Simple Summary
This review focuses on the classification, risk stratification, and metabolic characteristics of acute myeloid leukaemia (AML) and acute lymphoblastic leukaemia (ALL) and their importance in the advancement of treatment. It discusses current management strategies, including standard chemotherapy regimens and targeted therapies, while highlighting the challenges of drug toxicity and resistance. It also emphasises the importance of understanding metabolic vulnerabilities in leukaemia cells for developing more effective and personalised treatments. Special attention is given to asparaginase therapy and its mechanism of action, limitations, and potential for improvement in both ALL and AML treatment.
Abstract
Acute myeloid leukaemia (AML) and acute lymphoblastic leukaemia (ALL) remain significant challenges in haematological oncology. This review examines the pathophysiology, classification, and risk stratification of these aggressive malignancies, emphasising their impact on treatment strategies and prognosis. We discuss current standard-of-care treatments, including chemotherapy regimens and targeted therapies, while addressing the associated adverse effects and hypersensitivity reactions. Delving into the metabolic characteristics and vulnerabilities of leukaemia cells, the review highlights the key differences between lymphoid and myeloid leukaemia and how metabolic insights can be utilised for therapeutic purposes, with special focus on asparaginase therapy and its potential for improvement in both ALL and AML treatment. The review conveys the importance of personalised medicine approaches based on individual metabolic profiles and the challenges posed by metabolic heterogeneity and plasticity in leukaemia cells. Combining molecular and metabolic profiling can enhance and refine treatment strategies for acute leukaemia, potentially improving patient outcomes and quality of life. However, integrating these into routine clinical practice requires overcoming various practical, technical, and logistical issues.
1. Introduction
Acute leukaemia, particularly acute myeloid leukaemia (AML) and acute lymphoblastic leukaemia (ALL), pose significant challenges in the field of haematological malignancies. Acute leukaemia impacts a diverse range of age groups. AML is more prevalent in adults, with 3100 new cases diagnosed in the UK each year. Despite treatment advances, the five-year survival rate of AML is still only 15%, with approximately 2600 deaths in the UK annually [1]. ALL is the most common childhood cancer, peaking in children aged 3–4 years, with 790 new cases and 230 deaths annually in the UK [1].
This review provides a comprehensive overview of the cell lineage and molecular and metabolic characteristics of these aggressive malignancies. This underpins the heterogeneity of the disease and the requirement for treatment to be personalised and tailored to individual cases. Understanding the specific cellular dependencies and vulnerabilities is crucial for future clinical advancements [2,3,4,5,6,7,8,9,10]. Treatment of acute leukaemia involves intensive chemotherapy regimens and targeted therapies, associated with significant toxicities [11,12]. These adverse effects range from myelosuppression and gastrointestinal issues to cardiotoxicity, pancreatitis, and secondary malignancies, necessitating a need for less toxic and more targeted treatments [13]. L-asparaginase has been used as a key component of ALL treatment protocols for the past 50 years and is particularly effective in lymphoblastic leukaemia due to these cells typically lacking the enzyme asparagine synthetase (ASNS) [14]. Exploration of the dual action of asparaginase against both asparagine and glutamine highlights its potential utility across different leukaemia subtypes. Studies in metabolic characterisation reveal potential pathways for further individualised use of asparaginase as perhaps a more targeted treatment than currently considered.
The integration of molecular and metabolic profiling in personalised medicine is central to developing effective treatment strategies tailored to individual patients’ needs [15] that minimise toxicity and maximise efficacy. While challenges remain in the fight against acute leukaemia, ongoing research aimed at the comprehensive classification and metabolic profiling of ALL and AML is a promising approach for enhancing treatment efficacy and improving outcomes for patients.
2. Pathophysiology
AML is a rapidly progressing heterogenous myeloid neoplasm characterised by the clonal expansion of myeloid progenitor cells in the bone marrow and peripheral blood. This proliferation primarily stems from the accumulation of diverse genomic and cytogenetic abnormalities, resulting in ineffective erythropoiesis, megakaryopoiesis, organ infiltration and bone marrow failure, causing the inadequate production of red blood cells and platelets [16].
