Simple Summary
KMT2A partial tandem duplication (PTD) involves intragenic duplications within the KMT2A gene and has been associated with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). KMT2A PTD cannot be detected by conventional karyotyping or fluorescence in situ hybridization (FISH). In this study, we used optical genome mapping (OGM) to analyze the cytogenomic alterations in 1277 hematolymphoid neoplasms and identified KMT2A PTD exclusively in patients with myeloid neoplasms, including 35 (7%) with AML, 5 (2.2%) with MDS, and 5 (7.2%) with chronic myelomonocytic leukemia (CMML). Neoplasms with KMT2A PTD frequently exhibit a normal or non-complex karyotype and consistently harbor gene mutations involving epigenetic regulators, the FLT3/RAS signaling pathway, transcription factors, and spliceosome genes. Patients with KMT2A PTD are generally resistant to conventional chemotherapy, with the exception of those with de novo AML, which demonstrates a high remission rate. Patients with KMT2A PTD positive secondary AML and MDS had a poor outcome.
Abstract
KMT2A partial tandem duplication (PTD) involves intragenic KMT2A duplications and has been associated with poorer prognosis. In this study, we evaluated KMT2A PTD in 1277 patients with hematological malignancies using optical genome mapping (OGM). KMT2A PTD was detected in 35 patients with acute myeloid leukemia (AML) (7%), 5 patients with myelodysplastic syndrome (MDS) (2.2%), and 5 patients with chronic myelomonocytic leukemia (CMML) (7.1%). The PTDs varied in size, region, and copy number. An Archer RNA fusion assay confirmed KMT2A PTD in all 25 patients tested: 15 spanning exons 2 to 8 and 10 spanning exons 2 to 10. Most patients exhibited a normal (n = 21) or non-complex (n = 20) karyotype. The most common chromosomal abnormalities included loss of 20q or 7q and trisomy 11/gain of 11q. All patients had gene mutations, with FLT3 ITD and DNMT3A prevalent in AML and DNMT3A and RUNX1 common in MDS and CMML. Among patients who received treatment and had at least one follow-up bone marrow evaluation, 82% of those with de novo AML achieved complete remission after initial induction chemotherapy, whereas 90% of patients with secondary or refractory/relapsed AML showed refractory or partial responses. All but one patient with MDS and CMML were refractory to therapy. We conclude that OGM is an effective tool for detecting KMT2A PTD. Neoplasms with KMT2A PTD frequently harbor gene mutations and display normal or non-complex karyotypes. Patients with KMT2A PTD are generally refractory to conventional therapy, except for de novo AML.
1. Introduction
KMT2A (also known as MLL), located on chromosome 11q23, is frequently rearranged in pediatric and adult leukemias across different lineages [1]. Unlike KMT2A fusion events, KMT2A partial tandem duplication (PTD) is an intragenic in-frame duplication within the N-terminal region of KMT2A. The breakpoints often occur in flanking intronic sequences of exons 2 to 8 or 10 and are often facilitated by Alu elements [2,3,4]. This alteration occurs in approximately 5–10% of cases of acute myeloid leukemia (AML) [2] and 3–5% of myelodysplastic syndromes (MDS) [5]. KMT2A PTD is often associated with AML with a normal karyotype and is enriched in AML with trisomy 11 as a sole chromosome aberration [4]. Studies suggest that KMT2A-PTD alone is insufficient to cause AML and additional genetic aberrations are required for the development of KMT2A PTD-associated leukemia [6,7]. Multiple studies have shown that KMT2A PTD AML is often associated with FLT3 ITD and the RAS signaling pathway. It frequently co-occurs with other mutations in genes of epigenetic regulators (e.g., DNMT3A and IDH1/IDH2), transcription factors (e.g., RUNX1), and spliceosome genes (e.g., U2AF1 and SRSF2) [8,9,10].
The prognosis impact of KMT2A PTD AML remains controversial [11]. In MDS, KMT2A-PTD is associated with excess blasts, a high risk of AML transformation, and poorer overall survival [5,12,13]. It is recognized as a high-risk molecular feature in the International Prognostic Scoring System-Molecular (IPSS-M) [12]. To date, the study of KMT2A PTD in other hematological neoplasms is scarce, including chronic myelomonocytic leukemia (CMML).
KMT2A PTD has been shown to drive HOX overexpression in both mouse models and AML with KMT2A PTD [14,15], suggesting potential therapeutic implications, particularly for the use of menin inhibitors [16]. At our institution, we are conducting a phase 2 study of revumenib for relapsed/refractory (R/R) disease targeting high HOX expression, including cases with KMT2A PTD. Detecting KMT2A PTD may help identify patients eligible for menin inhibitor therapies.
Detecting KMT2A PTD is challenging because it is a cryptic intragenic alteration that is not detectable by conventional karyotyping or fluorescence in situ hybridization (FISH) analysis [3]. Additionally, these intragenic duplications exceed the amplification capacity of traditional polymerase chain reaction (PCR) strategies. Furthermore, a recent report described rare AML cases with both KMT2A rearrangement and KMT2A PTD, where KMT2A rearrangement likely represented the founder clone, further complicating the detection of KMT2A PTD [17]. Historically, methods such as Southern blot hybridization [18,19], reverse transcription (RT) PCR, complementary cDNA sequencing [2,3], and cytogenomic microarray [13] have been employed, but there is a growing interest in employing DNA- or RNA-based next-generation sequencing (NGS) assays for routine clinical testing to detect KMT2A PTD [20,21,22].
