
Interplay of Ferroptosis and Cuproptosis in Cancer: Dissecting Metal-Driven Mechanisms for Therapeutic Potentials

 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Iron/Copper in Cancers
2.1. Iron Dysregulation in Cancer Cells
2.2. Copper Dysregulation in Cancer Cells
2.3. Iron and Copper Labile Pool
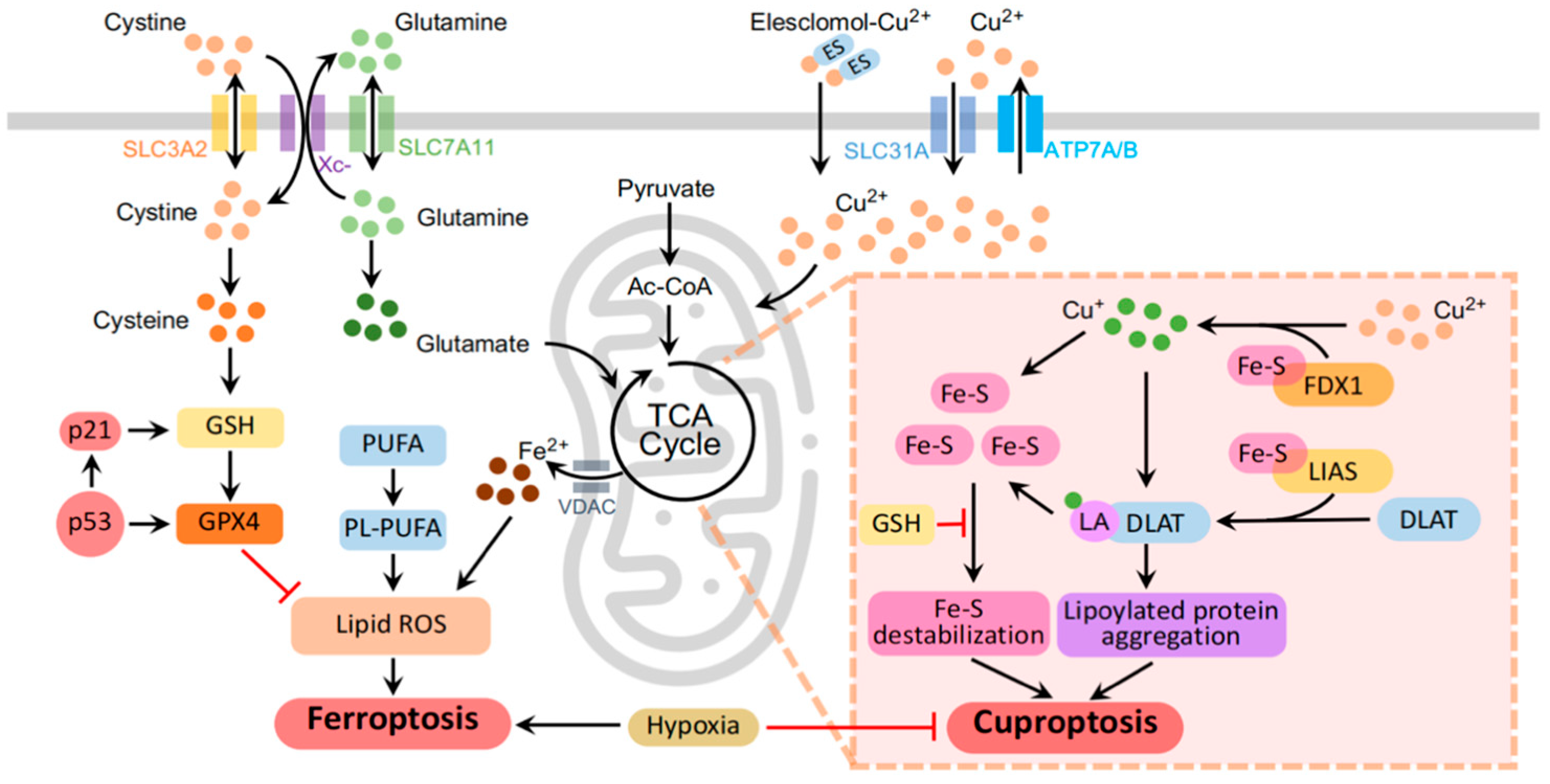
3. Ferroptosis and Cuproptosis
3.1. Ferroptosis
3.1.1. Iron Metabolism
3.1.2. Lipid Peroxidation and ROS
3.1.3. Key Molecules Governing Ferroptosis
3.2. Cuproptosis
3.2.1. Intracellular Copper Accumulation
3.2.2. Aggregation of Lipoylated Proteins and Destabilization of Fe-S Cluster Proteins
3.2.3. Oxidative Phosphorylation Disruption
4. The Role of Mitochondria in Ferroptosis and Cuproptosis
4.1. Mitochondria and Ferroptosis
4.2. Mitochondria and Cuproptosis
4.3. Mitochondrial Interplay in Ferroptosis and Cuproptosis
5. Targeting Ferroptosis/Cuproptosis to Overcome Drug Resistance
5.1. Drug Resistance Related to Iron/Copper
5.2. Persister Cells and CSCs
6. Clinical Trials Targeting Ferroptosis and Cuproptosis
7. Ferroptosis and Cuproptosis in Conventional Treatment
7.1. Ferroptosis as a Novel Approach
7.2. Cuproptosis as a Novel Approach
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Salnikow, K. Role of iron in cancer. Semin. Cancer Biol. 2021, 76, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Kang, R.; Klionsky, D.J.; Tang, D.; Liu, J.; Chen, X. Copper metabolism in cell death and autophagy. Autophagy 2023, 19, 2175–2195. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent Form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Li, Y.; Du, Y.; Zhou, Y.; Chen, Q.; Luo, Z.; Ren, Y.; Chen, X.; Chen, G. Iron and copper: Critical executioners of ferroptosis, cuproptosis and other forms of cell death. Cell Commun. Signal. 2023, 21, 327. [Google Scholar] [CrossRef]
- Wu, Z.-H.; Tang, Y.; Yu, H.; Li, H.-D. The role of ferroptosis in breast cancer patients: A comprehensive analysis. Cell Death Discov. 2021, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, X.; Ge, C.; Min, J.; Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022, 29, 467–480. [Google Scholar] [CrossRef]
- Li, L.; Qiu, C.; Hou, M.; Wang, X.; Huang, C.; Zou, J.; Liu, T.; Qu, J. Ferroptosis in Ovarian Cancer: A Novel Therapeutic Strategy. Front. Oncol. 2021, 11, 665945. [Google Scholar] [CrossRef]
- Jin, X.K.; Liang, J.L.; Zhang, S.M.; Huang, Q.X.; Zhang, S.K.; Liu, C.J.; Zhang, X.Z. Orchestrated copper-based nanoreactor for remodeling tumor microenvironment to amplify cuproptosis-mediated anti-tumor immunity in colorectal cancer. Mater. Today 2023, 68, 108–124. [Google Scholar] [CrossRef]
- Li, Q.; Wang, T.; Zhou, Y.; Shi, J. Cuproptosis in lung cancer: Mechanisms and therapeutic potential. Mol. Cell. Biochem. 2023, 1–13. [Google Scholar] [CrossRef]
- Wang, W.; Lu, K.; Jiang, X.; Wei, Q.; Zhu, L.; Wang, X.; Jin, H.; Feng, L. Ferroptosis inducers enhanced cuproptosis induced by copper ionophores in primary liver cancer. J. Exp. Clin. Cancer Res. 2023, 42, 142. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, D.; Liang, Q.; Yang, M.; Pan, Y.; Li, H. Cross-talk between cuproptosis and ferroptosis regulators defines the tumor microenvironment for the prediction of prognosis and therapies in lung adenocarcinoma. Front. Immunol. 2022, 13, 1029092. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, H.; Lai, Z. The Role of Ferroptosis and Cuproptosis in Curcumin against Hepatocellular Carcinoma. Molecules 2023, 28, 1623. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Quan, Y. Effect of tumor microenvironment on ferroptosis: Inhibition or promotion. Front. Oncol. 2023, 13, 1155511. [Google Scholar] [CrossRef]