ALL is an aggressive malignancy of B or T lymphoblasts, characterised by the uncontrolled proliferation of abnormal, immature lymphocytes and their progenitors. Environmental damage to DNA and genetic predisposition are among aetiologies that precede disease, causing lymphoid cells to undergo uncontrolled growth and spread throughout the body [17,18,19,20]. Like AML, this ultimately leads to the replacement of functional bone marrow elements and other lymphoid organs, leading to bone marrow failure. Furthermore, splenomegaly and hepatomegaly occur due to the sequestration of platelets and lymphocytes [21].
3. Classification and Risk Stratification, Informing Treatment and Prognosis
The World Health Organisation (WHO) classifies AML and ALL according to cell lineage, gene or chromosome changes, and cell differentiation (see Table 1). Acute leukaemia of ambiguous lineage (ALAL) and mixed-phenotype acute leukaemia (MPAL) have overlapping clinical and immunophenotypic features and are therefore grouped together. These have been found to share common molecular pathogenic mechanisms [22,23,24]. The separation of ALAL/MPAL allows for molecular classification, distinguishing those with genetic abnormalities from those defined by immunophenotyping. Cells may exhibit a pattern in which some leukaemia cells have myeloid features and others have lymphoid features, while some cells simultaneously display both myeloid and lymphoid features [25].

Table 1.
WHO classification of AML and ALL.
3.1. AML
AML is a heterogeneous disease requiring individualised cytogenetic and molecular characterisation (Table 1). Prognostic factors are subdivided into those related to either the patient or to the disease (Figure 1). The European LeukemiaNET (ELN) guidelines (2022) emphasise molecular characterisation and risk stratification as critical for prognostic classification and treatment strategy, categorising AML into favourable-, intermediate-, or adverse-risk groups (see Table 2) [28]. Despite advancements in therapeutic approaches, prognosis remains suboptimal, especially among older populations. Characterising fitness in the adult population is important when deciding treatment strategy.
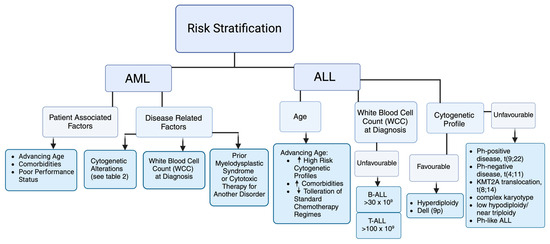
Figure 1.
Risk stratification in AML and ALL as per European LeukemiaNET (ELN) guidelines and the National Comprehensive Cancer Network (NCCN) [28,29].
Table 2.
ELN-recommended stratification of molecular and cytogenic alterations in AML.
3.2. ALL
Clinical factors and cytogenetic changes play an important role in risk stratification, which guides initial treatment regimen and allogeneic stem cell transplantation (Allo-SCT) (Figure 1 and Table 1). The Philadelphia chromosome, the t(9;22) cytogenetic aberration, has the greatest impact on prognosis and treatment. Prevalence in adult ALL can range from 15 to 30% and increases with age [30]. Response to initial therapy also predicts outcome. Patients are evaluated for minimal residual disease (MRD) using molecular techniques such as flow cytometry and PCR [31]. Bruggemann et al. re-stratified standard-risk patients to low risk (<10−4), intermediate risk, and high risk (>10−4), with relapse rates of 0%, 47%, and 94%, respectively, based on the persistence of elevated MRD, defined as >10−4 [32]. The National Comprehensive Cancer Network (NCCN) has developed recommendations to approach risk stratification [29]. The NCCN recognises that adolescent and young adults (AYAs) (those aged 15–39 years) may benefit from treatment with paediatric-inspired regimens, with adults (>40 years) considered separately [33]. Both age groups are then stratified into subgroups: high-risk Ph-positive (Philadelphia chromosome-positive ALL) and standard-risk Ph-negative. The Ph-negative subgroup can further be categorised as high, intermediate, or low risk (Table 3), with the 5-year overall survival rates based on risk categories being 5%, 34%, and 55% respectively [34].
Table 3.
Standard-risk pH-negative acute lymphoid leukaemia (ALL) subdivision.
4. Treating Acute Leukaemia
4.1. Drug Classification
Cancer chemotherapy drugs are divided into two main categories: non-targeted agents with broad specificity, and targeted drugs developed for specific molecular targets on cancer cells.
