Simple Summary
Despite recent advances in targeted therapy and immunotherapy, the treatment of lung cancer remains challenging due to its high metastatic potential, which adversely affects both the prognosis and the quality of life of these patients. Rictor, the scaffold protein of the mTORC2 complex, plays an important role in regulating essential cellular functions and promoting metastases through epithelial–mesenchymal transition. Amplification of the RICTOR gene and subsequent overexpression of the Rictor protein can activate mTORC2 and promote cell survival and migration. Recent studies suggest that RICTOR amplification and Rictor overexpression may serve as markers of mTORC2 activation. Potentially, they can also provide druggable targets for advanced therapy. Although these drugs are still in a preclinical phase, selective mTORC2 inhibitors are a promising approach to inhibit tumor cell migration and metastasis formation. This review highlights the importance of Rictor and mTORC2 as predictive markers and promising therapeutic targets in the treatment of lung cancer.
Abstract
Lung carcinoma is one of the most common cancer types for both men and women. Despite recent breakthroughs in targeted therapy and immunotherapy, it is characterized by a high metastatic rate, which can significantly affect quality of life and prognosis. Rictor (encoded by the RICTOR gene) is known as a scaffold protein for the multiprotein complex mTORC2. Among its diverse roles in regulating essential cellular functions, mTORC2 also facilitates epithelial–mesenchymal transition and metastasis formation. Amplification of the RICTOR gene and subsequent overexpression of the Rictor protein can result in the activation of mTORC2, which promotes cell survival and migration. Based on recent studies, RICTOR amplification or Rictor overexpression can serve as a marker for mTORC2 activation, which in turn provides a promising druggable target. Although selective inhibitors of Rictor and the Rictor-mTOR association are only in a preclinical phase, they seem to be potent novel approaches to reduce tumor cell migration and metastasis formation. Here, we summarize recent advances that support an important role for Rictor and mTORC2 as potential therapeutic targets in the treatment of lung cancer. This is a traditional (narrative) review based on Pubmed and Google Scholar searches for the following keywords: Rictor, RICTOR amplification, mTORC2, Rictor complexes, lung cancer, metastasis, progression, mTOR inhibitors.
1. Introduction—Lung Cancer and mTOR
Based on the GLOBOCAN 2020 estimates of cancer incidence and mortality, lung cancer is the second most common malignancy worldwide, with about 2.2 million new cases (11.4% of all cancer cases in 2020) [1]. Despite recent breakthroughs in targeted and immunotherapy, it remains the leading cause of cancer-related death (18% of all cancers) [1]. This mortality can be partly explained by the high metastatic potential of the disease, and the fact that most patients already have metastatic disease at diagnosis. At the time of diagnosis, 75% of tumors cannot be removed surgically, leaving systemic therapy and radiotherapy as the only treatment options for these patients [2,3]. These findings underscore the urgent need for new, effective therapeutic options that may also have an impact on the metastatic propensity of the tumor, which is an important determinant of the prognosis [4,5].
The most common histological subtypes of lung cancer are adenocarcinoma (ADC), squamous cell carcinoma (SCC), and small cell lung carcinoma (SCLC). From a clinical point of view, ADCs, SCCs, and LCCs comprise a group referred to as non-small cell lung cancer (NSCLC), accounting for 80–85% of all lung carcinomas. Small cell lung cancer (SCLC), although less common (15% of all cases), has a particularly aggressive clinical behavior; most patients present with distant metastases at the time of diagnosis [6,7]. While a wide range of treatment options (including targeted and immunotherapy or even surgical resection in the early stage) is available for NSCLCs, the therapeutic armamentarium for SCLC is more limited [8].
Many molecular pathomechanisms of lung cancer have been identified over the last two decades. Aside from the well-known genetic alterations, such as EGFR and KRAS mutations or ALK and ROS1 rearrangements in lung adenocarcinoma, mutations or copy number variations of the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway members also frequently occur in lung cancer and may also serve as therapeutic targets [9,10]. The most common genetic alterations of the mTOR pathway in lung cancer are shown in Figure 1.
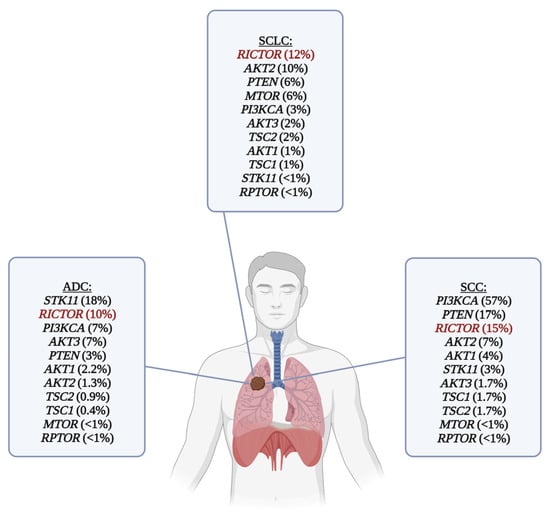
Figure 1.
Frequency of genetic alterations of the mTOR pathway in the most common histological subtypes of lung cancer. RICTOR amplification is the most common targetable genetic alteration in SCLC, and it is also one of the most frequent genetic alterations in lung ADC and SCC. RICTOR is highlighted with red color. For ADC and SCC, data (mutations and copy number alterations) were obtained from The Cancer Genom Atlas (Firehose Legacy) via www.cbioportal.org (downloaded on 20 December 2023). Only the alterations marked as putative drivers are shown in the picture. SCLC data were obtained from various studies [9,11,12,13,14,15], a subset of which [11,12,13,14] was also downloaded from www.cbioportal.org (downloaded on 20 December 2023). List of abbreviations: ADC, adenocarcinoma; SCC, squamous cell carcinoma; SCLC, small cell lung carcinoma.
The mechanistic/mammalian target of rapamycin (mTOR) is a serine/threonine kinase known as a master regulator of cellular metabolism. It is involved in integrating environmental signals (hormones, growth factors, oxygen, etc.) and the subsequent activation or inhibition of essential cellular pathways related to proliferation, growth, and survival. In the absence of adequate molecular signals or energy sources, it can also initiate cell survival mechanisms, such as autophagy, at the expense of cell growth [16]. The mTOR kinase is tightly linked to the PTEN/PI3K/Akt axis and other essential molecular pathways (e.g., LKB1-AMPK pathway), creating an extensive and complex signaling network [17,18]. Because of its diverse role in regulating essential cellular functions, alterations in mTOR signaling can lead to the development of a wide range of diseases, including metabolic, cardiovascular, or neurodegenerative disorders, accelerated aging, and cancer [19,20].
The mTOR kinase can be a part of two structurally and functionally distinct multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Both complexes share the common subunits mammalian lethal with SEC13 protein 8 (mLST8) and DEP domain-containing mTOR-interacting protein (DEPTOR). Specific protein subunits of mTORC1 are regulatory-associated proteins of mTOR (Raptor), acting as a scaffold protein and proline-rich Akt substrate of 40 kDa (PRAS40). mTORC2 has a rapamycin-insensitive companion of mTOR (Rictor) instead of Raptor, a mammalian stress-activated protein kinase-interacting protein 1 (mSin1), and a protein observed with Rictor 1 and 2 (Protor 1/2) [21]. Having Rictor as a scaffold unit creates a unique spatial structure for mTORC2, as the FKBP-rapamycin binding site is placed inside the molecule, rendering the mTORC2 inaccessible to the conventional mTOR inhibitor drug, rapamycin (also known as sirolimus) [22].