- Chang, W.; Li, H.; Zhong, L.; Zhu, T.; Chang, Z.; Ou, W.; Wang, S. Development of a copper metabolism-related gene signature in lung adenocarcinoma. Front. Immunol. 2022, 13, 1040668. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yang, Y.; Gao, Y.; He, J. Cuproptosis: Mechanisms and links with cancers. Mol. Cancer 2023, 22, 46. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef]
- Auerbach, M.; Macdougall, I.C.; Rodgers, G.M.; Deloughery, T.; Richards, T. The iron revolution: Keeping abreast of the developments in iron therapy. Am. J. Hematol. 2022, 97, 250–252. [Google Scholar] [CrossRef]
- Chen, Q.; Ma, X.; Xie, L.; Chen, W.; Xu, Z.; Song, E.; Song, Y. Iron-based nanoparticles for MR imaging-guided ferroptosis in combination with photodynamic therapy to enhance cancer treatment. Nanoscale 2021, 13, 4855–4870. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J. Iron Load Toxicity in Medicine: From Molecular and Cellular Aspects to Clinical Implications. Int. J. Mol. Sci. 2023, 24, 12928. [Google Scholar] [CrossRef] [PubMed]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Brady, D.C. Capturing copper to inhibit inflammation. Nat. Chem. Biol. 2023, 19, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.J.F.; Woolf, C.J.; Weiss, G.; Penninger, J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jin, D.; Zhou, S.; Dong, N.; Ji, Y.; An, P.; Wang, J.; Luo, Y.; Luo, J. Regulatory roles of copper metabolism and cuproptosis in human cancers. Front. Oncol. 2023, 13, 1123420. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and Cancer: 2020 Vision. Cancer Res. 2020, 80, 5435–5448. [Google Scholar] [CrossRef]
- Holbein, B.E.; Lehmann, C. Dysregulated Iron Homeostasis as Common Disease Etiology and Promising Therapeutic Target. Antioxidants 2023, 12, 671. [Google Scholar] [CrossRef]
- Rockfield, S.; Raffel, J.; Mehta, R.; Rehman, N.; Nanjundan, M. Iron overload and altered iron metabolism in ovarian cancer. Biol. Chem. 2017, 398, 995–1007. [Google Scholar] [CrossRef]
- Chang, V.C.; Cotterchio, M.; Khoo, E. Iron intake, body iron status, and risk of breast cancer: A systematic review and meta-analysis. BMC Cancer 2019, 19, 543. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Yanatori, I.; Hara, Y.; Nishina, S. Iron and liver cancer: An inseparable connection. FEBS J. 2022, 289, 7810–7829. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Berghe, T.V. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Müller, S.; Versini, A.; Sindikubwabo, F.; Belthier, G.; Niyomchon, S.; Pannequin, J.; Rodriguez, R. Metformin reveals a mitochondrial copper addiction of mesenchymal cancer cells. PLoS ONE 2018, 13, e0206764. [Google Scholar]
- Tang, X.; Yan, Z.; Miao, Y.; Ha, W.; Li, Z.; Yang, L.; Mi, D. Copper in cancer: From limiting nutrient to therapeutic target. Front. Oncol. 2023, 13, 1209156. [Google Scholar] [CrossRef]
- Cheng, B.; Tang, C.; Xie, J.; Zhou, Q.; Luo, T.; Wang, Q.; Huang, H. Cuproptosis illustrates tumor micro-environment features and predicts prostate cancer therapeutic sensitivity and prognosis. Life Sci. 2023, 325, 121659. [Google Scholar] [CrossRef]
- Safi, R.; Nelson, E.R.; Chitneni, S.K.; Franz, K.J.; George, D.J.; Zalutsky, M.R.; McDonnell, D.P. Copper signaling axis as a target for prostate cancer therapeutics. Cancer Res. 2014, 74, 5819–5831. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef]
- Anandhan, A.; Dodson, M.; Shakya, A.; Chen, J.; Liu, P.; Wei, Y.; Tan, H.; Wang, Q.; Jiang, Z.; Yang, K.; et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 2023, 9, eade9585. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Das, N.K.; Talukder, I.; Singhal, R.; Castillo, C.; Andren, A.; Mancias, J.D.; Lyssiotis, C.A.; Shah, Y.M. PTEN-induced kinase PINK1 supports colorectal cancer growth by regulating the labile iron pool. J. Biol. Chem. 2023, 299, 104691. [Google Scholar] [CrossRef] [PubMed]