Optical genome mapping (OGM) has emerged as a novel technology that enables high-resolution, comprehensive analysis of the whole genome, including intragenic gene duplications. To date, only one study has used OGM as an orthogonal method to confirm KMT2A PTD during the validation of a DNA-based NGS assay in a limited number of cases; the results suggested that OGM may be a superior tool for detecting KMT2A PTD compared to NGS [23].
In this study, we used OGM to evaluate the cytogenomic changes in a large cohort of patients with hematolymphoid tumors. KMT2A PTD was detected exclusively in myeloid neoplasms, including AML, MDS, and CMML. We analyzed the cytogenetic and molecular profiles of these neoplasms and further assessed the patients’ associated clinical characteristics, treatment responses, and outcomes.
2. Materials and Methods
2.1. Patients
We reviewed hematologic malignancies tested by OGM at our institution between 1 November 2022 and 15 October 2024 and collected the cases that showed KMT2A PTD. Of note, a part of these patients have been published previously [24]. Clinical information was retrieved from the electronic medical records. This study was approved by the Institutional Review Board of MD Anderson Cancer Center and was conducted in accord with the Declaration of Helsinki.
2.2. Conventional Chromosome Analysis
G-banded chromosomal analysis was performed as part of our routine diagnostic workup for hematological malignancies. Twenty Giemsa-banded metaphase cells were analyzed, and the results were reported using the International System for Human Cytogenetic Nomenclature (ISCN 2020). A complex karyotype was defined as ≥3 unrelated chromosomal abnormalities with at least one structural abnormality.
2.3. Optical Genome Mapping
OGM was performed on fresh peripheral blood or bone marrow aspirate specimens following the manufacturer’s protocol (Bionano Genomics, San Diego, CA, USA), as previously described [24]. In brief, ultra-high-molecular-weight genomic DNA (UHMW gDNA) was extracted from approximately 1.5 million cells and labeled using the Bionano Prep Direct Label and Stain (DLS) kit, which tags DNA with a fluorescent label and stains the DNA backbone. The labeled gDNA was imaged using the Bionano Saphyr, acquiring approximately 1500 Gb of DNA per sample. DNA molecules were then assembled into a genome with >400× coverage using the Bionano Solve software (version Solve3.8.2). Data analysis was performed using the Rare Variant Analysis Pipeline in Bionano Access (version 1.8.2), focusing on clinically relevant genes and loci based on the HemeTargets and hg38-primary_transcripts feature files [24]. KMT2A PTD was identified as an intragenic insertion and/or intragenic duplication of KMT2A, based on the patterns shown in Figure 1 and Figure S1. Although OGM identified an insertion, ins(11;?), in most cases, manual examination revealed that these insertions were duplications or triplications of genic fragments within KMT2A.
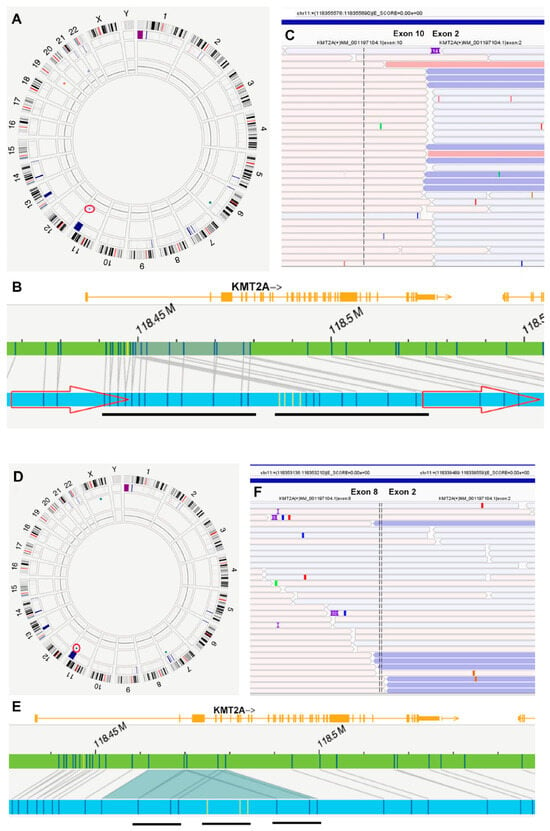
Figure 1.
Detection of KMT2A PTD by optical genome mapping (OGM) and Archer RNA fusion assay. A–C (Case #18): The OGM Circos plot (A) indicates a duplication (circled) on chromosome 11q23, with detail provided in Genome Browser (B). OGM identified this as dup(11)(q23.3q23.3), with coordinates RefStartPos 118,448,714 and RefEndPos 118,479,068, size 30,354 bp, and a VAF of 0.79. The duplicated sequence, verified by manual examination, is underlined. The RNA fusion assay demonstrates a fusion between exon 10 and exon 2 of KMT2A (C). (D–F) (Case #5): The OGM Circos plot (D) shows an insertion (circled), ins(11q23;?), on chromosome 11q23, with detail provided in the Genome Browser (E). The “unknown” inserted sequence, marked in yellow by OGM, has coordinates RefStartPos 118,461,867 and RefEndPos 118,479,068, size 31,007 bp, and a VAF of 0.78. Manual examination reveals an “insertion” and duplication of the same fragment, resulting in a triplication. The triplicated sequence is underlined. The RNA fusion assay shows a fusion between exon 8 and exon 2 of KMT2A (F).