4.2. Current Standard of Care
Treatment of ALL and AML typically follows an induction, maintenance, and consolidation regime. The induction phase typically involves intensive chemotherapy combination regimens and aims to achieve complete remission (Table 4 and Table 5). Targeted therapies are incorporated into induction protocols to improve outcomes in specific genetic and molecular subgroups (Table 6). Newly developed targeted therapies include small-molecule inhibitors such as those that target FLT3, IDH, and BCL-2, as well as immunotherapies including Chimeric Antigen Receptor (CAR) T-cell therapy and bispecific T-cell engagers (BiTEs). Additionally, there are antibody–drug conjugates and emerging Menin inhibitors which are currently under investigation for use in KMT2A rearrangements or NPM1 mutations in both ALL and AML. The optimal sequence and combination of these targeted agents with standard chemotherapy is an active area of research in AML [35,36]. Less intensive options are considered for older adults or those with significant comorbidities who cannot tolerate intensive chemotherapy.
Table 4.
Standard induction regimes for AML and ALL.
Table 5.
Non-targeted chemotherapy drugs used in the treatment of AML and ALL.
Table 6.
Targeted chemotherapy drugs used in the treatment of AML and ALL.
4.3. Adverse Reactions
Non-targeted anti-leukemic drugs are cytotoxic; suppress haematopoiesis; and can cause cutaneous eruptions, vascular damage, and lung and liver injury (Table 5). Current research into dose optimisation aims to adjust drug doses and schedules to maintain efficacy while minimising adverse effects [75]. Further attempts to decrease the overall toxicity of leukaemia treatments focuses on increasing the specificity of drugs. Targeting molecular pathways specific to leukaemia cells has potential to spare healthy cells and reduce systemic adverse effects by reducing the dose of traditional chemotherapy agents required [12]. Adverse effect profiles of targeted treatments tend to be more manageable; however, these include lengthened QT intervals, a conduction disorder of the heart, febrile neutropenia, cytopenia, infections, gastrointestinal issues, and skin disorders (Table 6). Additionally, targeted immune activation aims to harness the immune system to deliver precise treatment with reduced off-target effects [76].
4.4. Hypersensitivity Reactions
In chemotherapy treatment for acute leukaemia, various adverse effects may have an immune basis, including drug-induced thrombocytopenia, neutropenia, and anaemia; vascular disorders; liver injury; lung disease; and various dermatological manifestations (Table 5). Certain drugs cause specific hypersensitivity reactions, such as cytarabine, causing a Type IV, delayed, and T cell-mediated reaction and L-asparaginase, causing a Type I, IgE antibody-mediated reaction (Table 5 and Table 7).
Table 7.
Adverse effects of asparaginase therapy.
5. Metabolic Characteristics, Vulnerabilities, and Treatment Strategy
Metabolic vulnerabilities in leukaemia cells present significant opportunities for targeted therapies by exploiting the unique dependencies of these cells. Understanding and targeting the metabolic vulnerabilities of leukaemia cells offers a promising strategy for developing more effective and personalised treatments for leukaemia.
5.1. Key Differences in Metabolic Profiles Between Lymphoid and Myeloid Leukaemia Cells
5.1.1. Glycolysis and Oxidative Phosphorylation
Lymphoid leukaemia cells rely more on oxidative phosphorylation (OxPhos) rather than glycolysis, exhibiting a reduced glucose uptake in comparison to normal hematopoietic stem cells (HSCs), with increased mitochondrial respiration and reactive oxygen species (ROS) production [2,3]. Myeloid leukaemia cells show significant alterations in glycolytic pathways, characterised by a different set of metabolic adaptations that may include both glycolysis and OxPhos, depending on the specific subtype of myeloid leukaemia [4]. AML cells can alter the expression of glycolytic enzymes, upregulating glycolysis and switching between glycolysis and OxPhos depending on environmental conditions [85,86].
5.1.2. Amino Acid Metabolism
In lymphoid leukaemia cells, there is protective activity in glutathione metabolism, involving the overexpression of enzymes like glutamine dehydrogenase, which is crucial for glutathione synthesis [5,6,7]. Myeloid leukaemia cells also exhibit the aberrant regulation of glutathione, with significantly lower levels of reduced glutathione (GSH), higher levels of oxidised glutathione (GSSG) and reduced total glutathione. In addition, studies observe a decreased GSH-to-GSSG ratio in CD34+ AML cells [8].