While it is well-known that mTORC1 enables anabolic processes such as protein, lipid, and nucleotide synthesis or ribosome biogenesis and controls cellular metabolism, mTORC2 is also involved in the regulation of a wide range of cellular functions, including bioenergetic processes (see Figure 2) [23]. Although the upstream regulation of mTORC2 remains less well characterized compared to mTORC1, there is emerging evidence that several kinases, such as receptor tyrosine kinases, AMP-activated protein kinases, or small GTPases can act on mTORC2 or its scaffold protein Rictor to regulate the activation of mTORC2 [9,19,23]. Upon activation, mTORC2 can phosphorylate its downstream targets, which include Akt, serum glucocorticoid-regulated kinase 1, and protein kinase C. Of these, Akt protein seems to be one of the most important targets, which, upon phosphorylation (at Ser473), affects the expression of oncogenic transcription factors [17,23]. It has also been described that mTORC2 plays a crucial role in cell migration by regulating the actin cytoskeletal system and epithelial–mesenchymal transition (EMT), which are important aspects of tumor cell invasion and metastasis formation [23,24,25,26,27]. In various types of cancer, mTORC2 and Rictor have been implicated in the regulation of the tumor microenvironment through angiogenesis or stromal remodeling, interacting with vascular endothelial growth factor A, one of the key molecules in these processes [28].
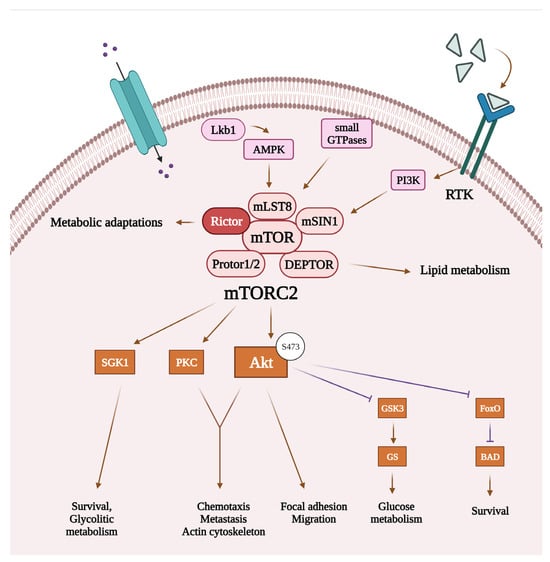
Figure 2.
Cellular functions regulated by mTORC2 and related signaling pathways. Recent studies suggest that mTORC2 can be activated by receptor tyrosine kinases through PI3K signaling, AMP-activated protein kinase, or small GTPases. Once activated, mTORC2’s main targets include Akt, SGK 1, and PKC. By activating its targets, mTORC2 can regulate bioenergetic processes and actin cytoskeleton reorganization, thereby promoting survival and migration of the tumor cells. List of abbreviations: Akt, protein kinase B; AMPK, AMP-activated protein kinase; BAD, BCL2 associated agonist of cell death; DEPTOR, domain-containing mTOR-interacting protein; FoxO, forkhead box protein O1; GS, glycogen synthase; GSK3, glycogen synthase kinase-3; mLST8, mammalian lethal with SEC13 protein 8; mSin1, mammalian stress-activated map kinase-interacting protein 1; mTOR, mammalian target of rapamycin; mTORC2, mTOR complex 2; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; Protor1/2, protein observed with Rictor 1/2; Rictor, rapamycin-insensitive companion of mTOR; RTK, receptor tyrosine kinase; SGK1, serum glucocorticoid-regulated kinase 1.
Hyperactivation of the mTOR complexes resulting from the genetic alterations of the members of the PI3K/Akt/mTOR signaling cascade is frequently seen in solid tumors, including the most common histological subtypes of lung cancer [9,22,29]. The prevalence of mTOR pathway alterations was 50–70% in NSCLCs (90% of ADCs, 40% of SCCs, and 60% of LCCs) and 36% in SCLCs [9,22,30]. Mutational hotspots for amino acid changes in the MTOR gene, encoding for the mTOR kinase, are 1799K, T1977R, V2006F, S2215Y, and R2505P, which could reduce the binding of DEPTOR, the inhibitory subunit of the mTOR complexes, and thus increase the phosphorylation and activation of target molecules, such as p70 S6 kinase and eukaryotic translation initiation factor 4E binding protein 1 [31,32]. Structural changes in the mTOR complexes can also negatively affect the binding to allosteric mTOR inhibitors (such as F2108L mutation of the MTOR gene in the FKBP–rapamycin binding (FRB) domain), leading to therapy resistance [33]. In addition to mutations affecting the mTOR kinase, impaired signaling can occur through genetic alterations involving various other genes encoding for proteins implicated in the PI3K/Akt/mTOR pathway (e.g., PIK3CA, PTEN, AKT1, AKT2, AKT3, TSC1, TSC2, STK11) [23] or affecting one of the subunits of the mTOR complexes (e.g., RICTOR) (see Figure 1) [34].
Identifying the specific molecular changes in the background of mTOR hyperactivation has undeniable clinical benefits, as these alterations could serve as therapeutic targets for selected patients [9,35]. As new therapeutic options have emerged, the need to characterize mTOR activity in malignant tumors has increased, particularly in cancers with elevated mTORC2 activity, which may not respond to traditional mTOR inhibitors that only target mTORC1 [36]. Evaluation of mTOR activity could be achieved by assessing the active phosphorylated form of mTOR (p-mTOR) or its phosphorylated target molecules (e.g., p-4EBP1, p-p70S6K, or p-S6) using immunohistochemistry (IHC). For mTORC2 activity, recent studies suggest that analysis of Rictor and p-Akt (Ser473) expression is the most effective method used to predict the response to mTORC1/2 inhibitors. The mTORC1/mTORC2 ratio can also be assessed by evaluating the ratio of their respective scaffold subunits, Raptor and Rictor, respectively. In summary, the optimal in situ marker combination for assessing mTOR activity in tumors may include p-mTOR, Raptor, and Rictor, as well as an additional target protein of mTORC1 (such as p-S6, p-70S6K, p-4EBP1) and mTORC2 (p-Akt (Ser473)) [37,38]. However, it is important to note that the detection of phosphorylated proteins in formalin-fixed paraffin-embedded sections can be difficult and unreliable in some cases due to the inconsistent sample processing, especially in clinical samples. For example, rapid and proper sample processing is particularly important for the detection of p-Akt (Ser473) protein because of the potential for rapid dephosphorylation [39].