2.4. Next-Generation Sequencing
DNA-based NGS for somatic mutations was performed on all cases as part of our routine diagnostic workup, targeting all exonic or “hotspot” regions of 81 genes commonly mutated in myeloid neoplasms, as previously described [24,25].
2.5. FLT3 ITD and TKD Analysis
A multiplex fluorescent-based PCR analysis followed by capillary electrophoresis was performed to detect FLT3 internal tandem duplication (ITD) and/or tyrosine kinase domain (TKD) mutations on DNA isolated from BM aspirate samples, as described previously [26].
2.6. Archer RNA Fusion Assay
The EndLeukemia RNA Translocation Assay is an NGS-based analysis interrogating 106 genes with the ability to detect targeted inter- and intragenic translocations using RNA extracted from peripheral blood or bone marrow aspirate specimens. Target enrichment is achieved through anchored multiplex PCR (AMP), and sequencing is performed bidirectionally on an Illumina sequencer. The resulting sequencing reads are aligned with the human reference genome GRCh37/hg19, and structural variants are identified using Archer Analysis Software v6.2.7. The analytical sensitivity of this assay is estimated at 100% in validation studies involving samples with at least 5% disease involvement. This assay detects KMT2A PTD as an abnormal KMT2A exonic junction between upstream and downstream exons.
3. Results
3.1. Patients
A total of 1277 patients with hematolymphoid tumors were evaluated by OGM during the study interval, including 855 patients with myeloid neoplasms and 422 patients with B- and T-cell neoplasms. The myeloid neoplasms included 502 AML, 228 MDS, 38 myeloproliferative neoplasms (MPN), and 87 MDS/MPN (including 70 CMML). KMT2A PTD was detected in 45 patients: 35 (7%) with AML, 5 (2.2%) with MDS, and 5 (7.1%) with CMML. The diagnosis and demographic features of the patients with KMT2A PTD are summarized in Table 1. There were 31 men and 14 women, with a median age of 68 years (range, 23–86). Among the AML patients, 14 were newly diagnosed (de novo), 14 were refractory/relapsed (R/R), and 7 had a prior history of myeloid neoplasm (secondary AML, sAML). The sAML cases were preceded by MDS (n = 5), CMML (n = 1), and polycythemia vera (n = 1) (Table 1). Of note, 11 (31%) AML cases showed myelomonocytic or monoblastic differentiation.

Table 1.
Demographic and clinicopathological features of neoplasms with KMT2A PTD.
3.2. Detection of KMT2A PTD by OGM and Confirmation by Archer RNA Fusion Assay
OGM detected an intragenic “insertion” of KMT2A in 41 cases and intragenic duplications of KMT2A in 4 cases, both corresponding to KMT2A PTD (Table 2, Figure 1, Figure S1 and Table S1). Although OGM often called these events an “insertion” of unknown material, ins(11;?)(q23.3;?), most cases were recognized as repeats of sequence based on the label patterns by manual review. A manual review showed that the PTD spanned from intron 1, exon 2, or intron 2 to intron 5 (or exon 6) in all cases. The number of repeats ranged from two to four (two repeats in 26 cases, five in 17 cases, and four in 2 cases) (Figure 1 and Figure S1 and Table S1). The variant allele frequency (VAF) ranged from 0.37 to 0.93, with a median of 0.82. Based on the VAF provided by OGM, the number of repeats (manually verified), and blast counts (used as a proxy for neoplastic cells or clonal size) in AML, the estimated fractional copy number of these “duplicated” regions ranged from 3 to 17.2. No significant differences in fractional copy number of PTD were observed among patients with de novo AML, R/R AML, or sAML. Due to uncertainty about clonal size in MDS and CMML, estimating the fraction copy number of PTD in these conditions was challenging.
Table 2.
Karyotype and optical genome mapping (OGM) findings. CNV: copy number variant; SV: structural variant. Please see more detailed information on structural variants in Table S1.
RNA fusion assays were performed on 25 cases, all showing KMT2A PTD: 15 showed PTD spanning from exon 2 to exon 8 (chr11:118,353,210::118,339,490), and 10 cases from exon 2 to exon 10 (chr11:118,355,690::118,339,490) (Figure 1). The discrepancy between OGM and the RNA Anchor assay at the exon level is primarily due to the absence of labeling sites between exons 6 and 16 of KMT2A in OGM (Figure S3). Additionally, KMT2A PTD was identified by RNA sequencing in one case (#29) at an outside hospital (Table 2).
3.3. Cytogenomic Abnormalities Detected by G-Banded Karyotyping and OGM
Among 44 patients with an available karyotype, 21 (48%) had a normal karyotype, 20 (45%) exhibited 1–2 abnormalities, and 3 (7%) had a complex karyotype. The complex karyotype cases included two AML patients: case #15 with a prior history of polycythemia vera and case #17 with a history of mantle cell lymphoma who developed therapy-related MDS and later progressed to AML. A third patient (#38) had refractory MDS with 17% blasts.
OGM identified KMT2A PTD as a sole abnormality in 20 patients and with additional cytogenomic abnormalities in 25 patients (Table 2). Additional structural variants (SVs) were detected in six patients, including intragenic insertions of RUNX1 ins(21;?)(q22.12;?) in two patients (#17, #37). Other SVs were found in single cases, such as chromoanagenesis in case #15. Copy number variants (CNVs) were detected in 23 cases, with loss of 20q (n = 7) or 7q (n = 6) and trisomy 11/gain of 11q (+11/+11q, n = 6) being the most common. Other recurrent CNVs included del(5q), trisomy 8, trisomy 13, del(9q) and del(12q), detected in two or more cases.
