer by directly targeting CCL8. Biomed. Pharmacother. 2019, 111, 891–900. [Google Scholar] [CrossRef]
- Dagouassat, M.; Suffee, N.; Hlawaty, H.; Haddad, O.; Charni, F.; Laguillier, C.; Vassy, R.; Martin, L.; Schischmanoff, P.O.; Gattegno, L.; et al. Monocyte chemoattractant protein-1 (MCP-1)/CCL2 secreted by hepatic myofibroblasts promotes migration and invasion of human hepatoma cells. Int. J. Cancer 2010, 126, 1095–1108. [Google Scholar] [CrossRef]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Huffman, A.P.; Lin, J.H.; Kim, S.I.; Byrne, K.T.; Vonderheide, R.H. CCL5 mediates CD40-driven CD4+ T cell tumor infiltration and immunity. JCI Insight 2020, 5, e137263. [Google Scholar] [CrossRef] [PubMed]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. CXC chemokines in cancer angiogenesis and metastases. Adv. Cancer Res. 2010, 106, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Clark-Lewis, I.; Dewald, B.; Geiser, T.; Moser, B.; Baggiolini, M. Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc. Natl. Acad. Sci. USA 1993, 90, 3574–3577. [Google Scholar] [CrossRef] [PubMed]
- Puneet, P.; Moochhala, S.; Bhatia, M. Chemokines in acute respiratory distress syndrome. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2005, 288, L3–L15. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Polverini, P.J.; Kunkel, S.L.; Arenberg, D.A.; Burdick, M.D.; Kasper, J.; Dzuiba, J.; Van Damme, J.; Walz, A.; Marriott, D. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J. Biol. Chem. 1995, 270, 27348–27357. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, L.; Ding, G.; Huang, H.; Cao, G.; Sun, X.; Lou, N.; Wei, Q.; Shen, T.; Xu, X.; et al. Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J. Immunother. Cancer 2020, 8, e000308. [Google Scholar] [CrossRef]
- Ludwig, A.; Weber, C. Transmembrane chemokines: Versatile ‘special agents’ in vascular inflammation. Thromb. Haemost. 2007, 97, 694–703. [Google Scholar] [CrossRef]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Hertwig, L.; Hamann, I.; Romero-Suarez, S.; Millward, J.M.; Pietrek, R.; Chanvillard, C.; Stuis, H.; Pollok, K.; Ransohoff, R.M.; Cardona, A.E.; et al. CX3CR1-dependent recruitment of mature NK cells into the central nervous system contributes to control autoimmune neuroinflammation. Eur. J. Immunol. 2016, 46, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Rooper, L.; Xie, J.; Kajdacsy-Balla, A.A.; Barbolina, M.V. Fractalkine Receptor CX3CR1 Is Expressed in Epithelial Ovarian Carcinoma Cells and Required for Motility and Adhesion to Peritoneal Mesothelial CellsCX3CR1 in Ovarian Carcinoma. Mol. Cancer Res. 2012, 10, 11–24. [Google Scholar] [CrossRef]
- Yao, X.; Qi, L.; Chen, X.; Du, J.; Zhang, Z.; Liu, S. Expression of CX3CR1 associates with cellular migration, metastasis, and prognosis in human clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 162–170. [Google Scholar] [CrossRef]
- Marchesi, F.; Piemonti, L.; Fedele, G.; Destro, A.; Roncalli, M.; Albarello, L.; Doglioni, C.; Anselmo, A.; Doni, A.; Bianchi, P.; et al. The chemokine receptor CX3CR1 is involved in the neural tropism and malignant behavior of pancreatic ductal adenocarcinoma. Cancer Res. 2008, 68, 9060–9069. [Google Scholar] [CrossRef] [PubMed]
- Celesti, G.; Di Caro, G.; Bianchi, P.; Grizzi, F.; Marchesi, F.; Basso, G.; Rahal, D.; Delconte, G.; Catalano, M.; Cappello, P.; et al. Early expression of the fractalkine receptor CX3CR1 in pancreatic carcinogenesis. Br. J. Cancer 2013, 109, 2424–2433. [Google Scholar] [CrossRef]
- Legler, D.F.; Thelen, M. New insights in chemokine signaling. F1000Research 2018, 7, 95. [Google Scholar] [CrossRef]
- Laudanna, C.; Kim, J.Y.; Constantin, G.; Butcher, E.C. Rapid leukocyte integrin activation by chemokines. Immunol. Rev. 2002, 186, 37–46. [Google Scholar] [CrossRef]
- Weber, C.; Kitayama, J.; Springer, T.A. Differential regulation of beta 1 and beta 2 integrin avidity by chemoattractants in eosinophils. Proc. Natl. Acad. Sci. USA 1996, 93, 10939–10944. [Google Scholar] [CrossRef]
- Carveth, H.; Bohnsack, J.; McIntyre, T.; Baggiolini, M.; Prescott, S.; Zimmerman, G. Neutrophil activating factor (NAF) induces polymorphonuclear leukocyte adherence to endothelial cells and to subendothelial matrix proteins. Biochem. Biophys. Res. Commun. 1989, 162, 387–393. [Google Scholar] [CrossRef]
- Gerszten, R.E.; Garcia-Zepeda, E.A.; Lim, Y.-C.; Yoshida, M.; Ding, H.A.; Gimbrone, M.A., Jr.; Luster, A.D.; Luscinskas, F.W.; Rosenzweig, A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999, 398, 718–723. [Google Scholar] [CrossRef]
- Lloyd, A.R.; Oppenheim, J.J.; Kelvin, D.J.; Taub, D.D. Chemokines regulate T cell adherence to recombinant adhesion molecules and extracellular matrix proteins. J. Immunol. 1996, 156, 932–938. [Google Scholar] [CrossRef]