2. Rictor-mTOR Association—The mTORC2 Protein Complex and Its Functions
Rictor (encoded by the RICTOR gene) was identified as a novel component of mTORC2 in 2004 [40]. As a scaffolding subunit, its primary function is to guide the assembly of mTORC2 and maintain its structural integrity [17,20]. One of the well-known oncogenic alterations of the mTOR pathway is the RICTOR amplification, which has been shown to contribute to progression and metastasis in certain cancers through its association with the molecular signaling behind these processes (e.g., Wnt/β-katenin, MAPK/ERK pathways) [41]. Several studies have demonstrated both RICTOR amplification and Rictor overexpression in different types of cancer, including lung cancer [9,17,42,43,44,45,46,47]. Moreover, both RICTOR amplification and Rictor overexpression have been identified to be associated with poor prognosis and shorter survival [17,28,48]. In addition to amplification, a few other mechanisms (e.g., glucose-dependent acetylation or DNA methylation) have also been described to regulate Rictor expression and, therefore, mTORC2 activation [48,49]. However, further investigations are needed to reveal the exact role of these processes.
As previously mentioned, mTORC2 has an important role in the regulation of cell survival and organization of the actin cytoskeleton system. Moreover, besides the extensive roles of the LKB1/AMPK signaling and mTORC1 in the regulation of bioenergetic processes [50,51], mTORC2 has also been implicated in metabolic processes: its activation has been described to be enhanced by amino acid (especially glutamine) withdrawal or glucose starvation [52]. Recent studies have also elucidated that, besides mTORC1, mTORC2 also plays essential functions in the maintenance of metabolic homeostasis [53].
Emerging evidence suggests that the kinase activity of mTORC2 may also depend on the subcellular localization. Recruitment of mTORC2 to the plasma membrane plays an important role in its activation and enables the phosphorylation of one of its major downstream targets, Akt, which is also recruited to the plasma membrane through its pleckstrin homology domain [54]. In addition to its distribution along the plasma membrane, it has also been detected in the cytosol and the nuclei. In the cytosol, mTORC2 can be related to various subcellular structures, such as mitochondria, endosomes, lysosomes, Golgi complex, and endoplasmic reticulum [55,56]. A recent study on glioblastoma cells has suggested that mTORC2 translocates from the plasma membrane to the nuclear and perinuclear compartments when mTORC2 is inactivated [57]. However, there is still controversy over whether mTORC2 can translocate among different subcellular compartments or whether its location is static. The latter is in correlation with the findings that there are distinct forms of mTORC2, which are defined by specific isoforms of the components in the complex, particularly mSin1, and sensitivity to PI3K signaling [58]. This topic has become increasingly complex over the years, and the exact role of subcellular localization in the regulation and functions of mTORC2 remains to be elucidated.
3. Other Rictor-Containing Protein Complexes
Recent findings suggest that, in addition to mTORC2, Rictor may be part of other protein complexes with potential oncogenic or tumor-suppressing properties, further expanding its important role in tumorigenesis. One of the most studied partner proteins is the integrin-linked kinase (ILK), which interacts with β1-integrin. The Rictor/ILK complex has been reported to play an essential role in transforming growth factor beta-1 (TGFβ1)-mediated EMT in vitro, and the complex has also been shown to phosphorylate Akt on Ser473 in an mTOR-independent manner [59]. Other Rictor partners include Culin-1: its complex gains E3 ubiquitin-ligase activity and contributes to the degradation of the mTORC2 effector serum/glucocorticoid-induced kinase 1 [60]. Another Rictor-containing complex in human glioma cells consists of tetraspanin 8 and integrin a3. This complex was found to be required for the assembly and proper function of mTORC2 in glioblastoma cells [61]. Rictor can also interact with the myosin Myo1c and activate paxillin to influence cortical actin remodeling [62]. The co-localization of Rictor and protein kinase Cζ has also been reported, and their complex plays a pivotal role in cancer chemotaxis and metastasis formation in breast cancer [63]. The role of Rictor/F-box WD repeat containing 7 complexes with tumor suppressor properties has also been described in colorectal cancer cells, targeting the oncogenes c-Myc and cyclin E [64]. Moreover, in renal cancer, Rictor can interact with programmed cell death protein 4 at the expense of mTORC2, thereby reducing the metastatic ability of the tumor cells [65]. Although increasing evidence supports the essential role of the Rictor/ILK complex in the regulation of EMT [66,67,68], further studies are still needed to elucidate the clinical significance of these protein complexes. Taken together, it is hypothesized that Rictor acts as a universal scaffold protein and, therefore, can exert different biological functions independent of the mTORC2 complex [17,69].
4. The Role of Rictor in the Epithelial–Mesenchymal Transition and Migration of the Tumor Cells
EMT is a process by which epithelial cells undergo a phenotypic change to become motile mesenchymal cells capable of invasion. For this change to occur, cells have to lose their junctions (prominently E-cadherin) and apical–basal polarity, reorganize the cytoskeleton, and reprogram gene expression and signaling related to cell shape and motility [25]. Mesenchymal–epithelial transition (MET) describes the reverse process in which cells regain their epithelial properties. EMT occurs in physiological conditions such as embryogenesis and wound healing, but it has also been shown to play a crucial role in cancer progression and metastasis formation [69,70]. EMT promotes the dissociation, migration, and invasion of tumor cells, enabling them to metastasize to distant organs, where they revert to their original epithelial nature during MET. Since EMT is known to promote invasiveness and metastatic activity of the tumor, it is strongly correlated with a worse prognosis [71,72]. Several reports suggest that Rictor may play a central role in these processes (see Figure 3) [59,69,73,74,75].
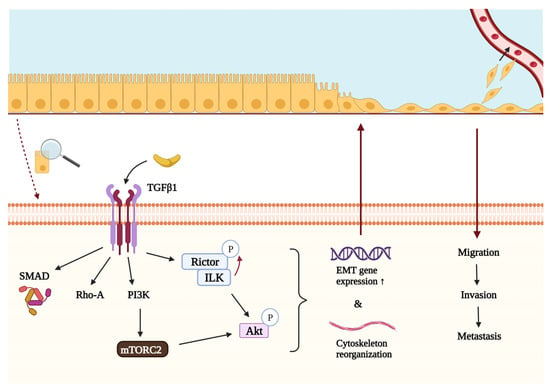
Figure 3.
The role of Rictor in the TGFβ-mediated epithelial–mesenchymal transition and metastasis formation. TGFβ1 is an important regulator of epithelial–mesenchymal transition and thus metastasis formation. In addition to Rho-A and Smad proteins, it can interact with PI3K, which activates mTORC2 and leads to the subsequent phosphorylation of Akt on Ser473 and initiation of downstream effector processes. Akt can also be phosphorylated by another protein complex containing Rictor and ILK. The activity of ILK is also required for the phosphorylation of Rictor in Thr1135, which can regulate the assembly of mTORC2. Once activated by either mTORC2 or the ILK/Rictor complex, Akt regulates cellular functions that can promote epithelial–mesenchymal transition and metastasis formation. List of abbreviations: Akt, protein kinase B; EMT, epithelial–mesenchymal transition; ILK, integrin-linked kinase; mTORC2, mTOR complex 2; PI3K, phosphatidylinositol 3-kinase; Rho-A, Ras homolog family member A; Rictor, rapamycin-insensitive companion of mTOR; SMAD, suppressor of mothers against decapentaplegic; TGFβ1, transforming growth factor β1.