3.4. Gene Mutations and FLT3 ITD
In the AML group, the most frequently affected pathways and genes included the following: epigenetic regulators (e.g., DNMT3A and IDH1/IDH2), observed in 91% of cases; the FLT3-RAS pathway (e.g., FLT3 ITD) in 65% of cases; transcription factors (e.g., RUNX1) in 45% of cases; spliceosome genes (e.g., U2AF1 and SRSF2) in 37% of cases; and cohesion genes (e.g., STAG2) in 20% of cases. Specifically, FLT3 ITD mutations were identified in 16 cases, while FLT3 TKD mutations appeared in 3 cases, including 1 case (#23) with both mutations. The FLT3 ITD ratio and VAF were notably higher in R/R AML compared to de novo and sAML cases. No TP53 mutations were detected in de novo AML, but these mutations were present in three sAML cases (#15, #16, and #19) and one R/R AML case (#30). Similarly, TET2 mutations were not detected in de novo AML but were presented in two sAML and five R/R AML cases (Table 3, Figure 2).
Table 3.
Gene mutations detected in patients with KMT2A-PTD. AR: allele ratio (ratio of the mutant ITD allele to the wild-type allele; VAF: variant allele frequency.
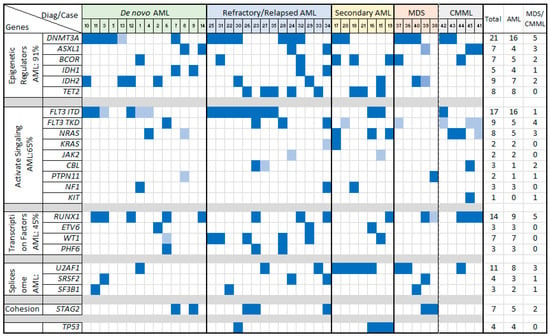
Figure 2.
Gene mutations in neoplasms with KMT2A-PTD, sorted by variant categories. Fills in dark blue: VAF ≥ 0.05; fills in light blue: VAF < 0.05.
In the MDS group, the most commonly affected pathways involved epigenetic regulators (e.g., DNMT3A) and spliceosome genes (e.g., U2AF1), observed in four out of five cases. Notably, one MDS case with a proliferative mutation in PTPN11 progressed to AML. In the CMML group, recurrently altered pathways included epigenetic regulators (e.g., DNMT3A) and FLT3 TKD/RAS kinase mutations, with all five cases showing these mutations. FLT3 TKD was more prevalent than FLT3 ITD in both MDS and CMML, although both mutations appeared as subclonal with low VAFs (Table 2, Figure 2).
3.5. Response to Treatments and Outcomes
Among the 35 patients with KMT2A PTD AML, 31 received chemotherapy, with some also receiving FLT3 or IDH2 inhibitors as part of their mainline treatments (Table 1).
Of the 14 patients with de novo AML, 11 received treatment and had follow-up. Nine patients responded well to treatments and achieved remission, while two showed a partial response. Five patients subsequently underwent allogeneic stem cell transplant (allo-SCT). By the end of the follow-up period, three patients had died (two from bone marrow failure and one from unknown causes), eight were alive in complete remission, two were still receiving induction chemotherapy, and one was lost to follow-up. The median overall survival (OS) was not reached.
Among the 14 patients with R/R AML, 9 were refractory to therapy, 3 exhibited partial response to treatments, and 2 responded well to treatments and achieved remission. By the end of follow-up, seven patients had died: three with persistent AML, two in remission, and two with no follow-up after the detection of KMT2A PTD, with a median OS of 19.2 months from diagnosis or 5.3 months from the detection of KMT2A PTD.
Of the seven patients with secondary AML, four were refractory and three showed partial response to treatments. By the end of follow-up, four had died and three had persistent AML, with a median OS of 4.5 months.
Among the five patients with MDS associated with KMT2A PTD, one patient died within a month of diagnosis due to multiple comorbidities. The remaining four patients received hypomethylating regimens, but all were refractory to treatment: one patient progressed to AML, and the other three had persistent disease. By the end of the follow-up period, three patients had died, and two were alive with persistent disease. The median overall survival was 8.5 months.
All five patients with CMML associated with KMT2A PTD received treatments. One patient (#41) responded to a combination of decitabine and venetoclax, followed by allo-SCT, and was in remission by the end of the follow-up. One patient (#43) progressed to AML and the other three were refractory to treatments. By the end of the follow-up, one patient had died, one was in remission, two patients had persistent CMML, and one had AML. The median overall survival was not reached.
4. Discussion
In this study, we used OGM to identify KMT2A PTD across a spectrum of hematological neoplasms. KMT2A PTD was detected exclusively in a subset of myeloid neoplasms, including AML, MDS, and CMML. OGM provides straightforward visualization of KMT2A PTD and is a reliable tool for detecting KMT2A PTD. Among these 45 cases, 25 were assessed using the Archer RNA fusion assay, and 1 was assessed by RNA sequencing. KMT2A PTD was confirmed in all 26 cases, demonstrating 100% concordance with RNA fusion assays. Conversely, cases that did not show KMT2A PTD by OGM were also negative by RNA fusion assays in those cases both assays were performed. In a previous study [23], KMT2A PTD was detected in 41 of 932 (4.4%) AML cases by using DNA-based NGS; of these 41 patients, 13 were also tested by OGM. OGM confirmed KMT2A PTD in 11 cases, whereas 2 cases initially shown by NGS to have KMT2A PTD were proven to be KMT2A rearranged by OGM. These findings underscore the high sensitivity and specificity of OGM for KMT2A PTD detection. Nonetheless, we observed certain limitations with OGM, including challenges in estimating PTD size, exon involvement, PTD copy number, and clarifying the complexity of PTD.