- Monti, P.; Leone, B.E.; Marchesi, F.; Balzano, G.; Zerbi, A.; Scaltrini, F.; Pasquali, C.; Calori, G.; Pessi, F.; Sperti, C. The CC chemokine MCP-1/CCL2 in pancreatic cancer progression: Regulation of expression and potential mechanisms of antimalignant activity. Cancer Res. 2003, 63, 7451–7461. [Google Scholar] [PubMed]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2020, 8, 607209. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3–CCL2 SignalingFAP via STAT3–CCL2 promote tumor immunosuppression. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deguchi, Y.; Wei, D.; Liu, F.; Moussalli, M.J.; Deguchi, E.; Li, D.; Wang, H.; Valentin, L.A.; Colby, J.K.; et al. Rapid acceleration of KRAS-mutant pancreatic carcinogenesis via remodeling of tumor immune microenvironment by PPARdelta. Nat. Commun. 2022, 13, 2665. [Google Scholar] [CrossRef] [PubMed]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W. CCL2 Mediates Cross-talk between Cancer Cells and Stromal Fibroblasts That Regulates Breast Cancer Stem CellsFibroblast-Derived CCL2 Induces Cancer Stem Cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef]
- Aldinucci, D.; Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm. 2014, 2014, 292376. [Google Scholar] [CrossRef]
- Wang, X.; Lang, M.; Zhao, T.; Feng, X.; Zheng, C.; Huang, C.; Hao, J.; Dong, J.; Luo, L.; Li, X.; et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3(+)Treg cells in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 3048–3058. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Wei, X.; Jiang, H.; Lan, C.; Yang, S.; Wang, H.; Yang, Y.; Tian, C.; Xu, Z.; et al. PD-L1 is a direct target of cancer-FOXP3 in pancreatic ductal adenocarcinoma (PDAC), and combined immunotherapy with antibodies against PD-L1 and CCL5 is effective in the treatment of PDAC. Signal Transduct. Target. Ther. 2020, 5, 38. [Google Scholar] [CrossRef]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.A.; Bae, S.; Lillard, J.W., Jr.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1323. [Google Scholar] [CrossRef]
- Makinoshima, H.; Dezawa, M. Pancreatic cancer cells activate CCL5 expression in mesenchymal stromal cells through the insulin-like growth factor-I pathway. FEBS Lett. 2009, 583, 3697–3703. [Google Scholar] [CrossRef]
- Matsuo, Y.; Raimondo, M.; Woodward, T.A.; Wallace, M.B.; Gill, K.R.; Tong, Z.; Burdick, M.D.; Yang, Z.; Strieter, R.M.; Hoffman, R.M.; et al. CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int. J. Cancer 2009, 125, 1027–1037. [Google Scholar] [CrossRef]
- Niu, N.; Shen, X.; Zhang, L.; Chen, Y.; Lu, P.; Yang, W.; Liu, M.; Shi, J.; Xu, D.; Tang, Y.; et al. Tumor Cell-Intrinsic SETD2 Deficiency Reprograms Neutrophils to Foster Immune Escape in Pancreatic Tumorigenesis. Adv. Sci. 2023, 10, e2202937. [Google Scholar] [CrossRef]
- Bianchi, A.; De Castro Silva, I.; Deshpande, N.U.; Singh, S.; Mehra, S.; Garrido, V.T.; Guo, X.; Nivelo, L.A.; Kolonias, D.S.; Saigh, S.J. Cell-autonomous Cxcl1 sustains tolerogenic circuitries and stromal inflammation via neutrophil-derived TNF in pancreatic cancer. Cancer Discov. 2023, 13, 1428–1453. [Google Scholar] [CrossRef]
- Deng, J.; Kang, Y.; Cheng, C.C.; Li, X.; Dai, B.; Katz, M.H.; Men, T.; Kim, M.P.; Koay, E.A.; Huang, H.; et al. DDR1-induced neutrophil extracellular traps drive pancreatic cancer metastasis. JCI Insight 2021, 6, e146133. [Google Scholar] [CrossRef]
- Wang, Z.Z.; Li, X.T.; Li, Q.J.; Zhou, J.X. Targeting CXCL5 in Pancreatic Cancer Cells Inhibits Cancer Xenograft Growth by Reducing Proliferation and Inhibiting EMT Progression. Dig. Dis. Sci. 2023, 68, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.; Furth, E.E.; Vonderheide, R.H. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2016, 4, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.-H.; Ma, Y.; Ang, C.-S.; Dumesny, C.; Huynh, N.; Yang, Y.; Wang, K.; Nikfarjam, M.; He, H. CXCL5 knockdown attenuated gemcitabine resistance of pancreatic cancer through regulation of cancer cells and tumour stroma. Am. J. Transl. Res. 2023, 15, 2676. [Google Scholar] [PubMed]
- Brat, D.J.; Bellail, A.C.; Van Meir, E.G. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-Oncol. 2005, 7, 122–133. [Google Scholar] [CrossRef]
- Huang, S.; Mills, L.; Mian, B.; Tellez, C.; McCarty, M.; Yang, X.-D.; Gudas, J.M.; Bar-Eli, M. Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am. J. Pathol. 2002, 161, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Varney, M.L.; Singh, R.K. Expression of interleukin 8 and its receptors in human colon carcinoma cells with different metastatic potentials. Clin. Cancer Res. 2001, 7, 3298–3304. [Google Scholar] [PubMed]
- Awaji, M.; Futakuchi, M.; Heavican, T.; Iqbal, J.; Singh, R.K. Cancer-Associated Fibroblasts Enhance Survival and Progression of the Aggressive Pancreatic Tumor Via FGF-2 and CXCL8. Cancer Microenviron. 2019, 12, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Peng, Y.; Wu, X.; Meng, S.; Yu, W.; Zhao, J.; Zhang, H.; Wang, J.; Li, W. CCL18 secreted from M2 macrophages promotes migration and invasion via the PI3K/Akt pathway in gallbladder cancer. Cell. Oncol. 2019, 42, 81–92. [Google Scholar] [CrossRef]