- Spangler, B.; Morgan, C.W.; Fontaine, S.D.; Wal, M.N.V.; Chang, C.J.; AWells, J.; Renslo, A.R. A reactivity-based probe of the intracellular labile ferrous iron pool. Nat. Chem. Biol. 2016, 12, 680–685. [Google Scholar] [CrossRef]
- Phiwchai, I.; Thongtem, T.; Thongtem, S.; Pilapong, C. Liver Cancer Cells Uptake Labile Iron via L-type Calcium Channel to Facilitate the Cancer Cell Proliferation. Cell Biochem. Biophys. 2021, 79, 133–139. [Google Scholar] [CrossRef]
- Ackerman, C.M.; Chang, C.J. Copper signaling in the brain and beyond. J. Biol. Chem. 2018, 293, 4628–4635. [Google Scholar] [CrossRef]
- Wang, H.-T.; Ju, J.; Wang, S.-C.; Zhang, Y.-H.; Liu, C.-Y.; Wang, T.; Yu, X.; Wang, F.; Cheng, X.-R.; Wang, K.; et al. Insights Into Ferroptosis, a Novel Target for the Therapy of Cancer. Front. Oncol. 2022, 12, 812534. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2023, 25, 133–155. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Li, J.; Zhang, Y.; Chang, Y.Z. Cellular iron metabolism and regulation. Adv. Exp. Med. Biol. 2019, 1173, 21–32. [Google Scholar] [PubMed]
- Rochette, L.; Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C. The iron-regulatory hormone hepcidin: A possible therapeutic target? Pharmacol. Ther. 2015, 146, 35–52. [Google Scholar] [CrossRef]
- Lv, H.; Shang, P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics 2018, 10, 899–916. [Google Scholar] [CrossRef]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2022, 20, 7–23. [Google Scholar] [CrossRef]
- Morales, M.; Xue, X. Targeting iron metabolism in cancer therapy. Theranostics 2021, 11, 8412–8429. [Google Scholar] [CrossRef]
- Yin, W.; Chang, J.; Sun, J.; Zhang, T.; Zhao, Y.; Li, Y.; Dong, H. Nanomedicine-mediated ferroptosis targeting strategies for synergistic cancer therapy. J. Mater. Chem. B 2023, 11, 1171–1190. [Google Scholar] [CrossRef]
- Gutteridge, J.M.; Halliwell, B. Mini-Review: Oxidative stress, redox stress or redox success? Biochem. Biophys. Res. Commun. 2018, 502, 183–186. [Google Scholar] [CrossRef]
- Zheng, Q.; Li, P.; Zhou, X.; Qiang, Y.; Fan, J.; Lin, Y.; Chen, Y.; Guo, J.; Wang, F.; Xue, H.; et al. Deficiency of the X-inactivation escaping gene KDM5C in clear cell renal cell carcinoma promotes tumorigenicity by reprogramming glycogen metabolism and inhibiting ferroptosis. Theranostics 2021, 11, 8674–8691. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Lee, J.-Y.; Oh, M.; Lee, E.-W. An integrated view of lipid metabolism in ferroptosis revisited via lipidomic analysis. Exp. Mol. Med. 2023, 55, 1620–1631. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-H.; Liu, T.; Wang, H.; Xue, L.-X.; Wang, J.-J. Fatty Acids Metabolism: The Bridge Between Ferroptosis and Ionizing Radiation. Front. Cell Dev. Biol. 2021, 9, 675617. [Google Scholar] [CrossRef] [PubMed]
- Gan, B. ACSL4, PUFA, and ferroptosis: New arsenal in anti-tumor immunity. Signal Transduct. Target. Ther. 2022, 7, 128. [Google Scholar] [CrossRef]
- Tang, D.; Kroemer, G. Peroxisome: The new player in ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 273. [Google Scholar] [CrossRef]
- Qu, G.; Liu, H.; Li, J.; Huang, S.; Zhao, N.; Zeng, L.; Deng, J. GPX4 is a key ferroptosis biomarker and correlated with immune cell populations and immune checkpoints in childhood sepsis. Sci. Rep. 2023, 13, 11358. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free. Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Li, F.J.; Long, H.Z.; Zhou, Z.W.; Luo, H.Y.; Xu, S.G.; Gao, L.C. System Xc−/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar]