Most of the cases in this cohort had a normal or non-complex karyotype. Trisomy 11 or +11q is an uncommon cytogenetic abnormality in myeloid neoplasms [27]; however, it is frequently observed in cases with KMT2A PTD; approximately 30% to 70% of myeloid neoplasms with +11/+11q cases harbor KMT2A PTD [19,28,29,30]. During this study period, we identified 15 myeloid neoplasms with +11/+11q, and 6 (40%) had KMT2A PTD. Conversely, KMT2A PTD appeared to be mutually exclusive with class-defining chromosomal or gene rearrangements, as has been reported in other studies [28,29]. Deletions of 7q and 20q are common cytogenetic abnormalities in myeloid neoplasms and were likewise observed frequently in cases with KMT2A PTD [29]. The additional cytogenetic abnormalities did not appear to affect patient survival in this cohort, as has been reported by others [30].
Gene mutations are very common in myeloid neoplasms with KMT2A PTD, detected in all (100%) cases in this cohort and in a similarly high percentage of cases in another study [30]. The frequent co-occurrence of additional mutations appears critical for leukemogenesis in KMT2A PTD. Mouse model studies have shown that KMT2A PTD alone is insufficient to induce leukemia [6]. For instance, DNMT3A mutations enhance the self-renewal capacity of KMT2A PTD-positive leukemic cells, conferring a proliferative advantage [31]. Furthermore, mice with both KMT2A PTD and FLT3 ITD develop acute leukemia, highlighting the role of cooperative mutations in leukemogenesis [6].
The most frequently mutated genes included DNMT3A, FLT3-ITD, RUNX1, U2AF1, STAG2, TET2, and IDH2; no cases carried NPM1 mutation. These results are in accord with other reports [9,10,28,30]. DNMT3A, the most frequently mutated gene, exhibited hotspot mutations (R882H/C) and stop-gain mutations, mirroring earlier observations. Recurrent hotspot mutations in IDH1 (R132C), IDH2 (R140Q), U2AF1 (S34F), and FLT3 (D835Y/V/E) are suggestive of gain-of-function effects. Conversely, mutations in genes such as ASXL1, TET2, RUNX1, WT1, STAG2, and PHF6 were predominantly frameshift or stop-gain mutations, indicative of loss-of-function mechanisms. Additionally, mutually exclusive relationships between mutations in IDH1/IDH2 and TET2, as well as STAG2 and U2AF1, were observed in our cohort, consistent with earlier studies [10].
The association of these gene mutations with patient overall survival (OS) remains controversial. Some investigators have found that DNMT3A mutations, especially non-R882 variants, correlate with shorter OS [9,21,28,30]. In our cohort, although patients with DNMT3A mutations had a slightly shorter OS compared to those without (12.4 vs. 15.6 months), this difference was not statistically significant. We also observed that mutations in TP53 and TET2 were not detected in de novo AML but presented in sAML and R/R AML, suggesting that these mutations were likely acquired during disease relapse or progression and may influence treatment response and outcomes.
Patients with de novo AML and KMT2D PTD generally show a good response to treatment, with a high (70~90%) remission rate after the first induction in this cohort and others reported previously [18,19]. But these patients are reported to experience a high relapse rate, up to 70% with a median disease-free survival of 7.75 months in one study [18]. In another study, these patients had a 100% relapse rate within a year [19]. In the de novo AML group of this study, no relapses were observed, which could be attributable to the relatively short follow-up time, different therapeutic regimens compared to previous studies, and the fact that more patients received allo-SCT after the first remission. Although KMT2A PTD in AML has been associated with poorer survival, it is not generally considered an independent prognostic factor [30,32]. Some factors may impact the survival of KMT2A PTD AML, such as the DNMT3A mutations mentioned above. Whether the clonal burden of KMT2A PTD or copy number of PTD contributes to survival or not remains controversial. One study of AML patients found that a high initial clonal burden of KMT2A PTD was the only independent factor influencing remission rates and clinical outcomes [33]; however, another study did not observe this effect [21]. In the de novo AML group in this study, three patients (#3, #7, and #10) died; all three patients showed DNMT3A mutations and had high PTD copy numbers (estimated to be 17, 8.6, and 9.7, respectively), although the sample size is small.
Studies have shown that KMT2A PTD in MDS is associated with inferior survival and a high risk of progression from MDS to sAML [5,12,20]. In our cohort, patients in both the MDS and sAML groups exhibited a high rate of treatment resistance (nearly 100%) and short survival. Studies have shown that KMT2A PTD can be acquired during transformation from MDS to AML [5]. MDS cases also have a lower copy ratio burden of KMT2A PTD compared to AML [20]. In the current study, we evaluated KMT2A PTD at a single time point, so it is unknown whether KMT2A PTD was acquired during disease progression. However, KMT2A PTD was detected more frequently in sAML, 7 of 26 (27%) cases, as compared with AML as a whole group (7%).