- Nie, Y.; Chen, J.; Huang, D.; Yao, Y.; Chen, J.; Ding, L.; Zeng, J.; Su, S.; Chao, X.; Su, F. Tumor-Associated Macrophages Promote Malignant Progression of Breast Phyllodes Tumors by Inducing Myofibroblast DifferentiationTAMs Promote Malignancy of Phyllodes Tumors. Cancer Res. 2017, 77, 3605–3618. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, J.S.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Pierson, K.C.; Pitts-Kiefer, A.; Fan, L.; Belkin, D.A.; Wang, C.Q.; Bhuvanendran, S.; Johnson-Huang, L.M. Tumor-associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J. Investig. Dermatol. 2011, 131, 1322–1330. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, B.; Sun, X.; Zhang, X.; Lv, S.; Li, H.; Wang, X.; Zhao, C.; Zhang, H.; Xie, X. CC chemokine ligand 18 (CCL18) promotes migration and invasion of lung cancer cells by binding to Nir1 through Nir1-ELMO1/DOC180 signaling pathway. Mol. Carcinog. 2016, 55, 2051–2062. [Google Scholar] [CrossRef]
- Islam, S.A.; Ling, M.F.; Leung, J.; Shreffler, W.G.; Luster, A.D. Identification of human CCR8 as a CCL18 receptor. J. Exp. Med. 2013, 210, 1889–1898. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Zhao, M.; Khaldoyanidi, S.K.; DiScipio, R.G. The chemokine CCL18 causes maturation of cultured monocytes to macrophages in the M2 spectrum. Immunology 2012, 135, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liao, J.; Liu, J.; Huang, D.; He, C.; Chen, F.; Yang, L.; Wu, W.; Chen, J.; Lin, L.; et al. Blocking the recruitment of naive CD4(+) T cells reverses immunosuppression in breast cancer. Cell Res. 2017, 27, 461–482. [Google Scholar] [CrossRef] [PubMed]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.; John, S.; Taams, L.S. CD4+ CD25+ Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Zhou, Q.; Zheng, S.; Li, G.; Lin, Q.; Wei, L.; Fu, Z.; Zhang, B.; Liu, Y.; Li, Z.; et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018, 9, 453. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Li, W.; Li, C.; Gao, Z.; Guo, K.; Song, S. CCL18 promotes epithelial-mesenchymal transition, invasion and migration of pancreatic cancer cells in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2015, 46, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Xiong, L.; Wu, W.; Li, S.; Liu, J.; Yang, L.; Lao, L.; Huang, P.; Zhang, M.; Chen, H.; et al. CCL18 signaling from tumor-associated macrophages activates fibroblasts to adopt a chemoresistance-inducing phenotype. Oncogene 2023, 42, 224–237. [Google Scholar] [CrossRef]
- Jablonska, J.; Wu, C.F.; Andzinski, L.; Leschner, S.; Weiss, S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-beta. Int. J. Cancer 2014, 134, 1346–1358. [Google Scholar] [CrossRef]
- Wolpe, S.D.; Sherry, B.; Juers, D.; Davatelis, G.; Yurt, R.W.; Cerami, A. Identification and characterization of macrophage inflammatory protein 2. Proc. Natl. Acad. Sci. USA 1989, 86, 612–616. [Google Scholar] [CrossRef]
- Li, J.L.; Lim, C.H.; Tay, F.W.; Goh, C.C.; Devi, S.; Malleret, B.; Lee, B.; Bakocevic, N.; Chong, S.Z.; Evrard, M. Neutrophils self-regulate immune complex-mediated cutaneous inflammation through CXCL2. J. Investig. Dermatol. 2016, 136, 416–424. [Google Scholar] [CrossRef]
- Takikawa, T.; Hamada, S.; Matsumoto, R.; Tanaka, Y.; Kataoka, F.; Sasaki, A.; Masamune, A. Senescent Human Pancreatic Stellate Cells Secrete CXCR2 Agonist CXCLs to Promote Proliferation and Migration of Human Pancreatic Cancer AsPC-1 and MIAPaCa-2 Cell Lines. Int. J. Mol. Sci. 2022, 23, 9275. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Tu, C.; Cheng, X.; Xu, Z.; Wang, X.; Shen, J.; Chai, K.; Chen, W. Inflammatory and senescent phenotype of pancreatic stellate cells induced by Sqstm1 downregulation facilitates pancreatic cancer progression. Int. J. Biol. Sci. 2019, 15, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Vucic, E.; Kurz, E.; Hajdu, C.; Bar-Sagi, D. Gain-of-function p53(R172H) mutation drives accumulation of neutrophils in pancreatic tumors, promoting resistance to immunotherapy. Cell Rep. 2021, 36, 109578. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Struyf, S.; Proost, P. The unique structural and functional features of CXCL12. Cell Mol. Immunol. 2018, 15, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Jones, D.; Borghesani, P.R.; Segal, R.A.; Nagasawa, T.; Kishimoto, T.; Bronson, R.T.; Springer, T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4-and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Ahirwar, D.K.; Nasser, M.W.; Ouseph, M.M.; Elbaz, M.; Cuitino, M.C.; Kladney, R.D.; Varikuti, S.; Kaul, K.; Satoskar, A.R.; Ramaswamy, B.; et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene 2018, 37, 4428–4442. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ge, H.; Shi, Y.; Yuan, F.; Yue, H. CAFs secrete CXCL12 to accelerate the progression and cisplatin resistance of colorectal cancer through promoting M2 polarization of macrophages. Med. Oncol. 2023, 40, 90. [Google Scholar] [CrossRef]