5.1.3. Lipid Metabolism
Lymphoid leukaemia cells demonstrate active lipid metabolism, with an accumulation of ceramide and lipoprotein lipases, making them susceptible to fatty acid oxidation (FAO) inhibitors. This is observed to be useful in cases of treatment resistance [9]. Myeloid leukaemia cells undergo the metabolic reprogramming of lipid metabolism crucial for tumorigenesis and disease progression, supporting processes such as invasion, metastasis, and abnormal signalling [10]. Pathway alterations and their implications can differ based on genetic and environmental factors. Leukaemia cells treated with L-asparaginase have been shown to react by reprogramming their metabolism to increase fatty acid oxidation (FAO) and autophagy to compensate for asparagine and glutamine depletion [87]. The pharmacological inhibition of FAO increases sensitivity to asparaginase in leukaemia cells, supporting the theory of their pro-survival effect and potential role in the mechanism of treatment resistance [87].
5.2. Key Insights into How These Vulnerabilities Are Being Utilised for Therapeutic Purposes
5.2.1. Targeting Specific Metabolic Pathways
Leukaemia cells exhibit altered metabolic pathways that support their rapid proliferation and survival. These include glycolysis, the oxidation of fatty acids, and the Krebs cycle. Targeting these pathways can disrupt the energy supply and biosynthetic processes crucial for leukaemia cell survival.
5.2.2. Leukaemia Stem Cells
Leukaemia stem cells (LSCs) resistant to conventional treatments have distinct metabolic preferences, such as heavy reliance on OXPHOS and specific amino acid metabolisms [88]. Inhibiting these pathways, for example, by targeting the enzyme nicotinamide phosphribosyltransferase (NAMPT), can reduce OXPHOS, with potential to eradicate resistant LSCs, while sparing normal cells [89].
5.2.3. Combination Therapies
Combining metabolic inhibitors with chemotherapy or immunotherapy can enhance therapeutic efficacy. Venetoclax combined with azacytidine disrupts energy metabolism in AML cells, targeting both blasts and LSCs, and restores sensitivity to treatment in relapsed or refractory cases of AML [90]. Additionally, 2-deoxy-D-glucose (2-DG) interferes with D-glucose metabolism, enhancing the anti-cancer effects of idarubicin (IDA) in IDA-resistant P388 leukaemia cells [91].
5.2.4. Personalised Medicine
Leukaemia cells display distinct metabolic states and adaptation mechanisms, serving as potential targets for treatment. However, the heterogeneity of the disease advocates for therapies to be tailored more individually. AML displays significant metabolic heterogeneity between patients within genetically distinct subclones [92,93,94]. The serine/threonine protein kinase PDK1 acts as a targetable determinant of different metabolic states in AML, functioning as a gatekeeper of glycolysis by phosphorylating and inactivating pyruvate dehydrogenase (PDH) [95]. Studies identified two main metabolic states in AML: PDK1-low, which is OXPHOS-driven, and PDK1-high, which is associated with low OXPHOS and an increase in stemness transcriptional signatures [85]. Insights into metabolic differences alongside clonal heterogeneity in AML allow for personalised, subclone-specific targeting strategies.
In addition to metabolic heterogeneity, the metabolic plasticity of leukaemia cells also presents a challenge in treatment design. With flexibility to undergo compensatory metabolic and energetic adaptations in response to the inhibition of metabolic pathways, leukaemia cells can adapt and survive when specific metabolic pathways are targeted therapeutically [77,96]. LSC plasticity in mixed-lineage leukaemia-rearranged B-lymphoblastic leukaemia (MLL-r B-ALL) allows the cells to switch lineages and emerge from differentiated populations, seen most frequently when under chemotherapy pressure [97]. AML stem cells can switch between a low-cycling chemotherapy-resistant state and an actively proliferating state [77]. This plasticity allows for interruption in drug efficacy, contributing to treatment resistance and disease progression.
6. Asparaginase as Targeted Treatment in Acute Leukaemia
In healthy cells, the non-essential amino acid, asparagine, can be synthesised by the enzymatic action of asparagine synthetase (ASNS) or obtained from the diet. Sufficient levels of cellular asparagine are required for DNA, RNA, and protein synthesis. Depleted asparagine levels ultimately lead to the activation of apoptotic cell death mechanisms (Figure 2).

Figure 2.
Mechanism of action of asparaginase in the treatment of leukaemia. ASNase: L-asparaginase; ASNS: L-asparagine synthetase; dashed arrows: reduced or missing.