- Lee, J.; Roh, J.-L. Targeting GPX4 in human cancer: Implications of ferroptosis induction for tackling cancer resilience. Cancer Lett. 2023, 559, 216119. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, W.; Zhang, W. Ferroptosis and the bidirectional regulatory factor p53. Cell Death Discov. 2023, 9, 197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, W. p53 in ferroptosis regulation: The new weapon for the old guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Tang, D.; Kroemer, G.; Ren, J. Ferroptosis in hepatocellular carcinoma: Mechanisms and targeted therapy. Br. J. Cancer 2023, 128, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, W.D.; Kim, S.K.; Moon, D.H.; Lee, S.J. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 2020, 11, 406. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Lei, G.; Zhang, Y.; Yan, Y.; Mao, C.; Kondiparthi, L.; Gan, B. A targetable CoQ-FSP1 axis drives ferroptosis-and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 2022, 13, 2206. [Google Scholar]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef]
- Chen, L.; Min, J.; Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target. Ther. 2022, 7, 378. [Google Scholar] [CrossRef]
- Tarin, M.; Babaie, M.; Eshghi, H.; Matin, M.M.; Saljooghi, A.S. Elesclomol, a copper-transporting therapeutic agent targeting mitochondria: From discovery to its novel applications. J. Transl. Med. 2023, 21, 745. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, Y.; Shi, H.; Peng, Y.; Fan, X.; Li, C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflügers Arch. Eur. J. Physiol. 2020, 472, 1415–1429. [Google Scholar] [CrossRef]
- Zheng, P.; Zhou, C.; Lu, L.; Liu, B.; Ding, Y. Elesclomol: A copper ionophore targeting mitochondrial metabolism for cancer therapy. J. Exp. Clin. Cancer Res. 2022, 41, 271. [Google Scholar] [CrossRef]
- La Fontaine, S.; Mercer, J.F. Trafficking of the copper-ATPases, ATP7A and ATP7B: Role in copper homeostasis. Arch. Biochem. Biophys. 2007, 463, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Chen, H.H.W.; Song, I.-S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83. [Google Scholar] [CrossRef]
- Li, Z.-H.; Zheng, R.; Chen, J.-T.; Jia, J.; Qiu, M. The role of copper transporter ATP7A in platinum-resistance of esophageal squamous cell cancer (ESCC). J. Cancer 2016, 7, 2085–2092. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef]
- ARowland, E.; Snowden, C.K.; Cristea, I.M. Protein lipoylation: An evolutionarily conserved metabolic regulator of health and disease. Curr. Opin. Chem. Biol. 2018, 42, 76–85. [Google Scholar] [CrossRef]
- Petronek, M.S.; Spitz, D.R.; Allen, B.G. Iron–Sulfur Cluster Biogenesis as a Critical Target in Cancer. Antioxidants 2021, 10, 1458. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Zhang, J.; Yang, Y.; Fleishman, J.S.; Wang, Y.; Wang, H. Cuproptosis: A novel therapeutic target for overcoming cancer drug resistance. Drug Resist. Updates 2024, 72, 101018. [Google Scholar]
- Xiong, C.; Ling, H.; Hao, Q.; Zhou, X. Cuproptosis: p53-regulated metabolic cell death? Cell Death Differ. 2023, 30, 876–884. [Google Scholar] [PubMed]
- Yang, L.; Yang, P.; Lip, G.Y.; Ren, J. Copper homeostasis and cuproptosis in cardiovascular disease therapeutics. Trends Pharmacol. Sci. 2023, 44, 573–585. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Wang, D.; Tian, Z.; Zhang, P.; Zhen, L.; Meng, Q.; Sun, B.; Xu, X.; Jia, T.; Li, S. The molecular mechanisms of cuproptosis and its relevance to cardiovascular disease. Biomed. Pharmacother. 2023, 163, 114830. [Google Scholar] [CrossRef]
- Zhang, S.; Xin, W.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13, 40. [Google Scholar] [CrossRef]
- Yang, P.; Li, J.; Zhang, T.; Ren, Y.; Zhang, Q.; Liu, R.; Li, H.; Hua, J.; Wang, W.-A.; Wang, J.; et al. Ionizing radiation-induced mitophagy promotes ferroptosis by increasing intracellular free fatty acids. Cell Death Differ. 2023, 30, 2432–2445. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [PubMed]
- Otasevic, V.; Vucetic, M.; Grigorov, I.; Martinovic, V.; Stancic, A. Ferroptosis in Different Pathological Contexts Seen through the Eyes of Mitochondria. Oxidative Med. Cell. Longev. 2021, 2021, 5537330. [Google Scholar] [CrossRef]