Studies on KMT2A PTD in CMML are very limited. In this cohort we report, KMT2A PTD was present in approximately 7% of CMML patients, a frequency similar to that observed in AML. Four of these five patients had KMT2A PTD as the sole cytogenetic abnormality, but all patients showed two or more gene mutations, most commonly involving RUNX1, DNMT3A, ASXL1, and NRAS (found in 2–3 cases). This group of patients tended to have high blast counts and was frequently refractory to treatment. However, further study is needed to clarify the associated clinicopathological features and outcomes due to the relatively small number of cases and short follow-up period.
5. Conclusions
OGM effectively detects KMT2A PTD, showing 100% concordance with RNA fusion assays when both assays were performed. KMT2A PTD is exclusively found in myeloid neoplasms, including AML, MDS, and CMML. Patients with KMT2A PTD-positive MDS and secondary AML seem to often show resistance to treatment and have poorer survival. Further studies are needed to better understand the clinical course and outcomes for patients with KMT2A PTD-positive CMML.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers16244193/s1, Figure S1: The Genome Browser illustrates various forms of KMT2A PTD, all labeled as ins(11q23;?) by OGM, differing in repeat number, size, and breakpoints. Repeated sequences are underlined. A (Case #38, MDS): OGM provides the coordinates RefStartPos 118,477,357 and RefEndPos 118,479,068, with a size 35,074 bp, and VAF of 0.39. Manual examination reveals a duplication with breakpoints within intron 1 and intron 5, spanning 5 labels. Table S1: Structural variants (insertion and duplication) detected by optical genome mapping, the breakpoints (RefStartPos, RefEndPos), length, VAF (variant allele frequency). * by manual exam; ** fractional copy number of KMT2A-PTD estimated based on VAF, number of repeats and the blast counts. Figure S2: OGM labels (corresponding to the CTTAAG motif) within KMT2A. A: The upper panel lists the 36 exons of KMT2A, while the lower panel shows the distribution of labels, with a higher concentration observed within intron 1. B: A higher magnification view focused on exons 2 to 17, highlighting an absence of labels between exons 6 and 16. B (Case #3, AML): OGM provides the coordinates RefStartPos 118,477,357 and RefEndPos 118,479,068, size 25,447 bp and VAF of 0.88. Manual examination reveals a duplication with breakpoints within intron 1 and intron 5, spanning 4 labels. C (Case #11, AML): OGM provides the coordinates RefStartPos 118,470,405 and RefEndPos 118,479,068, with a size of 16,189 bp and VAF of 0.97. Manual examination reveals a duplication with breakpoints within intron 2 and intron 5, spanning 3 labels. D (Case #42, CMML): OGM provides the coordinates RefStartPos 118,461,867 and RefEndPos 118,479,068, with a size of 47,562 bp and VAF of 0.82. Manual examination reveals a quadruple repeat with breakpoints within intron 1 and intron 5, spanning 3 labels.
Author Contributions
Conception and design: G.T.; data procurement and data analysis: G.T., Q.W., S.H. and S.L.; initial draft of manuscript: Q.W. and G.T.; manuscript preparation: Q.W., S.H., J.X., S.L., N.D., G.A.T., W.W., L.J.M. and G.T. All authors have read and agreed to the published version of the manuscript.
Funding
The authors received no specific funding for this work.
Institutional Review Board Statement
This study has been approved by the Institutional Review Board of MD Anderson Cancer Center (2021-0476). The study was performed in accordance with the Declaration of Helsinki.
Informed Consent Statement
This study is a retrospective study and has been approved by our Institutional Review Board (IRB, 2021-0476). Though informed consent was obtained from all subjects involved in the study, specific informed consent for this study has been waivered. In addition, due to the limitation of our institute policy, we have not been authorized to provide a blank copy of informed consent at this moment.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The authors are grateful to the optical genome mapping team in our cytogenetics laboratory at MDACC for their dedicated work.