L-asparaginase has been a key component of ALL treatment protocols for the past fifty years and has been found to be particularly effective in lymphoblastic leukaemia due to cells typically lacking ASNS [14]. Lymphoblastic leukaemia cells are also naturally susceptible to asparagine depletion. Cells unable to produce asparagine on their own are heavily dependent on extracellular sources. At sufficient activity levels, asparaginase depletes serum L-asparagine, eventually leading to leukaemic cell death.
6.1. Current Limitations of Asparaginase Therapy
Despite their effectiveness, the high toxicity and side effects of current L-asparaginase formulations limit optimal clinical application, with lack of tolerance often leading to treatment interruption and discontinuation (Table 7). Current studies aim to improve treatment adherence, to reduce treatment interruption, and to improve the quality of life of patients receiving this drug. Combining L-asparaginase with other drugs, such as FAOs, may improve efficacy while reducing adverse effects. The development of L-asparaginase variants with reduced L-glutaminase coactivity has been found to reduce acute toxicity compared to FDA-approved high L-glutaminase enzymes, while maintaining anti-leukaemic efficacy [98]. The short half-lives of current L-asparaginase formulations have prompted efforts to develop variants with longer half-lives, which correlate with lower immunogenicity and reduced toxicity, while maintaining efficacy [99]. Paediatric studies emphasise the importance of monitoring in the early detection of asparaginase-related toxicity, in addition to understanding risk factors for toxicity that can inform individualised treatment approaches [100]. Identified risk factors include the presence of the TEL-AML1 fusion gene, which is associated with a higher risk of clinical hypersensitivity. Additionally, T-cell ALL correlates with a higher risk of hepatotoxicity in comparison to B-cell ALL. Other identified factors include age, gender, response to treatment, and presence of central nervous system (CNS) infiltration at diagnosis.
6.2. Obstacles to Asparaginase Use in AML
AML cells are, on average, 7-fold more resistant to L-asparaginase than ALL cells [101]. This may be due to AML cells having variable expression of ASNS, in comparison to ALL cells. However, ASNS activity in blast cells of acute monocytic leukaemia (10% of AML cases) are reported to have the lowest median of ASNS activity, with activity varying over only a fourfold range [14]; therefore, certain subgroups of AML may show more promising outcomes when treated with L-asparaginase. Lysosomal cysteine protease cathepsin B (CTSB), asparaginyl endopeptidase, and neutralising antibodies may also play a role in resistance and sensitivity to L-asparaginase [102,103]. L-asparaginase should therefore be tested in combination with other drugs that counteract factors decreasing its efficacy, such as the inhibition of ASNS and/or CTSB with specific protease inhibitors. With its potential to downregulate transcription of the ASNS gene, cytarabine shows promising synergetic effects with L-asparaginase. For example, asparaginase intensification studies, consisting of a combination of high doses of cytarabine and asparaginase (with noted importance of the drug sequence, with asparaginase following cytarabine) was found to eliminate the benefit of prolonged maintenance therapy in childhood AML. This was accompanied by an overall improvement in survival and improved complete remission rate [104]. The efficacy of asparaginase was also observed when combined with methotrexate in both adult and paediatric refractory or relapsed AML [105,106]. L-asparaginase may have anti-leukaemic activity in AML cells in general, and leukaemic stem cells in particular, if the protective effect of the bone marrow microenvironment can be overcome. A proposed approach to counteract this protective effect on residual AML cells, is to interfere with the binding of residual cells to the bone marrow stroma with CXCR4 chemokine-receptor antagonists, such as plerixafor [107].
Although AML cells may have variable expression of ASNS, they are particularly susceptible to glutamine depletion [103]. While L-asparaginase’s primary target is asparagine, it has significant activity against glutamine (Figure 2). L-asparaginase’s primary activity hydrolyses L-asparagine into L-aspartic acid and ammonia, with secondary hydrolysis of L-glutamine into L-glutamic acid and ammonia (glutaminase activity). The glutaminase activity of FDA-approved L-asparaginases ranges from 2% to 10% of their primary asparaginase activity [98]. AML cells and LSCs rely heavily on glutamine metabolism for critical cellular processes and show increased sensitivity to glutamine depletion compared to normal cells [108]. Additionally, the genetic knockdown or pharmacologic inhibition of glutaminase negatively affects AML cells, while sparing normal CD34+ HSCs [109]. The glutaminase activity of L-asparaginase enhances the drug’s anticancer effects in ASNS-positive cancer cells [98]. Further studies have also found that the glutaminase activity of L-asparaginase was necessary for durable anticancer activity in vivo, even against ASNS-negative cancer types [110].