One of the most important mechanisms regulated by mTORC2 is cell migration via reorganization of the actin cytoskeleton, which may contribute to metastasis formation [76]. Recent studies on neutrophil migration have revealed that membrane tension can act on actin nucleation and polymerization to regulate protrusion formation. Increasing membrane tension activates a mechanosensitive signaling cascade and stimulates mTORC2, which controls a negative feedback mechanism in maintaining the homeostasis of membrane stretch [77,78]. It has also been shown in glioblastoma cells that RICTOR knockdown and subsequent inactivation of mTORC2 significantly decrease tumor cell migration by affecting the actin cytoskeleton and microtubule organization [57]. Thus, by integrating divergent cytoskeletal programs, mTORC2 is an essential regulator of cell polarity and migration and, therefore, metastasis formation.
TGFβ1 is a key regulator of growth, differentiation, and epithelial transformation in tumorigenesis, interacting with PI3K, Rho-A, and Smad proteins [25,59]. Another fundamental molecule during TGFβ1-induced EMT is the aforementioned ILK, which connects the cytoplasmic domains of β-integrins to the actin cytoskeleton. When interacting with Rictor, the ILK/Rictor complex can phosphorylate Akt on Ser473, initiating downstream effector processes [66]. It has been described that TGFβ1 can activate mTORC2 to enhance Rictor expression and ILK/Rictor complex formation in an ILK-dependent manner, thereby increasing Akt activation. ILK can also phosphorylate Rictor on Thr1135, a site that is also responsive to growth factor stimuli, PI3K activity, and ribosomal protein S6 kinase 1 activity, which is a downstream effector of mTORC1 (Figure 3). In vitro knockdown of ILK and Rictor suppresses TGFβ1-induced EMT in tumor cells, highlighting the fundamental role of the ILK/Rictor complex in mediating this process. It was also found that the ILK/Rictor complex forms in cancer cells but not in normal cells, making it a promising target for cancer-specific therapy that is not harmful to normal cells [17,59].
Increased ribosome biogenesis regulated by the mTORC2 is also an important factor of EMT. In the G1/S cell cycle arrest during EMT, Rictor has been found to allocate to the nucleoli to associate with the newly formed ribosomes. The mTORC2 complex can also bind to ribosomes, contributing to mesenchymal gene expression [79]. In summary, Rictor is a crucial effector of TGFβ1-induced EMT not only through mTORC2 activation but also individually and as part of the Rictor/ILK complex [59].
5. RICTOR Amplification and/or Rictor Overexpression in Lung Cancer
Identification of RICTOR amplification serves as a predictive marker for effective inhibition of the PI3K/mTOR/Akt pathway, particularly increased mTORC2 activity. The gold standard method for detecting RICTOR amplification is fluorescence in situ hybridization (FISH). Moreover, next-generation sequencing (NGS) or Droplet Digital PCR (ddPCR) can detect the RICTOR copy number variations. In situ, Rictor expression can be assessed by IHC or immunocytochemistry, depending on the sample type [38,80].
RICTOR amplification has been found in 10% of lung ADCs, 16% of lung SCCs, and 23.6% of lung neuroendocrine tumors [17]. In lung ADCs, Rictor overexpression has been described in 37% of the primary cases [37]. RICTOR amplification has been identified as one of the most common targetable genetic alterations in SCLC, with a prevalence ranging from 6 to 15% [9,81]. Moreover, a positive correlation between the presence of RICTOR amplification and Rictor expression has been described in multiple studies [28,38,46]. In some cases, Rictor overexpression has been found at a higher rate than RICTOR amplification, which can be explained by other genetic alterations in the mTOR pathway, epigenetic changes, or miRNA expression aberrations in the tumor cells [82,83].
In the clinical setting, patients with RICTOR-amplified SCLC are expected to have shorter overall survival [81]. Similar results have been found in a study investigating the expression of mTOR pathway markers in lung ADC: Rictor protein expression has been assessed in primary and brain metastatic lung ADCs; Rictor and Rictor/mTOR levels have been increased in the brain metastases, and elevated Rictor expression has also been associated with a higher stage of the primary tumor [17,37]. Similar trends have been observed in SCLC, as Rictor expression has been found to be significantly higher in brain metastases compared to primary tumors and lymph node metastases [38]. In contrast to the above-mentioned study by Sakre et al. [81], this study could not confirm the association between the presence of RICTOR amplification and unfavorable clinical outcome. However, the high expression of both Rictor and p-Akt was associated with shorter overall survival [38]. Moreover, p-Akt and Rictor could also serve as surrogate markers to identify lung cancer patients with an increased probability of responding to double mTORC1/2 inhibitors [47].
These findings further highlight the importance of detecting RICTOR amplification or Rictor overexpression in lung cancer, as they may serve as markers of aggressive clinical behavior and therapy sensitivity, especially in cases in which RICTOR amplification is the only targetable oncogenic driver [38].
6. Selective Inhibitors of mTORC2—Targeting the Association of Rictor and mTOR
Over the past decade, a large number of PI3K/Akt/mTOR pathway inhibitors have been developed (Figure 4), but despite the many ongoing clinical trials, the number of approved drugs remains low. Inadequate patient selection due to a lack of predictive markers may explain this phenomenon. By studying certain genetic alterations (loss-of-function mutation of PTEN, AKT mutation, and RICTOR amplification) or the overexpression of the activated pathway molecules (such as p-S6 or p-Akt), the increased efficacy of mTOR inhibitors could be anticipated [84].
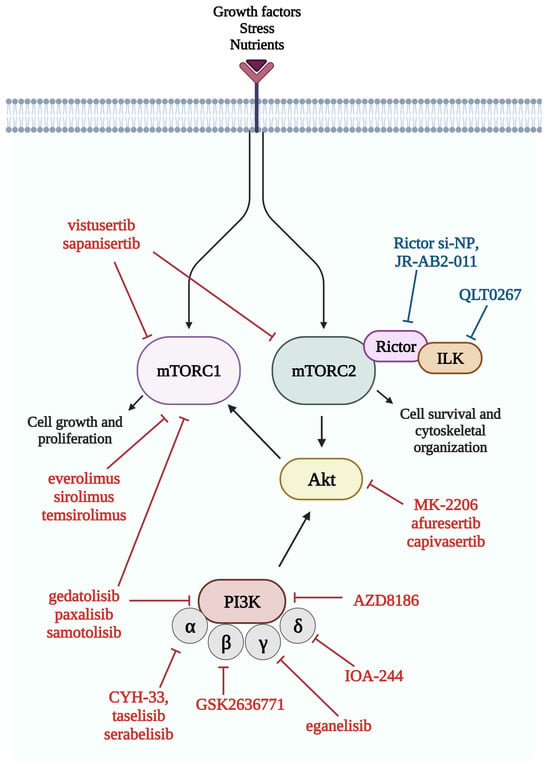
Figure 4.
Members of the mTOR pathway and their inhibitors. Several inhibitors of the mTOR pathway have been developed in recent years, and most of them are currently studied in clinical trials. Red color indicates inhibitors, which are currently studied in clinical trials, while blue color indicates inhibitors in the preclinical phase. List of abbreviations: Akt, protein kinase B; ILK, integrin-linked kinase; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; PI3K, phosphatidylinositol 3-kinase; Rictor, rapamycin-insensitive companion of mTOR.