- Li, J.; Jia, Y.-C.; Ding, Y.-X.; Bai, J.; Cao, F.; Li, F. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int. J. Biol. Sci. 2023, 19, 2756–2771. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2022, 24, 449. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, J.; Kuang, F.; Chen, X.; Zeh, H.J.; Kang, R.; Kroemer, G.; Xie, Y.; Tang, D. PDK4 dictates metabolic resistance to ferroptosis by suppressing pyruvate oxidation and fatty acid synthesis. Cell Rep. 2021, 34, 108767. [Google Scholar] [CrossRef]
- Yuan, H.-J.; Xue, Y.-T.; Liu, Y. Cuproptosis, the novel therapeutic mechanism for heart failure: A narrative review. Cardiovasc. Diagn. Ther. 2022, 12, 681–692. [Google Scholar] [CrossRef]
- Oh, S.J.; Ikeda, M.; Ide, T.; Hur, K.Y.; Lee, M.S. Mitochondrial event as an ultimate step in ferroptosis. Cell Death Discov. 2022, 8, 414. [Google Scholar]
- Panda, S.P.; Kesharwani, A. Micronutrients/miRs/ATP networking in mitochondria: Clinical intervention with ferroptosis, cuproptosis, and calcium burden. Mitochondrion 2023, 71, 1–16. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, W.; Razak, S.R.A.; Han, T.; Ahmad, N.H.; Li, X. Ferroptosis as a potential target for cancer therapy. Cell Death Dis. 2023, 14, 460. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Z.; Ruan, S.; Yan, Q.; Chen, Y.; Cui, J.; Wang, X.; Huang, S.; Hou, B. Regulation of iron metabolism and ferroptosis in cancer stem cells. Front. Oncol. 2023, 13, 1251561. [Google Scholar] [CrossRef]
- Nie, Z.; Chen, M.; Gao, Y.; Huang, D.; Cao, H.; Peng, Y.; Guo, N.; Wang, F.; Zhang, S. Ferroptosis and Tumor Drug Resistance: Current Status and Major Challenges. Front. Pharmacol. 2022, 13, 879317. [Google Scholar] [CrossRef]
- Xia, X.; Fan, X.; Zhao, M.; Zhu, P. The Relationship between Ferroptosis and Tumors: A Novel Landscape for Therapeutic Approach. Curr. Gene Ther. 2019, 19, 117–124. [Google Scholar] [CrossRef]
- Kazan, H.H.; Urfali-Mamatoglu, C.; Gunduz, U. Iron metabolism and drug resistance in cancer. BioMetals 2017, 30, 629–641. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, S.; Zhu, C.; Shen, J. The role of ferroptosis in esophageal cancer. Cancer Cell Int. 2022, 22, 266. [Google Scholar] [CrossRef]
- Majumder, S.; Chatterjee, S.; Pal, S.; Biswas, J.; Efferth, T.; Choudhuri, S.K. The role of copper in drug-resistant murine and human tumors. BioMetals 2009, 22, 377–384. [Google Scholar] [CrossRef]
- Lu, Y.; Pan, Q.; Gao, W.; Pu, Y.; He, B. Reversal of cisplatin chemotherapy resistance by glutathione-resistant copper-based nanomedicine via cuproptosis. J. Mater. Chem. B 2022, 10, 6296–6306. [Google Scholar] [PubMed]
- Cabanos, H.F.; Hata, A.N. Emerging Insights into Targeted Therapy-Tolerant Persister Cells in Cancer. Cancers 2021, 13, 2666. [Google Scholar] [CrossRef] [PubMed]
- Kuşoğlu, A.; Avcı, B. Cancer stem cells: A brief review of the current status. Gene 2019, 681, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Bhattacharyya, S. Cancer Stem Cells: Metabolic Characterization for Targeted Cancer Therapy. Front. Oncol. 2021, 11, 756888. [Google Scholar] [CrossRef]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer 2022, 22, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Schreiber, S.L.; Conrad, M. Persister cancer cells: Iron addiction and vulnerability to ferroptosis. Mol. Cell 2022, 82, 728–740. [Google Scholar] [CrossRef]
- Schwartz, A.J.; Goyert, J.W.; Solanki, S.; Kerk, S.A.; Chen, B.; Castillo, C.; Shah, Y.M. Hepcidin sequesters iron to sustain nucleotide metabolism and mitochondrial function in colorectal cancer epithelial cells. Nat. Metab. 2021, 3, 969–982. [Google Scholar]
- Phipps, O.; Brookes, M.J.; Al-Hassi, H.O. Iron deficiency, immunology, and colorectal cancer. Nutr. Rev. 2021, 79, 88–97. [Google Scholar]