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Meyer, C.; Larghero, P.; Almeida Lopes, B.; Burmeister Gröger, D.; Sutton, R.; Venn, N.C.; Cazzaniga, G.; Corral Abascal, L.; Tsaur, G. The KMT2A Recombinome of Acute Leukemias in 2023. Leukemia 2023, 37, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- Steudel, C.; Wermke, M.; Schaich, M.; Schäkel, U.; Illmer, T.; Ehninger, G.; Thiede, C. Comparative Analysis of MLL Partial Tandem Duplication and FLT3 Internal Tandem Duplication Mutations in 956 Adult Patients with Acute Myeloid Leukemia. Genes Chromosomes Cancer 2003, 37, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Shiah, H.S.; Kuo, Y.Y.; Tang, J.L.; Huang, S.Y.; Yao, M.; Tsay, W.; Chen, Y.C.; Wang, C.H.; Shen, M.C.; Lin, D.T.; et al. Clinical and Biological Implications of Partial Tandem Duplication of the MLL Gene in Acute Myeloid Leukemia without Chromosomal Abnormalities at 11q23. Leukemia 2002, 16, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Basecke, J.; Whelan, J.T.; Griesinger, F.; Bertrand, F.E. The MLL Partial Tandem Duplication in Acute Myeloid Leukaemia. Br. J. Haematol. 2006, 135, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Dicker, F.; Haferlach, C.; Sundermann, J.; Wendland, N.; Weiss, T.; Kern, W.; Haferlach, T.; Schnittger, S. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 Patients with MDS or Secondary AML. Leukemia 2010, 24, 1528–1532. [Google Scholar] [CrossRef]
- Zorko, N.A.; Bernot, K.M.; Whitman, S.P.; Siebenaler, R.F.; Ahmed, E.H.; Marcucci, G.G.; Yanes, D.A.; McConnell, K.K.; Mao, C.; Kalu, C.; et al. Mll Partial Tandem Duplication and Flt3 Internal Tandem Duplication in a Double Knock-In Mouse Recapitulates Features of Counterpart Human Acute Myeloid Leukemias. Blood 2012, 120, 1130–1136. [Google Scholar] [CrossRef]
- Schnittger, S.; Wörmann, B.; Hiddemann, W.; Griesinger, F. Partial Tandem Duplications of the MLL Gene Are Detectable in Peripheral Blood and Bone Marrow of Nearly All Healthy Donors. Blood 1998, 92, 1728–1734. [Google Scholar] [CrossRef]
- Testa, U.; Riccioni, R.; Militi, S.; Coccia, E.; Stellacci, E.; Samoggia, P.; Latagliata, R.; Mariani, G.; Rossini, A.; Battistini, A.; et al. Elevated Expression of IL-3Ralpha in Acute Myelogenous Leukemia is Associated with Enhanced Blast Proliferation, Increased Cellularity, and Poor Prognosis. Blood 2002, 100, 2980–2988. [Google Scholar] [CrossRef]
- Hinai, A.; Pratcorona, M.; Grob, T.; Kavelaars, F.G.; Bussaglia, E.; Sanders, M.A.; Nomdedeu, J.; Valk, P.J.M. The Landscape of KMT2A-PTD AML: Concurrent Mutations, Gene Expression Signatures, and Clinical Outcome. Hemasphere 2019, 3, e181. [Google Scholar] [CrossRef]
- Sun, Q.Y.; Ding, L.W.; Tan, K.T.; Chien, W.; Mayakonda, A.; Lin, D.C.; Loh, X.Y.; Xiao, J.F.; Meggendorfer, M.; Alpermann, T.; et al. Ordering of Mutations in Acute Myeloid Leukemia with Partial Tandem Duplication of MLL (MLL-PTD). Leukemia 2017, 31, 1–10. [Google Scholar] [CrossRef]
- Ye, W.; Ma, M.; Wu, X.; Deng, J.; Liu, X.; Zheng, X.; Gong, Y. Prognostic Significance of KMT2A-PTD in Patients with Acute Myeloid Leukaemia: A Systematic Review and Meta-Analysis. BMJ Open 2023, 13, e062376. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.M.; Dewar, R.; Burke, P.W.; Shao, L. Partial Tandem Duplication of KMT2A (MLL) May Predict a Subset of Myelodysplastic Syndrome with Unique Characteristics and Poor Outcome. Haematologica 2018, 103, e131–e134. [Google Scholar] [CrossRef] [PubMed]
- Dorrance, A.M.; Liu, S.; Yuan, W.; Becknell, B.; Arnoczky, K.J.; Guimond, M.; Strout, M.P.; Feng, L.; Nakamura, T.; Yu, L.; et al. Mll Partial Tandem Duplication Induces Aberrant Hox Expression In Vivo via Specific Epigenetic Alterations. J. Clin. Invest. 2006, 116, 2707–2716. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Sun, J.Z.; Liu, F.; Zhang, H.; Ma, Y. Higher Expression Levels of the HOXA9 Gene, Closely Associated with MLL-PTD and EZH2 Mutations, Predict Inferior Outcome in Acute Myeloid Leukemia. OncoTargets Ther. 2016, 9, 711–722. [Google Scholar] [CrossRef]
- Issa, G.C.; Ravandi, F.; DiNardo, C.D.; Jabbour, E.; Kantarjian, H.M.; Andreeff, M. Therapeutic Implications of Menin Inhibition in Acute Leukemias. Leukemia 2021, 35, 2482–2495. [Google Scholar] [CrossRef]
- Huang, J.; Zhu, Y.; Li, J.; Yang, G.; Zhang, S. The KMT2A Rearrangement is an Early Event Prior to KMT2A-PTD in AML Patients with Both Molecular Aberrations. Ann. Hematol. 2023, 102, 495–497. [Google Scholar] [CrossRef]
- Dohner, K.; Tobis, K.; Ulrich, R.; Fröhling, S.; Benner, A.; Schlenk, R.F.; Döhner, H. Prognostic Significance of Partial Tandem Duplications of the MLL Gene in Adult Patients 16 to 60 Years Old with Acute Myeloid Leukemia and Normal Cytogenetics: A Study of the Acute Myeloid Leukemia Study Group Ulm. J. Clin. Oncol. 2002, 20, 3254–3261. [Google Scholar] [CrossRef]