6.3. Metabolic Characteristics and Sensitivity to Asparaginase Treatment
Leukaemia cells cluster into distinct groups based on their metabolic profiles, which correlate with their response to asparaginase treatment. In a recent study describing the relationship between the metabolic profile of leukaemia cells and their sensitivity to commonly used anti-leukaemic drugs, cells sensitive to asparaginase were ranked in a lower glycolytic cluster [15]. Indeed, a significant correlation was observed between higher ATP-linked respiration, lower basal mitochondrial membrane potential, and increased sensitivity to asparaginase. No similar correlation was found for other cytostatic drugs tested [15]. The findings suggest that the metabolic profile of leukaemia cells plays a crucial role in determining the efficacy of asparaginase treatment, and that metabolic profiling could be used to predict asparaginase sensitivity in leukaemia patients. Thus, patients with higher glycolytic activity might benefit from alternative or additional treatments to asparaginase. Targeting specific metabolic pathways (e.g., glycolysis or mitochondrial respiration) could potentially enhance the efficacy of asparaginase treatment, especially in less sensitive patients who would otherwise be more likely to fail the conventional therapy without having any other detectable risk factors. Characterising leukaemic cell and blast metabolic state, at the time of diagnosis, may help identify patients who are particularly sensitive, or with lower sensitivity to asparaginase, and offer a potential pathway for personalised treatment strategies in leukaemia therapy.
However, the integration of metabolic profiling into routine clinical practice is hindered by several complex practical challenges. The techniques used to evaluate cellular metabolism include sophisticated stable isotope tracing and biochemical assays, which can be difficult to standardise and are not ideally suited to high-throughput clinical and diagnostic labs and are not currently used within UK National Health Services. More recent methods like extracellular flux analysis using Seahorse Analysers offer high throughput but require meticulous optimisation and set up which is why they are more commonly found in research settings. The cost of these technologies may also be a prohibiting factor for clinical laboratories along with the requirement for highly trained staff. Also, there is no single gold-standard test for metabolic diagnostics as metabolic activity often involves a sequence of events along a gradient of pathway flux. Consequently, it is likely that multiple tests will be necessary to accurately profile metabolic characteristics, posing challenges in terms of costs, accessibility, and standardisation.
7. Conclusions
The treatment landscape for acute leukaemia is evolving due to advances in our understanding of their cellular and metabolic characteristics, and the targeting of these for therapeutic benefit. Statistics surrounding these diseases highlight the urgency for improved treatment options. With AML resulting in over 2600 deaths each year and a mere 15% five-year survival rate despite treatment advances, and ALL being the most common childhood cancer, it is evident that more effective and better tolerated therapies are critically needed.
The toxicity profile of current leukaemia treatments remains a significant challenge, impacting patient quality of life and limiting therapeutic options; however, the exploration of metabolic characteristics reveals potential pathways for targeted therapies. The effectiveness of L-asparaginase in treating ALL exemplifies how understanding metabolic dependencies can inform treatment strategies. Individual susceptibility to asparaginase therapy has potential to inform its utilisation across the subtypes of leukaemia on a case-by-case basis, perhaps changing the outlook on asparaginase as an increasingly targeted therapy for both ALL and AML. This prompts the question of whether additional screening prior to therapy is necessary, which highlights the need to better understand the practical limitations of integrating these investigations into clinical practice, and may also involve considering the potential outsourcing of clinical investigations.
Continued collaboration among researchers, clinicians, and patients will be essential to navigate these scientific insights into clinical practice. While challenges remain in the fight against acute leukaemia, ongoing research into more personalised medicine offers promising avenues for enhancing treatment efficacy, while reducing toxicity, and provides hope for improved outcomes for all patients affected by AML and ALL.
Author Contributions
Conceptualisation, C.W. and S.R.C.; methodology, C.W., E.J. and R.S.; investigation, C.W.; resources, S.R.C. and C.W.; writing—original draft preparation, C.W.; writing—review and editing, C.W., E.J., R.S. and S.R.C.; visualisation, C.W.; supervision, E.J. and R.S.; acquisition, C.W. and S.R.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the UK Biotechnology and Biological Sciences Research Council (BBSRC) under grant numbers BB/J004529/1, BB/R012490/1, and BBS/E/F000PR10355 (S.R.C.). C.W. was supported by a student bursary from Big C Cancer Charity, registered charity number 281730.