The most well-known mTOR inhibitor, rapamycin, was originally isolated as an antibiotic and was only later recognized as an immunosuppressant and anti-tumoral agent when a transplant patient with kidney cancer received rapamycin treatment [85,86]. Following the discovery of its target, the mTOR complex, rapamycin (or sirolimus), and its analogs (everolimus, temsirolimus) became therapeutic options for patients with various types of cancer (e.g., breast, kidney, or central nervous system tumors), but clinical results have been highly variable. Moreover, the effect of most mTOR inhibitors in monotherapy is lacking [9,22,84,87,88]. In regard to mTORC1 inhibitors, this phenomenon can be explained by the existence of feedback loop mechanisms; inhibition of the mTORC1/S6K1-mediated negative feedback paradoxically increases PI3K and mTORC2 activity, leading to phosphorylation of Akt [89]. As described, rapamycin binds mTOR through its FKBP domain, which is unavailable in the mTORC2 complex due to structural dissimilarity, so the effect of rapamycin analogs in targeting mTORC2 hyperactivated tumors warrants further investigation [90].
Double kinase inhibitors inhibiting the activity of both mTORC1 and mTORC2 (e.g., CC-115, sapanisertib, vistusertib) and dual kinase inhibitors inhibiting the activity of mTOR kinase and other members of the signaling pathway (e.g., PI3K, such as edatolisib, paxalisib, samatolisib) are under investigation. Akt inhibitors (afuresertib, capivasertib, ipatasertib, TAS-117, triciribin, uprosertib, and in lung cancer: MK-2206) are also being studied in phase II and III trials for the treatment of advanced cancers (NCT01147211). In lung cancer specifically, gedatolisib (a dual inhibitor of PI3K and mTOR) is being tested in a phase I trial in combination with the CDK4/6 inhibitor palbociclib in NSCLC (NCT03065062). mTOR inhibitors combined with immune checkpoint inhibitors are also being evaluated in NSCLC (NCT04348292). The MEK inhibitor selumetinib is being studied in both NSCLC and SCLC in combination with vistusertib and temsirolimus (NCT02583542). In a phase I study, the combination of vistusertib and paclitaxel has been found to increase clinical response in SCC [91].
Specific targeting of mTORC2 while preserving mTORC1 activity remains a pharmacological challenge, although drugs based on siRNA technology (Rictor si-NP) or small molecule inhibitors targeting the mTOR-Rictor interaction (JR-AB2-011) are being investigated in the preclinical setting [92,93]. mTOR inhibitors and their combination with other targeted therapies currently under clinical investigation in lung cancer are shown in Table 1.

Table 1.
Selected clinical trials evaluating inhibitors of the PI3k/Akt/mTOR pathway in lung cancer.
Among lung neoplasms, sporadic lymphangioleiomyomatosis (LAM) is the only entity for which an mTOR inhibitor is approved as a first-line treatment option [94]. The FDA approved sirolimus for the treatment of LAM in 2015, which has served as the cornerstone of therapy ever since. However, it has also been noted that not all subgroups of LAM patients respond well to conventional mTOR inhibition; moreover, intolerable side effects can lead to treatment discontinuation [95,96]. Thus, there is an urgent need to explore alternative therapeutic options for this disease, as well. In addition to mTORC1, evidence for increased mTORC2 activity has also been found in LAM cells, suggesting a potential clinical benefit from the use of dual kinase inhibitors [97]. A study investigating vistusertib in animal models has shown that it inhibits EMT and tumor progression in tuberous sclerosis-associated tumors; however, clinical data on the efficacy of vistusertib in these tumors are not yet available [98].
Despite the clear association between hyperactivation of the PI3K/Akt/mTOR pathway and poor prognosis in many tumor types, including lung cancer, the efficacy of mTOR inhibitors as monotherapy or in combinations with other targeted therapies is unsatisfactory. Furthermore, frequent and sometimes severe side effects, which require careful management by clinicians, continue to hinder its widespread and efficient use [36]. However, the combination of mTOR inhibitors with chemotherapy or radiotherapy can counteract the development of resistance mechanisms and lower the incidence of unwanted side effects. New therapeutic regimens are currently being investigated to increase the efficacy of mTOR inhibitors in inhibiting tumor progression and improving survival [87].
As the understanding of genetic changes leading to the development of lung cancer increases and as predictive markers are being discovered, there is an increasing trend in clinical trials that patients are selected based on the molecular characteristics of the tumor. As an example, vistusertib monotherapy has been tested in specifically selected patients with SCLC who have a confirmed RICTOR amplification. In this study, four patients have been declared eligible, two of whom had their survival extended by almost one year following vistusertib administration [99]. These findings represent a promising avenue for developing novel therapies and individualized treatment [100].
7. Conclusions and Future Perspectives
Despite recent breakthroughs in targeted and immunotherapy of lung cancer, the prognosis for most patients remains poor [1]. Metastasis formation is one of the major determinants of survival and can also significantly affect life quality of the patients [2]. As a part of mTORC2, Rictor has been shown to be an important regulator of EMT and migration of the cancer cells by regulating the actin cytoskeleton, which in turn can facilitate the metastatic ability of the tumor [24,25].
Inhibition of mTORC2 seems to be a promising target in preventing or decreasing metastasis formation [93]. However, emerging results on different Rictor-containing complexes and the role of subcellular localization in the regulation of mTORC2 [55,59] can influence and further complicate the results in both preclinical and clinical studies and still need to be elucidated.
Inhibitors targeting the mTOR kinase, thereby inhibiting the activity of both mTORC1 and mTORC2, are currently studied in clinical trials for the treatment of different histological types of lung cancer (Table 1), but the results are variable. The lack of biomarker-based patient selection could be one of the main reasons behind the inefficacy of these inhibitors in previous studies. However, the start of several biomarker-based clinical trials in recent years is promising and may lead to the clinical translation of these drugs. Selective inhibitors of mTORC2 are also available in preclinical trials with encouraging results.
In conclusion, despite the increasing number of various inhibitors of the mTOR pathway tested in clinical trials, they have not yet achieved a breakthrough in the treatment of lung cancer. Identifying predictive biomarkers may improve the clinical translation of these inhibitors. To predict the response to mTORC2 or mTORC1/C2 inhibitors, RICTOR amplification and overexpression of Rictor and p-Akt are currently considered the best markers [80]. A recent study analyzing RICTOR amplification by next-generation sequencing, droplet digital PCR, and fluorescence in situ hybridization, as well as Rictor and p-Akt expression by immunohistochemistry on a large set of tumors of various origins has demonstrated the superiority of the traditional fluorescence in situ hybridization in the detection of RICTOR amplification. However, other molecular methods as well as Rictor and p-Akt immunohistochemistry can be used to select patients for further analysis [38,80]. Analyzing the mTORC2 activity of the tumor may also predict the metastatic potential of the cancer cells and thus the prognosis, thereby guiding treatment decisions.
Author Contributions
Conceptualization, D.S., F.S., I.K., A.S. and A.K.; methodology, D.S., F.S. and I.K.; data curation, D.S. and F.S.; writing—original draft preparation, D.S., F.S. and I.K.; writing—review and editing, A.K., A.S., J.P. and D.M.; visualization, D.S., F.S. and D.M.; funding acquisition, A.S. and I.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a grant from the Hungarian National Research, Development and Innovation Office (NKFI-FK-128404, AS) and the ÚNKP-23-4-II New National Excellence Program of the Ministry for Culture and Innovation from the Source of the National Research, Development, and Innovation Fund (ÚNKP-23-4-II-SE-9, IK).