- Li, D.; Qu, C.; Ning, Z.; Wang, H.; Zang, K.; Zhuang, L.; Chen, L.; Wang, P.; Meng, Z. Radiation promotes epithelial-to-mesenchymal transition and invasion of pancreatic cancer cell by activating carcinoma-associated fibroblasts. Am. J. Cancer Res. 2016, 6, 2192. [Google Scholar]
- Bleul, C.C.; Fuhlbrigge, R.C.; Casasnovas, J.M.; Aiuti, A.; Springer, T.A. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 1996, 184, 1101–1109. [Google Scholar] [CrossRef]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef]
- Ratajczak, M.; Zuba-Surma, E.; Kucia, M.; Reca, R.; Wojakowski, W.; Ratajczak, J. The pleiotropic effects of the SDF-1–CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia 2006, 20, 1915–1924. [Google Scholar] [CrossRef]
- Morimoto, M.; Matsuo, Y.; Koide, S.; Tsuboi, K.; Shamoto, T.; Sato, T.; Saito, K.; Takahashi, H.; Takeyama, H. Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: Effect of CXCR4 antagonists. BMC Cancer 2016, 16, 305. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine SDF-1α signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015, 6, 3085. [Google Scholar] [CrossRef]
- Shen, B.; Zheng, M.Q.; Lu, J.W.; Jiang, Q.; Wang, T.H.; Huang, X.E. CXCL12-CXCR4 promotes proliferation and invasion of pancreatic cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 5403–5408. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, Z.; Chen, X.; Duan, W.; Lei, J.; Zong, L.; Li, X.; Sheng, L.; Ma, J.; Han, L. Stromal-derived factor-1α/CXCL12-CXCR4 chemotactic pathway promotes perineural invasion in pancreatic cancer. Oncotarget 2015, 6, 4717. [Google Scholar] [CrossRef] [PubMed]
- Samara, G.J.; Lawrence, D.M.; Chiarelli, C.J.; Valentino, M.D.; Lyubsky, S.; Zucker, S.; Vaday, G.G. CXCR4-mediated adhesion and MMP-9 secretion in head and neck squamous cell carcinoma. Cancer Lett. 2004, 214, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Määttä, M.; Soini, Y.; Liakka, A.; Autio-Harmainen, H. Differential expression of matrix metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in hepatocellular and pancreatic adenocarcinoma: Implications for tumor progression and clinical prognosis. Clin. Cancer Res. 2000, 6, 2726–2734. [Google Scholar] [PubMed]
- McQuibban, G.A.; Butler, G.S.; Gong, J.H.; Bendall, L.; Power, C.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J. Biol. Chem. 2001, 276, 43503–43508. [Google Scholar] [CrossRef] [PubMed]
- Frederick, M.J.; Henderson, Y.; Xu, X.; Deavers, M.T.; Sahin, A.A.; Wu, H.; Lewis, D.E.; El-Naggar, A.K.; Clayman, G.L. In vivo expression of the novel CXC chemokine BRAK in normal and cancerous human tissue. Am. J. Pathol. 2000, 156, 1937–1950. [Google Scholar] [CrossRef] [PubMed]
- Hromas, R.; Broxmeyer, H.E.; Kim, C.; Nakshatri, H.; Christopherson II, K.; Azam, M.; Hou, Y.-H. Cloning of BRAK, a novel divergent CXC chemokine preferentially expressed in normal versus malignant cells. Biochem. Biophys. Res. Commun. 1999, 255, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Shurin, G.V.; Ferris, R.; Tourkova, I.L.; Perez, L.; Lokshin, A.; Balkir, L.; Collins, B.; Chatta, G.S.; Shurin, M.R. Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J. Immunol. 2005, 174, 5490–5498. [Google Scholar] [CrossRef]
- Kurth, I.; Willimann, K.; Schaerli, P.; Hunziker, T.; Clark-Lewis, I.; Moser, B. Monocyte selectivity and tissue localization suggests a role for breast and kidney–expressed chemokine (BRAK) in macrophage development. J. Exp. Med. 2001, 194, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Starnes, T.; Rasila, K.K.; Robertson, M.J.; Brahmi, Z.; Dahl, R.; Christopherson, K.; Hromas, R. The chemokine CXCL14 (BRAK) stimulates activated NK cell migration: Implications for the downregulation of CXCL14 in malignancy. Exp. Hematol. 2006, 34, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Luo, J.; Isaacs, W.B.; Jarrard, D.F. Modulation of CXCL14 (BRAK) expression in prostate cancer. Prostate 2005, 64, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Wente, M.N.; Mayer, C.; Gaida, M.M.; Michalski, C.W.; Giese, T.; Bergmann, F.; Giese, N.A.; Büchler, M.W.; Friess, H. CXCL14 expression and potential function in pancreatic cancer. Cancer Lett. 2008, 259, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef]
- Sjöberg, E.; Meyrath, M.; Milde, L.; Herrera, M.; Lövrot, J.; Hägerstrand, D.; Frings, O.; Bartish, M.; Rolny, C.; Sonnhammer, E. A Novel ACKR2-Dependent Role of Fibroblast-Derived CXCL14 in Epithelial-to-Mesenchymal Transition and Metastasis of Breast CancerFibroblast CXCL14/ACKR2/NOS1 Signaling in Breast Cancer. Clin. Cancer Res. 2019, 25, 3702–3717. [Google Scholar] [CrossRef]
- Augsten, M.; Hägglöf, C.; Olsson, E.; Stolz, C.; Tsagozis, P.; Levchenko, T.; Frederick, M.J.; Borg, Å.; Micke, P.; Egevad, L. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi-modal stimulator of prostate tumor growth. Proc. Natl. Acad. Sci. USA 2009, 106, 3414–3419. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.B.; Meyronet, D.; Hervieu, V.; Frederick, M.J.; Bergsland, E.; Bergers, G. Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep. 2015, 11, 577–591. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Ware, M.B.; El-Rayes, B.F.; Lesinski, G.B. Mirage or long-awaited oasis: Reinvigorating T-cell responses in pancreatic cancer. J. Immunother. Cancer 2020, 8, e001100. [Google Scholar] [CrossRef]