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Ozdian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 2017, 552, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, F.; Chen, J.; Chan, S.; He, Y.; Liu, W.; Zhang, G. Disulfiram/Copper Induces Antitumor Activity against both Nasopharyngeal Cancer Cells and Cancer-Associated Fibroblasts through ROS/MAPK and Ferroptosis Pathways. Cancers 2020, 12, 138. [Google Scholar]
- Mynott, R.L.; Habib, A.; Best, O.G.; Wallington-Gates, C.T. Ferroptosis in Haematological Malignancies and Associated Therapeutic Nanotechnologies. Int. J. Mol. Sci. 2023, 24, 7661. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef]
- Yee, P.P.; Wei, Y.; Kim, S.-Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, S.; He, G.; Chen, T.; Li, X.; Liang, Y.; Wu, W.; Weng, L.; Feng, J.; Gao, Z.; Yang, K. Emerging role of ferroptosis in glioblastoma: Therapeutic opportunities and challenges. Front. Mol. Biosci. 2022, 9, 974156. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. The Art of War: Ferroptosis and Pancreatic Cancer. Front. Pharmacol. 2021, 12, 773909. [Google Scholar] [CrossRef]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Zeh, H.J.; Kang, R.; Tang, D. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat. Commun. 2020, 11, 6339. [Google Scholar] [PubMed]
- Kim, J.W.; Kim, M.-J.; Han, T.-H.; Lee, J.-Y.; Kim, S.; Kim, H.; Oh, K.-J.; Kim, W.K.; Han, B.-S.; Bae, K.-H.; et al. FSP1 confers ferroptosis resistance in KEAP1 mutant non-small cell lung carcinoma in NRF2-dependent and -independent manner. Cell Death Dis. 2023, 14, 567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.; Li, Q.; Xu, A.; Hu, Y.; Sun, C. Ferroptosis in hematological malignancies and its potential network with abnormal tumor metabolism. Biomed. Pharmacother. 2022, 148, 112747. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fang, Z.-M.; Yi, X.; Wei, X.; Jiang, D.-S. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. 2023, 14, 205. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Wang, Y.; Lin, Y.; Mao, Q.; Feng, J.; Gao, G.; Jiang, J. Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci. Ther. 2019, 25, 465–475. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Liu, M.-R.; Shi, C.; Song, Q.-Y.; Kang, M.-J.; Jiang, X.; Liu, H.; Pei, D.-S. Sorafenib induces ferroptosis by promoting TRIM54-mediated FSP1 ubiquitination and degradation in hepatocellular carcinoma. Hepatol. Commun. 2023, 7, e0246. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Kołat, D.; Kałuzińska-Kołat, Ż.; Celik, I.; Kontek, R. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells 2023, 12, 659. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, Q.; Lin, X.; Shu, P.; Gao, X.; Shen, K. Imatinib induces ferroptosis in gastrointestinal stromal tumors by promoting STUB1-mediated GPX4 ubiquitination. Cell Death Dis. 2023, 14, 839. [Google Scholar] [CrossRef]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [PubMed]
- Chen, P.; Li, X.; Zhang, R.; Liu, S.; Xiang, Y.; Zhang, M.; Xie, T. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 2020, 10, 5107. [Google Scholar]
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2020, 384, 121390. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Z.; Liu, K.; Jiang, D.; Sun, X.; Mao, Y.; Li, S.; Ye, D. Circulating Copper and Liver Cancer. Biol. Trace Elem. Res. 2023, 201, 4649–4656. [Google Scholar] [CrossRef] [PubMed]
- Ramchandani, D.; Berisa, M.; Tavarez, D.A.; Li, Z.; Miele, M.; Bai, Y.; Lee, S.B.; Ban, Y.; Dephoure, N.; Hendrickson, R.C.; et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat. Commun. 2021, 12, 7311. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Tumor Type | Agent | Role | Phases | Indication | Refs |
|---|---|---|---|---|---|
| Breast cancer | Sorafenib | System Xc- inhibitors | phase II | Metastatic BC | NCT00101400 |