Data Availability Statement
A publicly available dataset was analyzed in this study, The Cancer Genom Atlas (Firehose Legacy), via www.cbioportal.org (downloaded on 20 December 2023).
Acknowledgments
Figures were prepared using BioRender (https://biorender.com, accessed on 20 January 2024). Full access was covered by the Department of Pathology and Experimental Cancer Research, Semmelweis University institutional license.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Alexander, M.; Kim, S.Y.; Cheng, H. Update 2020: Management of Non-Small Cell Lung Cancer. Lung 2020, 198, 897–907. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 765. [Google Scholar] [CrossRef]
- Cheng, T.Y.; Cramb, S.M.; Baade, P.D.; Youlden, D.R.; Nwogu, C.; Reid, M.E. The International Epidemiology of Lung Cancer: Latest Trends, Disparities, and Tumor Characteristics. J. Thorac. Oncol. 2016, 11, 1653–1671. [Google Scholar] [CrossRef]
- Bade, B.C.; Dela Cruz, C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef]
- Nicholson, A.G.; Tsao, M.S.; Beasley, M.B.; Borczuk, A.C.; Brambilla, E.; Cooper, W.A.; Dacic, S.; Jain, D.; Kerr, K.M.; Lantuejoul, S.; et al. The 2021 WHO Classification of Lung Tumors: Impact of Advances Since 2015. J. Thorac. Oncol. 2022, 17, 362–387. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Riely, G.J. New pathologic classification of lung cancer: Relevance for clinical practice and clinical trials. J. Clin. Oncol. 2013, 31, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Caliman, E.; Fancelli, S.; Petroni, G.; Gatta Michelet, M.R.; Cosso, F.; Ottanelli, C.; Mazzoni, F.; Voltolini, L.; Pillozzi, S.; Antonuzzo, L. Challenges in the treatment of small cell lung cancer in the era of immunotherapy and molecular classification. Lung Cancer 2023, 175, 88–100. [Google Scholar] [CrossRef]
- Krencz, I.; Sebestyen, A.; Khoor, A. mTOR in Lung Neoplasms. Pathol. Oncol. Res. 2020, 26, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.J.; Razi, S.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/mTOR pathway in lung cancer; oncogenic alterations, therapeutic opportunities, challenges, and a glance at the application of nanoparticles. Transl. Oncol. 2022, 18, 101364. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; de Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef]
- Umemura, S.; Mimaki, S.; Makinoshima, H.; Tada, S.; Ishii, G.; Ohmatsu, H.; Niho, S.; Yoh, K.; Matsumoto, S.; Takahashi, A.; et al. Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. J. Thorac. Oncol. 2014, 9, 1324–1331. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Gkountakos, A.; Pilotto, S.; Mafficini, A.; Vicentini, C.; Simbolo, M.; Milella, M.; Tortora, G.; Scarpa, A.; Bria, E.; Corbo, V. Unmasking the impact of Rictor in cancer: Novel insights of mTORC2 complex. Carcinogenesis 2018, 39, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- Huang, S. mTOR Signaling in Metabolism and Cancer. Cells 2020, 9, 2278. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 4, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, H.; Qu, T.; Luo, J.; An, P.; Ren, F.; Luo, Y.; Li, Y. mTORC2: A multifaceted regulator of autophagy. Cell Commun. Signal. 2023, 21, 4. [Google Scholar] [CrossRef]
- Senoo, H.; Kamimura, Y.; Kimura, R.; Nakajima, A.; Sawai, S.; Sesaki, H.; Iijima, M. Phosphorylated Rho-GDP directly activates mTORC2 kinase towards AKT through dimerization with Ras-GTP to regulate cell migration. Nat. Cell Biol. 2019, 21, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Jebali, A.; Dumaz, N. The role of RICTOR downstream of receptor tyrosine kinase in cancers. Mol. Cancer 2018, 17, 39. [Google Scholar] [CrossRef]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Sebestyén, A.; Dankó, T.; Sztankovics, D.; Moldvai, D.; Raffay, R.; Cervi, C.; Krencz, I.; Zsiros, V.; Jeney, A.; Petővári, G. The role of metabolic ecosystem in cancer progression—Metabolic plasticity and mTOR hyperactivity in tumor tissues. Cancer Metastasis Rev. 2021, 40, 989–1033. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kawazu, M.; Yasuda, T.; Soda, M.; Ueno, T.; Kojima, S.; Yashiro, M.; Yoshino, I.; Ishikawa, Y.; Sai, E.; et al. Transforming somatic mutations of mammalian target of rapamycin kinase in human cancer. Cancer Sci. 2015, 106, 1687–1692. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef]
- Zhao, D.; Jiang, M.; Zhang, X.; Hou, H. The role of RICTOR amplification in targeted therapy and drug resistance. Mol. Med. 2020, 26, 20. [Google Scholar] [CrossRef] [PubMed]
- Iksen; Pothongsrisit, S.; Pongrakhananon, V. Targeting the PI3K/AKT/mTOR Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products. Molecules 2021, 26, 4100. [Google Scholar] [CrossRef]
- Ali, E.S.; Mitra, K.; Akter, S.; Ramproshad, S.; Mondal, B.; Khan, I.N.; Islam, M.T.; Sharifi-Rad, J.; Calina, D.; Cho, W.C. Recent advances and limitations of mTOR inhibitors in the treatment of cancer. Cancer Cell Int. 2022, 22, 284. [Google Scholar] [CrossRef]
- Krencz, I.; Sebestyén, A.; Fábián, K.; Márk, Á.; Moldvay, J.; Khoor, A.; Kopper, L.; Pápay, J. Expression of mTORC1/2-related proteins in primary and brain metastatic lung adenocarcinoma. Hum. Pathol. 2017, 62, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Krencz, I.; Sebestyen, A.; Papay, J.; Lou, Y.; Lutz, G.F.; Majewicz, T.L.; Khoor, A. Correlation between immunohistochemistry and RICTOR fluorescence in situ hybridization amplification in small cell lung carcinoma. Hum. Pathol. 2019, 93, 74–80. [Google Scholar] [CrossRef]
- Baker, A.F.; Dragovich, T.; Ihle, N.T.; Williams, R.; Fenoglio-Preiser, C.; Powis, G. Stability of phosphoprotein as a biological marker of tumor signaling. Clin. Cancer Res. 2005, 11, 4338–4340. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Jiang, W.J.; Feng, R.X.; Liu, J.T.; Fan, L.L.; Wang, H.; Sun, G.P. RICTOR expression in esophageal squamous cell carcinoma and its clinical significance. Med. Oncol. 2017, 34, 32. [Google Scholar] [CrossRef] [PubMed]