- Romero, J.M.; Grunwald, B.; Jang, G.H.; Bavi, P.P.; Jhaveri, A.; Masoomian, M.; Fischer, S.E.; Zhang, A.; Denroche, R.E.; Lungu, I.M.; et al. A Four-Chemokine Signature Is Associated with a T-cell-Inflamed Phenotype in Primary and Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 1997–2010. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Vonderhaar, E.P.; Barnekow, N.S.; McAllister, D.; McOlash, L.; Eid, M.A.; Riese, M.J.; Tarakanova, V.L.; Johnson, B.D.; Dwinell, M.B. STING Activated Tumor-Intrinsic Type I Interferon Signaling Promotes CXCR3 Dependent Antitumor Immunity in Pancreatic Cancer. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 41–58. [Google Scholar] [CrossRef]
- Gorchs, L.; Oosthoek, M.; Yucel-Lindberg, T.; Moro, C.F.; Kaipe, H. Chemokine Receptor Expression on T Cells Is Modulated by CAFs and Chemokines Affect the Spatial Distribution of T Cells in Pancreatic Tumors. Cancers 2022, 14, 3826. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.; Huang, J. CD8(+) T Cells and NK Cells: Parallel and Complementary Soldiers of Immunotherapy. Curr. Opin. Chem. Eng. 2018, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Lim, S.A.; Kim, J.; Jeon, S.; Shin, M.H.; Kwon, J.; Kim, T.J.; Im, K.; Han, Y.; Kwon, W.; Kim, S.W.; et al. Defective Localization With Impaired Tumor Cytotoxicity Contributes to the Immune Escape of NK Cells in Pancreatic Cancer Patients. Front. Immunol. 2019, 10, 496. [Google Scholar] [CrossRef] [PubMed]
- Chibaya, L.; Murphy, K.C.; DeMarco, K.D.; Gopalan, S.; Liu, H.; Parikh, C.N.; Lopez-Diaz, Y.; Faulkner, M.; Li, J.; Morris, J.P.; et al. EZH2 inhibition remodels the inflammatory senescence-associated secretory phenotype to potentiate pancreatic cancer immune surveillance. Nat. Cancer 2023, 4, 872–892. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Ernst, M. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Therapeutic Opportunities and Clinical Challenges. Cancers 2021, 13, 2860. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cai, Y.; Liu, L.; Wu, Y.; Xiong, X. Crucial biological functions of CCL7 in cancer. PeerJ 2018, 6, e4928. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Rubie, C.; Frick, V.O.; Ghadjar, P.; Wagner, M.; Grimm, H.; Vicinus, B.; Justinger, C.; Graeber, S.; Schilling, M.K. CCL20/CCR6 expression profile in pancreatic cancer. J. Transl. Med. 2010, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Phillipson, M.; Kubes, P. The neutrophil in vascular inflammation. Nat. Med. 2011, 17, 1381–1390. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-β:“N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.-G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Mishalian, I.; Singhal, S.; Cheng, G.; Kapoor, V.; Horng, W.; Fridlender, G.; Bayuh, R.; Worthen, G.S. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS ONE 2012, 7, e31524. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Wang, W.; Liu, L.; Xu, H.; Wu, C.; Xiang, J.; Xu, J.; Liu, C.; Long, J.; Ni, Q.; Yu, X. Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. J. Br. Surg. 2016, 103, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ijichi, H.; Takahashi, R.; Miyabayashi, K.; Fujiwara, H.; Yamada, T.; Kato, H.; Nakatsuka, T.; Tanaka, Y.; Tateishi, K. Blocking CXCLs–CXCR2 axis in tumor–stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune-inflammatory microenvironment. Oncogenesis 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Wheatley-Price, P.; Asomaning, K.; Reid, A.; Zhai, R.; Su, L.; Zhou, W.; Zhu, A.; Ryan, D.P.; Christiani, D.C.; Liu, G. Myeloperoxidase and superoxide dismutase polymorphisms are associated with an increased risk of developing pancreatic adenocarcinoma. Cancer 2008, 112, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Gaida, M.M.; Steffen, T.G.; Günther, F.; Tschaharganeh, D.F.; Felix, K.; Bergmann, F.; Schirmacher, P.; Hänsch, G.M. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur. J. Immunol. 2012, 42, 3369–3380. [Google Scholar] [CrossRef]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, W.; Mathew, E.; Bednar, F.; Wan, S.; Collins, M.A.; Evans, R.A.; Welling, T.H.; Vonderheide, R.H.; di Magliano, M.P. CD4+ T lymphocyte ablation prevents pancreatic carcinogenesis in mice. Cancer Immunol. Res. 2014, 2, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, U.K.; Moore, T.T.; Joo, H.-G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.C.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.S.; Linehan, D.C. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Gorchs, L.; Kaipe, H. Interactions between Cancer-Associated Fibroblasts and T Cells in the Pancreatic Tumor Microenvironment and the Role of Chemokines. Cancers 2021, 13, 2995. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.A.; Marubayashi, S.; Jorapur, A.; Jacobson, S.; Zibinsky, M.; Robles, O.; Hu, D.X.; Jackson, J.J.; Pookot, D.; Sanchez, J.; et al. Tumors establish resistance to immunotherapy by regulating T(reg) recruitment via CCR4. J. Immunother. Cancer 2020, 8, e000764. [Google Scholar] [CrossRef] [PubMed]