| Artesunate | Iron oxidizing agents | phase I | Metastatic BC | NCT00764036 | |
| Neratinib | Iron activators | phase II | HER2 positive BC | NCT00300781 | |
| phase II | ER positive BC | NCT05933395 | |||
| DFO | Iron chelators | phase II | Metastatic Triple Negative BC | NCT05300958 | |
| Colon cancer | Mebendazole | System Xc- inhibitors | phase III | Stage IV CRC | NCT03925662 |
| Sulfasalazine | System Xc- inhibitors | phase III | Metastatic CRC | NCT06134388 | |
| Sorafenib | System Xc- inhibitors | phase II | EGFR positive metastatic CRC with mutant KRAS | NCT00326495 | |
| Curcuminoids | Combined indirect GPX4 inhibitors and HO-1 inducers | NA | Colorectal mucosa of subjects with previously resected adenomatous clonic polyps | NCT00118989 | |
| phase I | CRC | NCT00027495 | |||
| Artesunate | Iron oxidizing agents | phase II | Stage II/III CRC | NCT02633098 NCT03093129 | |
| Liver cancer | Lcaritin | Others | phase II | Unresectable, non-metastatic HCC | NCT05903456 |
| Mebendazole/ Lenvatinib | System Xc- inhibitors | NA | Cirrhotics with advanced HCC | NCT04443049 | |
| Ferumoxytol | Iron replacement product | NA | Primary and metastatic liver tumors and hepatic cirrhosis eligible for liver SBRT | NCT04682847 | |
| DFO | Iron chelators | phase I | Unresectable HCC | NCT03652467 | |
| Gastric cancer | Sorafenib | System Xc- inhibitors | phase I | Unresectable, recurrent GC | NCT00663741 |
| phase II | Advanced GC | NCT01187212 | |||
| Lung cancer | Sorafenib | System Xc- inhibitors | phase II | Relapsed NSCLC | NCT00098254 |
| phase II | Extensive stage SCLC | NCT00182689 | |||
| Cisplatin | GSH inhibitor | Phase IV | Advanced Squamous NSCLC | NCT05312840 | |
| Phase IV | Unresectable advanced, metastatic or recurrent non-squamous NSCLC | NCT02316327 | |||
| Glioma | L-alanosine | System Xc- inhibitors | phase I/II | High-grade progressive or recurrent malignant gliomas | NCT00075894 |
| Mebendazole | System Xc- inhibitors | phase I | high-grade glioma | NCT01729260 | |
| Prostate cancer | Quercetin/ Genistein | Others | NA | Rising Prostate-specific Antigen | NCT01538316 |
| Sorafenib | System Xc- inhibitors | phase I | Metastatic, androgen-independent PC | NCT00090545 | |
| phase II | Metastatic or recurrent PC | NCT00093457 | |||
| DFO | Iron chelators | phase I/II | STEAP1 positive PCC | NCT01774071 | |
| Head and neck cancer | Sorafenib | System Xc- inhibitors | phase II | Recurrent and/or metastatic head and neck cancer | NCT00199160 |
| 5-aminolevulinic acid | Combined indirect GPX4 inhibitors and HO-1 inducers | phase II | Newly diagnosed or recurrent malignancies | NCT05101798 | |
| Ovarian cancer | Withaferin A | GPX4 inhibitors | phase I/II | Recurrent OC | NCT05610735 |
| Neratinib | Iron activators | phase I | Platinum-resistant OC | NCT04502602 | |
| Thyroid cancer | Sorafenib | System Xc- inhibitors | phase II | metastatic or recurrent TC | NCT02084732 |
| Melanoma | Buthionine sulfoximine | GCL inhibitor | phase I | Persistent or recurrent stage III malignant melanoma | NCT00661336 |
| Tumor Type | Agent | Role | Phases | Indication | Refs |
|---|---|---|---|---|---|
| Breast cancer | Disulfiram | Copper supplementation | phase II | Metastatic BC upon failure of conventional systemic and/or locoregional therapies | NCT03323346 |
| phase II | CTC_EMT positive refractory metastatic hormone receptor positive, HER2 negative BC | NCT04265274 | |||
| ATN-224 | Copper depletion | phase II | Recurrent or advanced, oestrogen and progesterone receptor positive BC | NCT00674557 | |
| Tetrathiomolybdate | Copper depletion | phase II | BC with moderate-to-high risk of recurrence | NCT00195091 | |
| phase I/II | High risk for relapse triple negative BC | NCT06134375 | |||
| Disulfiram | Copper supplementation | phase II | Metastatic BC upon failure of conventional systemic and/or locoregional therapies | NCT03323346 | |