Data Availability Statement
No new data were created or analysed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cancer Research UK. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics-for-the-uk (accessed on 17 November 2024).
- Chen, C.; Hao, X.; Lai, X.; Liu, L.; Zhu, J.; Shao, H.; Huang, D.; Gu, H.; Zhang, T.; Yu, Z.; et al. Oxidative phosphorylation enhances the leukemogenic capacity and resistance to chemotherapy of B cell acute lymphoblastic leukemia. Sci. Adv. 2021, 7, eabd6280. [Google Scholar] [CrossRef]
- Jia, L.; Gribben, J.G. Dangerous power: Mitochondria in CLL cells. Blood 2014, 123, 2596–2597. [Google Scholar] [CrossRef][Green Version]
- Baker, F.; Polat, I.H.; Abou-El-Ardat, K.; Alshamleh, I.; Thoelken, M.; Hymon, D.; Gubas, A.; Koschade, S.E.; Vischedyk, J.B.; Kaulich, M.; et al. Metabolic Rewiring Is Essential for AML Cell Survival to Overcome Autophagy Inhibition by Loss of ATG3. Cancers 2021, 13, 6142. [Google Scholar] [CrossRef]
- Galicia-Vázquez, G.; Smith, S.; Aloyz, R. Del11q-positive CLL lymphocytes exhibit altered glutamine metabolism and differential response to GLS1 and glucose metabolism inhibition. Blood Cancer J. 2018, 8, 13. [Google Scholar] [CrossRef]
- Mayer, R.L.; Schwarzmeier, J.D.; Gerner, M.C.; Bileck, A.; Mader, J.C.; Meier-Menches, S.M.; Gerner, S.M.; Schmetterer, K.G.; Pukrop, T.; Reichle, A.; et al. Proteomics and metabolomics identify molecular mechanisms of aging potentially predisposing for chronic lymphocytic leukemia. Mol. Cell. Proteom. 2018, 17, 290–303. [Google Scholar] [CrossRef]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Callahan, K.P.; Balys, M.; Ashton, J.M.; Neering, S.J.; Lagadinou, E.D.; Corbett, C.; Ye, H.; Liesveld, J.L.; et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem. 2013, 288, 33542–33558. [Google Scholar] [CrossRef]
- Thurgood, L.A.; Best, O.G.; Rowland, A.; Lower, K.M.; Brooks, D.A.; Kuss, B.J. Lipid uptake in chronic lymphocytic leukemia. Exp. Hematol. 2022, 106, 58–67. [Google Scholar] [CrossRef]
- Li, D.; Liang, J.; Yang, W.; Guo, W.; Song, W.; Zhang, W.; Wu, X.; He, B. A distinct lipid metabolism signature of acute myeloid leukemia with prognostic value. Front. Oncol. 2022, 12, 876981. [Google Scholar] [CrossRef]
- Schilstra, C.E.; McCleary, K.; Fardell, J.E.; Donoghoe, M.W.; McCormack, E.; Kotecha, R.S.; Lourenco, R.D.A.; Ramachandran, S.; Cockcroft, R.; Conyers, R.; et al. Prospective longitudinal evaluation of treatment-related toxicity and health-related quality of life during the first year of treatment for pediatric acute lymphoblastic leukemia. BMC Cancer 2022, 22, 985. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Schmiegelow, K.; Levinsen, M.F.; Attarbaschi, A.; Baruchel, A.; Devidas, M.; Escherich, G.; Gibson, B.; Heydrich, C.; Horibe, K.; Ishida, Y.; et al. Second malignant neoplasms after treatment of childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2013, 31, 2469–2476. [Google Scholar] [CrossRef]
- Dübbers, A.; Würthwein, G.; Müller, H.J.; Schulze-Westhoff, P.; Winkelhorst, M.; Kurzknabe, E.; Lanvers, C.; Pieters, R.; Kaspers, G.J.; Creutzig, U.; et al. Asparagine synthetase activity in paediatric acute leukaemias: AML-M5 subtype shows lowest activity. Br. J. Haematol. 2000, 109, 427–429. [Google Scholar] [CrossRef]