- Wazir, U.; Newbold, R.F.; Jiang, W.G.; Sharma, A.K.; Mokbel, K. Prognostic and therapeutic implications of mTORC1 and Rictor expression in human breast cancer. Oncol. Rep. 2013, 29, 1969–1974. [Google Scholar] [CrossRef] [PubMed]
- Joechle, K.; Guenzle, J.; Hellerbrand, C.; Strnad, P.; Cramer, T.; Neumann, U.P.; Lang, S.A. Role of mammalian target of rapamycin complex 2 in primary and secondary liver cancer. World J. Gastrointest. Oncol. 2021, 13, 1632–1647. [Google Scholar] [CrossRef]
- Jebali, A.; Battistella, M.; Lebbé, C.; Dumaz, N. RICTOR Affects Melanoma Tumorigenesis and Its Re-sistance to Targeted Therapy. Biomedicines 2021, 9, 1498. [Google Scholar] [CrossRef] [PubMed]
- Bang, H.; Ahn, S.; Kim, E.J.; Kim, S.T.; Park, H.Y.; Lee, J.; Kim, K.M. Correlation between RICTOR overexpression and amplification in advanced solid tumors. Pathol.-Res. Pract. 2020, 216, 152734. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zou, Y.; Ross, J.S.; Wang, K.; Liu, X.; Halmos, B.; Ali, S.M.; Liu, H.; Verma, A.; Montagna, C.; et al. RICTOR Amplification Defines a Novel Subset of Patients with Lung Cancer Who May Benefit from Treatment with mTORC1/2 Inhibitors. Cancer Discov. 2015, 5, 1262–1270. [Google Scholar] [CrossRef]
- Sun, Y.; Li, R.; Nong, B.; Songyang, Z.; Wang, X.; Ma, W.; Zhou, Q. A Comprehensive Pan-Cancer Analysis of the Potential Biological Functions and Prognosis Values of RICTOR. Genes 2023, 14, 1280. [Google Scholar] [CrossRef]
- Masui, K.; Tanaka, K.; Ikegami, S.; Villa, G.R.; Yang, H.; Yong, W.H.; Cloughesy, T.F.; Yamagata, K.; Arai, N.; Cavenee, W.K.; et al. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc. Natl. Acad. Sci. USA 2015, 112, 9406–9411. [Google Scholar] [CrossRef]
- Goul, C.; Peruzzo, R.; Zoncu, R. The molecular basis of nutrient sensing and signalling by mTORC1 in metabolism regulation and disease. Nat. Rev. Mol. Cell Biol. 2023, 24, 857–875. [Google Scholar] [CrossRef]
- Simcox, J.; Lamming, D.W. The central moTOR of metabolism. Dev. Cell 2022, 57, 691–706. [Google Scholar] [CrossRef]
- Moloughney, J.G.; Kim, P.K.; Vega-Cotto, N.M.; Wu, C.C.; Zhang, S.; Adlam, M.; Lynch, T.; Chou, P.C.; Rabinowitz, J.D.; Werlen, G.; et al. mTORC2 Responds to Glutamine Catabolite Levels to Modulate the Hexosamine Biosynthesis Enzyme GFAT1. Mol. Cell 2016, 63, 811–826. [Google Scholar] [CrossRef]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Luo, Y.; Xu, W.; Li, G.; Cui, W. Weighing In on mTOR Complex 2 Signaling: The Expanding Role in Cell Metabolism. Oxidative Med. Cell. Longev. 2018, 2018, 7838647. [Google Scholar] [CrossRef] [PubMed]
- Ebner, M.; Sinkovics, B.; Szczygieł, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, J.R.; Fritzen, A.M.; James, D.E.; Jensen, T.E.; Kleinert, M.; Richter, E.A. Growth Factor-Dependent and -Independent Activation of mTORC2. Trends Endocrinol. Metab. 2020, 31, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Chantaravisoot, N.; Wongkongkathep, P.; Kalpongnukul, N.; Pacharakullanon, N.; Kaewsapsak, P.; Ariyachet, C.; Loo, J.A.; Tamanoi, F.; Pisitkun, T. mTORC2 interactome and localization determine aggressiveness of high-grade glioma cells through association with gelsolin. Sci. Rep. 2023, 13, 7037. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Serrano, I.; McDonald, P.C.; Lock, F.E.; Dedhar, S. Role of the integrin-linked kinase (ILK)/Rictor complex in TGFβ-1-induced epithelial-mesenchymal transition (EMT). Oncogene 2013, 32, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wan, L.; Inuzuka, H.; Berg, A.H.; Tseng, A.; Zhai, B.; Shaik, S.; Bennett, E.; Tron, A.E.; Gasser, J.A.; et al. Rictor forms a complex with Cullin-1 to promote SGK1 ubiquitination and destruction. Mol. Cell 2010, 39, 797–808. [Google Scholar] [CrossRef]
- Pan, S.J.; Zhan, S.K.; Pan, Y.X.; Liu, W.; Bian, L.G.; Sun, B.; Sun, Q.F. Tetraspanin 8-rictor-integrin α3 complex is required for glioma cell migration. Int. J. Mol. Sci. 2015, 16, 5363–5374. [Google Scholar] [CrossRef] [PubMed]
- Hagan, G.N.; Lin, Y.; Magnuson, M.A.; Avruch, J.; Czech, M.P. A Rictor-Myo1c complex participates in dynamic cortical actin events in 3T3-L1 adipocytes. Mol. Cell. Biol. 2008, 28, 4215–4226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, X.; Li, M.; Chen, P.; Zhang, B.; Guo, H.; Cao, W.; Wei, X.; Cao, X.; Hao, X.; et al. mTOR complex component Rictor interacts with PKCzeta and regulates cancer cell metastasis. Cancer Res. 2010, 70, 9360–9370. [Google Scholar] [CrossRef]
- Guo, Z.; Zhou, Y.; Evers, B.M.; Wang, Q. Rictor regulates FBXW7-dependent c-Myc and cyclin E degradation in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2012, 418, 426–432. [Google Scholar] [CrossRef]
- Bera, A.; Das, F.; Ghosh-Choudhury, N.; Kasinath, B.S.; Abboud, H.E.; Choudhury, G.G. microRNA-21-induced dissociation of PDCD4 from rictor contributes to Akt-IKKβ-mTORC1 axis to regulate renal cancer cell invasion. Exp. Cell Res. 2014, 328, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Tsirtsaki, K.; Gkretsi, V. The focal adhesion protein Integrin-Linked Kinase (ILK) as an important player in breast cancer pathogenesis. Cell Adhes. Migr. 2020, 14, 204–213. [Google Scholar] [CrossRef]
- Górska, A.; Mazur, A.J. Integrin-linked kinase (ILK): The known vs. the unknown and perspectives. Cell. Mol. Life Sci. 2022, 79, 100. [Google Scholar] [CrossRef] [PubMed]
- Almasabi, S.; Ahmed, A.U.; Boyd, R.; Williams, B.R.G. A Potential Role for Integrin-Linked Kinase in Colorectal Cancer Growth and Progression via Regulating Senescence and Immunity. Front. Genet. 2021, 12, 638558. [Google Scholar] [CrossRef]