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009, 69, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, G.M.; Knott, M.M.; Vetter, V.K.; Rapp, M.; Haubner, S.; Fesseler, J.; Kuhnemuth, B.; Layritz, P.; Thaler, R.; Kruger, S.; et al. Cancer cell-derived IL-1alpha induces CCL22 and the recruitment of regulatory T cells. Oncoimmunology 2016, 5, e1175794. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhou, W.; Chen, R.; Xiang, B.; Zhou, S.; Lan, L. CXCL10 is a Tumor Microenvironment and Immune Infiltration Related Prognostic Biomarker in Pancreatic Adenocarcinoma. Front. Mol. Biosci. 2021, 8, 611508. [Google Scholar] [CrossRef]
- Cannon, A.; Thompson, C.M.; Maurer, H.C.; Atri, P.; Bhatia, R.; West, S.; Ghersi, D.; Olive, K.P.; Kumar, S.; Batra, S.K. CXCR3 and Cognate Ligands are Associated with Immune Cell Alteration and Aggressiveness of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2020, 26, 6051–6063. [Google Scholar] [CrossRef]
- Lunardi, S.; Jamieson, N.B.; Lim, S.Y.; Griffiths, K.L.; Carvalho-Gaspar, M.; Al-Assar, O.; Yameen, S.; Carter, R.C.; McKay, C.J.; Spoletini, G. IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival. Oncotarget 2014, 5, 11064. [Google Scholar] [CrossRef]
- Hoerning, A.; Koss, K.; Datta, D.; Boneschansker, L.; Jones, C.N.; Wong, I.Y.; Irimia, D.; Calzadilla, K.; Benitez, F.; Hoyer, P.F.; et al. Subsets of human CD4(+) regulatory T cells express the peripheral homing receptor CXCR3. Eur. J. Immunol. 2011, 41, 2291–2302. [Google Scholar] [CrossRef]
- Lin, Y.N.; Schmidt, M.O.; Sharif, G.M.; Vietsch, E.E.; Kiliti, A.J.; Barefoot, M.E.; Riegel, A.T.; Wellstein, A. Impaired CXCL12 signaling contributes to resistance of pancreatic cancer subpopulations to T cell-mediated cytotoxicity. Oncoimmunology 2022, 11, 2027136. [Google Scholar] [CrossRef] [PubMed]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. IFNgamma and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016, 6, 400–413. [Google Scholar] [CrossRef]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef]
- Wu, M.; Han, J.; Wu, H.; Liu, Z. Proteasome-dependent senescent tumor cells mediate immunosuppression through CCL20 secretion and M2 polarization in pancreatic ductal adenocarcinoma. Front. Immunol. 2023, 14, 1216376. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Wang, F.; Zhang, K.; Chen, Z. Comprehensive analysis of prognostic value and immune infiltration of CXC chemokines in pancreatic cancer. BMC Med. Genom. 2022, 15, 96. [Google Scholar] [CrossRef]
- Hiraoka, N.; Yamazaki-Itoh, R.; Ino, Y.; Mizuguchi, Y.; Yamada, T.; Hirohashi, S.; Kanai, Y. CXCL17 and ICAM2 are associated with a potential anti-tumor immune response in early intraepithelial stages of human pancreatic carcinogenesis. Gastroenterology 2011, 140, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Q.; Peng, J.; Wang, M.; Li, T.; Liu, J.; Cui, M.; Zhang, X.; Gao, X.; Liao, Q.; et al. CXCL5 overexpression predicts a poor prognosis in pancreatic ductal adenocarcinoma and is correlated with immune cell infiltration. J. Cancer 2020, 11, 2371–2381. [Google Scholar] [CrossRef]
- Fang, Y.; Saiyin, H.; Zhao, X.; Wu, Y.; Han, X.; Lou, W. IL-8–Positive Tumor-Infiltrating Inflammatory Cells Are a Novel Prognostic Marker in Pancreatic Ductal Adenocarcinoma Patients. Pancreas 2016, 45, 671–678. [Google Scholar] [CrossRef]
- Qian, L.; Yu, S.; Yin, C.; Zhu, B.; Chen, Z.; Meng, Z.; Wang, P. Plasma IFN-gamma-inducible chemokines CXCL9 and CXCL10 correlate with survival and chemotherapeutic efficacy in advanced pancreatic ductal adenocarcinoma. Pancreatology 2019, 19, 340–345. [Google Scholar] [CrossRef]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef]
- D’Alterio, C.; Giardino, A.; Scognamiglio, G.; Butturini, G.; Portella, L.; Guardascione, G.; Frigerio, I.; Montella, M.; Gobbo, S.; Martignoni, G.; et al. CXCR4-CXCL12-CXCR7 and PD-1/PD-L1 in Pancreatic Cancer: CXCL12 Predicts Survival of Radically Resected Patients. Cells 2022, 11, 3340. [Google Scholar] [CrossRef]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Chen, J.; Ma, H.; Shao, Z.; Chen, H.; Jin, G. High expression of CX3CL1/CX3CR1 axis predicts a poor prognosis of pancreatic ductal adenocarcinoma. J. Gastrointest. Surg. 2012, 16, 1493–1498. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef]