| phase II | CTC_EMT positive refractory metastatic hormone receptor positive, HER2 negative BC | NCT04265274 | |||
| Prostate cancer | Disulfiram | Copper supplementation | NA | Recurrent PC | NCT01118741 |
| Phase Ib | Metastatic castrate-resistant PC | NCT02963051 | |||
| Elesclomol | Copper supplementation | phase I | Metastatic castration refractory PC | NCT00808418 | |
| ATN-224 | Copper depletion | phase II | Relapsed, early-stage PC not on hormone therapy | NCT00405574 | |
| phase II | Hormone refractory PC | NCT00150995 | |||
| Tetrathiomolybdate | Copper depletion | phase II | Hormone refractory PC | NCT00150995 | |
| Lung cancer | Disulfiram | Copper supplementation | phase II/III | Advanced NSCLC | NCT00312819 |
| Elesclomol | Copper supplementation | phase I/II | Chemotherapy naive patients with stage IIIB or stage IV NSCLC | NCT00088088 | |
| Tetrathiomolybdate | Copper depletion | phase I | Chemo-naive metastatic or recurrent NSCLC | NCT01837329 | |
| phase I | Stage I-IIIB NSCLC | NCT00560495 | |||
| Melanoma | Disulfiram | Copper supplementation | phase I/II | Metastatic melanoma | NCT00256230 |
| Phase Ib | Metastatic melanoma | NCT00571116 | |||
| Elesclomol | Copper supplementation | phase III | Metastatic melanoma | NCT00522834 | |
| phase I/II | Metastatic melanoma | NCT00084214 | |||
| ATN-224 | Copper depletion | phase II | Advanced melanoma | NCT00383851 | |
| Trientine | Copper depletion | phase I | BRAF-mutated metastatic melanoma | NCT02068079 | |
| Glioblastoma | Disulfiram | Copper supplementation | phase I/II | Suspected glioblastoma or recurrent glioblastoma undergoing surgical resection | NCT03151772 NCT02715609 |
| phase II | Recurrent glioblastoma | NCT03034135 | |||
| phase II | Newly diagnosed GBM | NCT01777919 | |||
| phase II | Unmethylated GBM | NCT03363659 | |||
| phase II/III | Recurrent GBM receiving alkylating chemotherapy | NCT02678975 | |||
| Gastric cancer | Disulfiram | Copper supplementation | NA | Advanced GC | NCT05667415 |
| Pancreatic cancer | Disulfiram | Copper supplementation | phase II | Metastatic PC with rising CA19-9 on Abraxane-Gemzar, FOLFIRINOX, or Gemcitabine | NCT03714555 |
| Liver cancer | Tetrathiomolybdate | Copper depletion | phase II | HCC | NCT00006332 |
| Colon cancer | Tetrathiomolybdate | Copper depletion | phase II | Metastatic CRC | NCT00176774 |
| Ovarian cancer | Trientine | Copper depletion | phase I/II | Latinum-resistant/refractory epithelial ovarian cancer | NCT03480750 |
| Head and neck cancer | Penicillamine | Copper depletion | phase II | Recurrent head and neck cancer | NCT06103617 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Li, J.; Liu, J.; Chan, K.-Y.; Lee, H.-S.; Lin, K.N.; Wang, C.-C.; Lau, T.-S. Interplay of Ferroptosis and Cuproptosis in Cancer: Dissecting Metal-Driven Mechanisms for Therapeutic Potentials. Cancers 2024, 16, 512. https://doi.org/10.3390/cancers16030512
Wang J, Li J, Liu J, Chan K-Y, Lee H-S, Lin KN, Wang C-C, Lau T-S. Interplay of Ferroptosis and Cuproptosis in Cancer: Dissecting Metal-Driven Mechanisms for Therapeutic Potentials. Cancers. 2024; 16(3):512. https://doi.org/10.3390/cancers16030512
Chicago/Turabian StyleWang, Jinjiang, Jiaxi Li, Jiao Liu, Kit-Ying Chan, Ho-Sze Lee, Kenneth Nansheng Lin, Chi-Chiu Wang, and Tat-San Lau. 2024. "Interplay of Ferroptosis and Cuproptosis in Cancer: Dissecting Metal-Driven Mechanisms for Therapeutic Potentials" Cancers 16, no. 3: 512. https://doi.org/10.3390/cancers16030512
APA StyleWang, J., Li, J., Liu, J., Chan, K.-Y., Lee, H.-S., Lin, K. N., Wang, C.-C., & Lau, T.-S. (2024). Interplay of Ferroptosis and Cuproptosis in Cancer: Dissecting Metal-Driven Mechanisms for Therapeutic Potentials. Cancers, 16(3), 512. https://doi.org/10.3390/cancers16030512