- Zhou, H.; Guan, Q.; Hou, X.; Liu, L.; Zhou, L.; Li, W.; Liu, H. Epithelial-mesenchymal reprogramming by KLF4-regulated Rictor expression contributes to metastasis of non-small cell lung cancer cells. Int. J. Biol. Sci. 2022, 18, 4869–4883. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Tanaka, Y.; Terai, Y.; Kawaguchi, H.; Fujiwara, S.; Yoo, S.; Tsunetoh, S.; Takai, M.; Kanemura, M.; Tanabe, A.; Ohmichi, M. Prognostic impact of EMT (epithelial-mesenchymal-transition)-related protein expression in endometrial cancer. Cancer Biol. Ther. 2013, 14, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- El Shamieh, S.; Saleh, F.; Moussa, S.; Kattan, J.; Farhat, F. RICTOR gene amplification is correlated with metastasis and therapeutic resistance in triple-negative breast cancer. Pharmacogenomics 2018, 19, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.M.; Dietrich, P.; Hackl, C.; Guenzle, J.; Bronsert, P.; Wagner, C.; Fichtner-Feigl, S.; Schlitt, H.J.; Geissler, E.K.; Hellerbrand, C.; et al. Inhibition of mTORC2/RICTOR Impairs Melanoma Hepatic Metastasis. Neoplasia 2018, 20, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Gulhati, P.; Bowen, K.A.; Liu, J.; Stevens, P.D.; Rychahou, P.G.; Chen, M.; Lee, E.Y.; Weiss, H.L.; O’Connor, K.L.; Gao, T.; et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011, 71, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Thomson, A.W. The “other” mTOR complex: New insights into mTORC2 immunobiology and their implications. Am. J. Transplant. 2019, 19, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Diz-Muñoz, A.; Thurley, K.; Chintamen, S.; Altschuler, S.J.; Wu, L.F.; Fletcher, D.A.; Weiner, O.D. Membrane Tension Acts through PLD2 and mTORC2 to Limit Actin Network Assembly during Neutrophil Migration. PLoS Biol. 2016, 14, e1002474. [Google Scholar] [CrossRef]
- Saha, S.; Town, J.P.; Weiner, O.D. Mechanosensitive mTORC2 independently coordinates leading and trailing edge polarity programs during neutrophil migration. Mol. Biol. Cell 2023, 34, ar35. [Google Scholar] [CrossRef]
- Prakash, V.; Carson, B.B.; Feenstra, J.M.; Dass, R.A.; Sekyrova, P.; Hoshino, A.; Petersen, J.; Guo, Y.; Parks, M.M.; Kurylo, C.M.; et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat. Commun. 2019, 10, 2110. [Google Scholar] [CrossRef]
- Sztankovics, D.; Krencz, I.; Moldvai, D.; Dankó, T.; Nagy, Á.; Nagy, N.; Bedics, G.; Rókusz, A.; Papp, G.; Tőkés, A.M.; et al. Novel RICTOR amplification harbouring entities: FISH validation of RICTOR amplification in tumour tissue after next-generation sequencing. Sci. Rep. 2023, 13, 19610. [Google Scholar] [CrossRef] [PubMed]
- Sakre, N.; Wildey, G.; Behtaj, M.; Kresak, A.; Yang, M.; Fu, P.; Dowlati, A. RICTOR amplification identifies a subgroup in small cell lung cancer and predicts response to drugs targeting mTOR. Oncotarget 2017, 8, 5992–6002. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, A.; Kozaki, K.; Tsuruta, T.; Furuta, M.; Morita, K.; Imoto, I.; Omura, K.; Inazawa, J. The tumor suppressive microRNA miR-218 targets the mTOR component Rictor and inhibits AKT phosphorylation in oral cancer. Cancer Res. 2011, 71, 5765–5778. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.P.; Sehgal, S.N.; Vézina, C. Activity of rapamycin (AY-22,989) against transplanted tumors. J. Antibiot. 1984, 37, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.N.; Baker, H.; Vézina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. 1975, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef]
- Basu, B.; Krebs, M.G.; Sundar, R.; Wilson, R.H.; Spicer, J.; Jones, R.; Brada, M.; Talbot, D.C.; Steele, N.; Ingles Garces, A.H.; et al. Vistusertib (dual m-TORC1/2 inhibitor) in combination with paclitaxel in patients with high-grade serous ovarian and squamous non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Werfel, T.A.; Wang, S.; Jackson, M.A.; Kavanaugh, T.E.; Joly, M.M.; Lee, L.H.; Hicks, D.J.; Sanchez, V.; Ericsson, P.G.; Kilchrist, K.V.; et al. Selective mTORC2 Inhibitor Therapeutically Blocks Breast Cancer Cell Growth and Survival. Cancer Res. 2018, 78, 1845–1858. [Google Scholar] [CrossRef] [PubMed]
- Guenzle, J.; Akasaka, H.; Joechle, K.; Reichardt, W.; Venkatasamy, A.; Hoeppner, J.; Hellerbrand, C.; Fichtner-Feigl, S.; Lang, S.A. Pharmacological Inhibition of mTORC2 Reduces Migration and Metastasis in Melanoma. Int. J. Mol. Sci. 2020, 22, 30. [Google Scholar] [CrossRef] [PubMed]
- Bee, J.; Fuller, S.; Miller, S.; Johnson, S.R. Lung function response and side effects to rapamycin for lymphangioleiomyomatosis: A prospective national cohort study. Thorax 2018, 73, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Revilla-López, E.; Berastegui, C.; Méndez, A.; Sáez-Giménez, B.; Ruiz de Miguel, V.; López-Meseguer, M.; Monforte, V.; Bravo, C.; Pujana, M.A.; Ramon, M.A.; et al. Long-term results of sirolimus treatment in lymphangioleiomyomatosis: A single referral centre experience. Sci. Rep. 2021, 11, 10171. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Mikami, A.; Kitamura, N.; Seyama, K.; Inoue, Y.; Nagai, K.; Suzuki, M.; Moriyama, H.; Akasaka, K.; Tazawa, R.; et al. Efficacy and Safety of Long-Term Sirolimus Therapy for Asian Patients with Lymphangioleiomyomatosis. Ann. Am. Thorac. Soc. 2016, 13, 1912–1922. [Google Scholar] [CrossRef]
- Krencz, I.; Sebestyen, A.; Papay, J.; Jeney, A.; Hujber, Z.; Burger, C.D.; Keller, C.A.; Khoor, A. In situ analysis of mTORC1/2 and cellular metabolism-related proteins in human Lymphangioleiomyomatosis. Hum. Pathol. 2018, 79, 199–207. [Google Scholar] [CrossRef]
- Jones, A.T.; Yang, J.; Narov, K.; Henske, E.P.; Sampson, J.R.; Shen, M.H. Allosteric and ATP-Competitive Inhibitors of mTOR Effectively Suppress Tumor Progression-Associated Epithelial-Mesenchymal Transition in the Kidneys of Tsc2+/- Mice. Neoplasia 2019, 21, 731–739. [Google Scholar] [CrossRef]
- Park, S.; Shim, J.; Mortimer, P.G.S.; Smith, S.A.; Godin, R.E.; Hollingsworth, S.J.; Kim, H.J.; Jung, H.A.; Sun, J.M.; Park, W.Y.; et al. Biomarker-driven phase 2 umbrella trial study for patients with recurrent small cell lung cancer failing platinum-based chemotherapy. Cancer 2020, 126, 4002–4012. [Google Scholar] [CrossRef]
- Zou, Y.; Zheng, H.; Ning, Y.; Yang, Y.; Wen, Q.; Fan, S. New insights into the important roles of phase seperation in the targeted therapy of lung cancer. Cell Biosci. 2023, 13, 150. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).