- Ho, T.T.B.; Nasti, A.; Seki, A.; Komura, T.; Inui, H.; Kozaka, T.; Kitamura, Y.; Shiba, K.; Yamashita, T.; Yamashita, T.; et al. Combination of gemcitabine and anti-PD-1 antibody enhances the anticancer effect of M1 macrophages and the Th1 response in a murine model of pancreatic cancer liver metastasis. J. Immunother. Cancer 2020, 8, e001367. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; McAllister, D.; Vonderhaar, E.P.; Palen, K.; Riese, M.J.; Gershan, J.; Johnson, B.D.; Dwinell, M.B. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J. Immunother. Cancer 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Pang, N.; Shi, J.; Qin, L.; Chen, A.; Tang, Y.; Yang, H.; Huang, Y.; Wu, Q.; Li, X.; He, B.; et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J. Hematol. Oncol. 2021, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Mackall, C.L. Interleukin-7: Master regulator of peripheral T-cell homeostasis? Trends Immunol. 2001, 22, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Nagira, M.; Imai, T.; Baba, M.; Takagi, S.; Tabira, Y.; Akagi, J.; Nomiyama, H.; Yoshie, O. EBI1-ligand chemokine (ELC) attracts a broad spectrum of lymphocytes: Activated T cells strongly up-regulate CCR7 and efficiently migrate toward ELC. Int. Immunol. 1998, 10, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W. CXCR1-or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [PubMed]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef] [PubMed]
- Lesch, S.; Blumenberg, V.; Stoiber, S.; Gottschlich, A.; Ogonek, J.; Cadilha, B.L.; Dantes, Z.; Rataj, F.; Dorman, K.; Lutz, J. T cells armed with CXC chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 2021, 5, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Kremer, V.; Ligtenberg, M.A.; Zendehdel, R.; Seitz, C.; Duivenvoorden, A.; Wennerberg, E.; Colón, E.; Scherman-Plogell, A.-H.; Lundqvist, A. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. Immunother. Cancer 2017, 5, 73. [Google Scholar] [CrossRef]
- Müller, N.; Michen, S.; Tietze, S.; Töpfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J. Immunother. 2015, 38, 197. [Google Scholar] [CrossRef]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.H.; Byrne, K.T.; Vonderheide, R.H. Immunotherapy and prevention of pancreatic cancer. Trends Cancer 2018, 4, 418–428. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.B.; Kim, V.M.; Muth, S.T.; Saung, M.T.; Lokker, N.; Blouw, B.; Armstrong, T.D.; Jaffee, E.M.; Tsujikawa, T.; Coussens, L.M.; et al. Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 5351–5363. [Google Scholar] [CrossRef]
- Liang, M. Oncorine, the world first oncolytic virus medicine and its update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef]
- Veinalde, R.; Pidelaserra-Marti, G.; Moulin, C.; Tan, C.L.; Schafer, T.E.; Kang, N.; Ball, C.R.; Leichsenring, J.; Stenzinger, A.; Kaderali, L.; et al. Virotherapy combined with anti-PD-1 transiently reshapes the tumor immune environment and induces anti-tumor immunity in a preclinical PDAC model. Front. Immunol. 2022, 13, 1096162. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Wilkinson, G.A.; Eng, K.H.; Fields, P.; Raber, P.; Moseley, J.L.; Cheetham, K.; Coffey, M.; Nuovo, G.; Kalinski, P.; et al. Pembrolizumab in Combination with the Oncolytic Virus Pelareorep and Chemotherapy in Patients with Advanced Pancreatic Adenocarcinoma: A Phase Ib Study. Clin. Cancer Res. 2020, 26, 71–81. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Investig. New Drugs 2020, 38, 800–811. [Google Scholar] [CrossRef]
- Linehan, D.; Noel, M.S.; Hezel, A.F.; Wang-Gillam, A.; Eskens, F.; Sleijfer, S.; Desar, I.M.; Erdkamp, F.; Wilmink, J.; Diehl, J. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: Correlation with blood monocyte counts. J. Clin. Oncol. 2018, 36, 92. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Papadopoulos, K.; Fong, P.C.; Patnaik, A.; Messiou, C.; Olmos, D.; Wang, G.; Tromp, B.J.; Puchalski, T.A.; Balkwill, F.; et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 1041–1050. [Google Scholar] [CrossRef]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A Phase I Study of the Anti-CC Chemokine Receptor 4 Antibody, Mogamulizumab, in Combination with Nivolumab in Patients with Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef]
- Zamarin, D.; Hamid, O.; Nayak-Kapoor, A.; Sahebjam, S.; Sznol, M.; Collaku, A.; Fox, F.E.; Marshall, M.A.; Hong, D.S. Mogamulizumab in Combination with Durvalumab or Tremelimumab in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2020, 26, 4531–4541. [Google Scholar] [CrossRef]
- Hong, D.S.; Rixe, O.; Chiu, V.K.; Forde, P.M.; Dragovich, T.; Lou, Y.; Nayak-Kapoor, A.; Leidner, R.; Atkins, J.N.; Collaku, A.; et al. Mogamulizumab in Combination with Nivolumab in a Phase I/II Study of Patients with Locally Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2022, 28, 479–488. [Google Scholar] [CrossRef]
- Lieberman-Blum, S.S.; Fung, H.B.; Bandres, J.C. Maraviroc: A CCR5-receptor antagonist for the treatment of HIV-1 infection. Clin. Ther. 2008, 30, 1228–1250. [Google Scholar] [CrossRef]
- Christenson, E.; Lim, S.J.; Wang, H.; Ferguson, A.; Parkinson, R.; Cetasaan, Y.; Rodriguez, C.; Burkhart, R.; De Jesus-Acosta, A.; He, J. Nivolumab and a CCR2/CCR5 dual antagonist (BMS-813160) with or without GVAX for locally advanced pancreatic ductal adenocarcinomas: Results of phase I study. J. Clin. Oncol. 2023, 41, 730. [Google Scholar] [CrossRef]