- Schnittger, S.; Kinkelin, U.; Schoch, C.; Heinecke, A.; Haase, D.; Haferlach, T.; Büchner, T.; Wörmann, B.; Hiddemann, W.; Griesinger, F. Screening for MLL Tandem Duplication in 387 Unselected Patients with AML Identify a Prognostically Unfavorable Subset of AML. Leukemia 2000, 14, 796–804. [Google Scholar] [CrossRef]
- Tsai, H.K.; Gibson, C.J.; Murdock, H.M.; Davineni, P.; Harris, M.H.; Wang, E.S.; Gondek, L.P.; Kim, A.S.; Nardi, V.; Lindsley, R.C. Allelic Complexity of KMT2A Partial Tandem Duplications in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Blood Adv. 2022, 6, 4236–4240. [Google Scholar] [CrossRef]
- Dai, B.; Yu, H.; Ma, T.; Lei, Y.; Wang, J.; Zhang, Y.; Lu, J.; Yan, H.; Jiang, L.; Chen, B. The Application of Targeted RNA Sequencing for KMT2A-Partial Tandem Duplication Identification and Integrated Analysis of Molecular Characterization in Acute Myeloid Leukemia. J. Mol. Diagn. 2021, 23, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- McKerrell, T.; Moreno, T.; Ponstingl, H.; Bolli, N.; Dias, J.M.L.; Tischler, G.; Colonna, V.; Manasse, B.; Bench, A.; Bloxham, D.; et al. Development and Validation of a Comprehensive Genomic Diagnostic Tool for Myeloid Malignancies. Blood 2016, 128, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Seto, A.; Downs, G.; King, O.; Salehi-Rad, S.; Baptista, A.; Chin, K.; Grenier, S.; Nwachukwu, B.; Tierens, A.; Minden, M.D.; et al. Genomic Characterization of Partial Tandem Duplication Involving the KMT2A Gene in Adult Acute Myeloid Leukemia. Cancers 2024, 16, 1693. [Google Scholar] [CrossRef] [PubMed]
- Loghavi, S.; Wei, Q.; Ravandi, F.; Quesada, A.E.; Routbort, M.J.; Hu, S.; Toruner, G.A.; Wang, S.A.; Wang, W.; Miranda, R.N.; et al. Optical Genome Mapping Improves the Accuracy of Classification, Risk Stratification, and Personalized Treatment Strategies for Patients with Acute Myeloid Leukemia. Am. J. Hematol. 2024, 10, 1959–1968. [Google Scholar] [CrossRef]
- Ok, C.Y.; Loghavi, S.; Sui, D.; Wei, P.; Kanagal-Shamanna, R.; Yin, C.C.; Zuo, Z.; Routbort, M.J.; Tang, G.; Tang, Z.; et al. Persistent IDH1/2 Mutations in Remission Can Predict Relapse in Patients with Acute Myeloid Leukemia. Haematologica 2019, 104, 305–311. [Google Scholar] [CrossRef]
- Warren, M.; Luthra, R.; Yin, C.C.; Ravandi, F.; Cortes, J.E.; Kantarjian, H.M.; Medeiros, L.J.; Zuo, Z. Clinical Impact of Change of FLT3 Mutation Status in Acute Myeloid Leukemia Patients. Mod. Pathol. 2012, 25, 1405–1412. [Google Scholar] [CrossRef]
- Wang, S.A.; Jabbar, K.; Lu, G.; Chen, S.S.; Galili, N.; Vega, F.; Jones, D.; Raza, A.; Kantarjian, H.; Garcia-Manero, G.; et al. Trisomy 11 in Myelodysplastic Syndromes Defines a Unique Group of Disease with Aggressive Clinicopathologic Features. Leukemia 2010, 24, 740–747. [Google Scholar] [CrossRef][Green Version]
- Eisfeld, A.K.; Kohlschmidt, J.; Mrózek, K.; Blachly, J.S.; Nicolet, D.; Kroll, K.; Orwick, S.; Carroll, A.J.; Stone, R.M.; de la Chapelle, A.; et al. Adult Acute Myeloid Leukemia with Trisomy 11 as the Sole Abnormality is Characterized by the Presence of Five Distinct Gene Mutations: MLL-PTD, DNMT3A, U2AF1, FLT3-ITD and IDH2. Leukemia 2016, 30, 2254–2258. [Google Scholar] [CrossRef]
- Caligiuri, M.A.; Strout, M.P.; Schichman, S.A.; Mrózek, K.; Arthur, D.C.; Herzig, G.P.; Baer, M.R.; Schiffer, C.A.; Heinonen, K.; Knuutila, S.; et al. Partial Tandem Duplication of ALL1 as a Recurrent Molecular Defect in Acute Myeloid Leukemia with Trisomy 11. Cancer Res. 1996, 56, 1418–1425. [Google Scholar]
- Vetro, C.; Haferlach, T.; Meggendorfer, M.; Stengel, A.; Jeromin, S.; Kern, W.; Haferlach, C. Cytogenetic and Molecular Genetic Characterization of KMT2A-PTD Positive Acute Myeloid Leukemia in Comparison to KMT2A-Rearranged Acute Myeloid Leukemia. Cancer Genet. 2020, 240, 15–22. [Google Scholar] [CrossRef]
- Bera, R.; Chiu, M.C.; Huang, Y.J.; Huang, G.; Lee, Y.S.; Shih, L.Y. DNMT3A Mutants Provide Proliferating Advantage with Augmentation of Self-Renewal Activity in the Pathogenesis of AML in KMT2A-PTD-Positive Leukemic Cells. Oncogenesis 2020, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Zhao, X.S.; Qin, Y.Z.; Zhu, H.H.; Jia, J.S.; Jiang, Q.; Wang, J.; Zhao, T.; Huang, X.J.; Jiang, H. The Initial Level of MLL-Partial Tandem Duplication Affects the Clinical Outcomes in Patients with Acute Myeloid Leukemia. Leuk. Lymphoma 2018, 59, 967–972. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).