- Dunne, R.F.; Ullman, N.A.; Belt, B.A.; Ruffolo, L.I.; Burchard, P.; Hezel, A.F.; Zittel, J.; Wang, W.; Ramsdale, E.E.; Kaul, V. A phase I study to evaluate the safety and tolerability of SX-682 in combination with PD-1 inhibitor as maintenance therapy for unresectable pancreatic adenocarcinoma. J. Clin. Oncol. 2022, 40, TPS631. [Google Scholar] [CrossRef]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Johnson, T.I.; Bax, L.; et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Barreto, C.M.N.; Ledesma, D.A.; Gite, S.; Solis, L.M.; Jiang, M.; Pandurengan, R.K.; Wolff, R.; Javle, M.; Pant, S.; Varadhachary, G. Abstract CT204: High cytotoxic T-cell or polymorphonuclear cell infiltrates in the tumor microenvironment correlate with responses to BL8040 plus pembrolizumab combination therapy in metastatic pancreatic tumors: Scientific correlates of a phase II clinical trial. Cancer Res. 2020, 80, CT204. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Williams, A.; Schreiber, J.; Hohmann, N.; Pruefer, U.; Krauss, J.; Jager, D.; Fromming, A.; Beyer, D.; Eulberg, D.; et al. Combined inhibition of CXCL12 and PD-1 in MSS colorectal and pancreatic cancer: Modulation of the microenvironment and clinical effects. J. Immunother. Cancer 2021, 9, e002505. [Google Scholar] [CrossRef]
- Lee, J.; Kang, T.H.; Yoo, W.; Choi, H.; Jo, S.; Kong, K.; Lee, S.R.; Kim, S.U.; Kim, J.S.; Cho, D.; et al. An Antibody Designed to Improve Adoptive NK-Cell Therapy Inhibits Pancreatic Cancer Progression in a Murine Model. Cancer Immunol. Res. 2019, 7, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef]
- King, D.A.; Smith, A.R.; Pineda, G.; Nakano, M.; Michelini, F.; Goedegebuure, S.P.; Thyparambil, S.; Liao, W.L.; McCormick, A.; Ju, J.; et al. Complete Remission of Widely Metastatic Human Epidermal Growth Factor Receptor 2-Amplified Pancreatic Adenocarcinoma After Precision Immune and Targeted Therapy With Description of Sequencing and Organoid Correlates. JCO Precis. Oncol. 2023, 7, e2100489. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Alderson, K.L.; Sondel, P.M. Clinical cancer therapy by NK cells via antibody-dependent cell-mediated cytotoxicity. J. Biomed. Biotechnol. 2011, 2011, 379123. [Google Scholar] [CrossRef] [PubMed]
- Safran, H.; Iannitti, D.; Ramanathan, R.; Schwartz, J.D.; Steinhoff, M.; Nauman, C.; Hesketh, P.; Rathore, R.; Wolff, R.; Tantravahi, U. Herceptin and gemcitabine for metastatic pancreatic cancers that overexpress HER-2/neu. Cancer Investig. 2004, 22, 706–712. [Google Scholar] [CrossRef]
- Beelen, N.A.; Aberle, M.R.; Bruno, V.; Olde Damink, S.W.M.; Bos, G.M.J.; Rensen, S.S.; Wieten, L. Antibody-dependent cellular cytotoxicity-inducing antibodies enhance the natural killer cell anti-cancer response against patient-derived pancreatic cancer organoids. Front. Immunol. 2023, 14, 1133796. [Google Scholar] [CrossRef]
- Haller, S.D.; Monaco, M.L.; Essani, K. The Present Status of Immuno-Oncolytic Viruses in the Treatment of Pancreatic Cancer. Viruses 2020, 12, 1318. [Google Scholar] [CrossRef]
- Kirchhammer, N.; Trefny, M.P.; Natoli, M.; Brücher, D.; Smith, S.N.; Werner, F.; Koch, V.; Schreiner, D.; Bartoszek, E.; Buchi, M. NK cells with tissue-resident traits shape response to immunotherapy by inducing adaptive antitumor immunity. Sci. Transl. Med. 2022, 14, eabm9043. [Google Scholar] [CrossRef] [PubMed]
- Alva, A.; Daniels, G.A.; Wong, M.K.; Kaufman, H.L.; Morse, M.A.; McDermott, D.F.; Clark, J.I.; Agarwala, S.S.; Miletello, G.; Logan, T.F.; et al. Contemporary experience with high-dose interleukin-2 therapy and impact on survival in patients with metastatic melanoma and metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2016, 65, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, C.; Pandit, H.; Nagy, B.A.; Stellas, D.; Jensen, S.M.; Bear, J.; Cam, M.; Valentin, A.; Fox, B.A.; Felber, B.K.; et al. Heterodimeric IL-15 delays tumor growth and promotes intratumoral CTL and dendritic cell accumulation by a cytokine network involving XCL1, IFN-gamma, CXCL9 and CXCL10. J. Immunother. Cancer 2020, 8, 000599. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lekan, A.A.; Weiner, L.M. The Role of Chemokines in Orchestrating the Immune Response to Pancreatic Ductal Adenocarcinoma. Cancers 2024, 16, 559. https://doi.org/10.3390/cancers16030559
Lekan AA, Weiner LM. The Role of Chemokines in Orchestrating the Immune Response to Pancreatic Ductal Adenocarcinoma. Cancers. 2024; 16(3):559. https://doi.org/10.3390/cancers16030559
Chicago/Turabian StyleLekan, Alexander A., and Louis M. Weiner. 2024. "The Role of Chemokines in Orchestrating the Immune Response to Pancreatic Ductal Adenocarcinoma" Cancers 16, no. 3: 559. https://doi.org/10.3390/cancers16030559
APA StyleLekan, A. A., & Weiner, L. M. (2024). The Role of Chemokines in Orchestrating the Immune Response to Pancreatic Ductal Adenocarcinoma. Cancers, 16(3), 559. https://doi.org/10.3390/cancers16030559