- Hlozkova, K.; Pecinova, A.; Alquezar-Artieda, N.; Pajuelo-Reguera, D.; Simcikova, M.; Hovorkova, L.; Rejlova, K.; Zaliova, M.; Mracek, T.; Kolenova, A.; et al. Metabolic profile of leukemia cells influences treatment efficacy of L-asparaginase. BMC Cancer 2020, 20, 526. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Cobaleda, C.; Godley, L.A.; Nichols, K.E.; Wlodarski, M.W.; Sanchez-Garcia, I. Insights into the Molecular Mechanisms of Genetic Predisposition to Hematopoietic Malignancies: The Importance of Gene–Environment Interactions. Cancer Discov. 2024, 14, 396–405. [Google Scholar] [CrossRef]
- Kachuri, L.; Jeon, S.; DeWan, A.T.; Metayer, C.; Ma, X.; Witte, J.S.; Chiang, C.W.K.; Wiemels, J.L.; de Smith, A.J. Genetic determinants of blood-cell traits influence susceptibility to childhood acute lymphoblastic leukemia. Am. J. Hum. Genet. 2021, 108, 1823–1835. [Google Scholar] [CrossRef]
- Rosendahl Huber, A.; Van Hoeck, A.; Van Boxtel, R. The Mutagenic Impact of Environmental Exposures in Human Cells and Cancer: Imprints Through Time. Front. Genet. 2021, 12, 760039. [Google Scholar] [CrossRef]
- Clark, A.D.; Nair, N.; Anderson, A.E.; Thalayasingam, N.; Naamane, N.; Skelton, A.J.; Diboll, J.; Barton, A.; Eyre, S.; Isaacs, J.D.; et al. Lymphocyte DNA methylation mediates genetic risk at shared immune-mediated disease loci. J. Allergy Clin. Immunol. 2020, 145, 1438–1451. [Google Scholar] [CrossRef]
- Wu, C.; Ning, H.; Liu, M.; Lin, J.; Luo, S.; Zhu, W.; Xu, J.; Wu, W.-C.; Liang, J.; Shao, C.-K.; et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J. Clin. Investig. 2018, 128, 3425–3438. [Google Scholar] [CrossRef]
- Wang, W.; Beird, H.; Kroll, C.J.; Hu, S.; Bueso-Ramos, C.E.; Fang, H.; Tang, G.; Tang, Z.; Wang, F.; Takahashi, K.; et al. T(6;14)(q25;q32) involves BCL11B and is highly associated with mixed-phenotype acute leukemia, T/myeloid. Leukemia 2020, 34, 2509–2512. [Google Scholar] [CrossRef]
- Di Giacomo, D.; La Starza, R.; Gorello, P.; Pellanera, F.; Kalender Atak, Z.; De Keersmaecker, K.; Pierini, V.; Harrison, C.J.; Arniani, S.; Moretti, M.; et al. 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T-lymphoid and myeloid immature acute leukemia. Blood 2021, 138, 773–784. [Google Scholar] [CrossRef]
- Montefiori, L.E.; Bendig, S.; Gu, Z.; Chen, X.; Pölönen, P.; Ma, X.; Murison, A.; Zeng, A.; Garcia-Prat, L.; Dickerson, K.; et al. Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage-Ambiguous Stem Cell Leukemia. Cancer Discov. 2021, 11, 2846–2867. [Google Scholar] [CrossRef]
- Alexander, T.B.; Gu, Z.; Iacobucci, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018, 562, 373–379. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Alvarnas, J.C.; Brown, P.A.; Aoun, P.; Ballen, K.K.; Barta, S.K.; Borate, U.; Boyer, M.W.; Burke, P.W.; Cassaday, R.; Castro, J.E.; et al. Acute Lymphoblastic Leukemia, Version 2.2015. J. Natl. Compr. Cancer Netw. 2015, 13, 1240–1279. [Google Scholar] [CrossRef]
- Faderl, S.; Kantarjian, H.M.; Talpaz, M.; Estrov, Z. Clinical Significance of Cytogenetic Abnormalities in Adult Acute Lymphoblastic Leukemia. Blood 1998, 91, 3995–4019. [Google Scholar] [CrossRef]
- van Dongen, J.J.; van der Velden, V.H.; Brüggemann, M.; Orfao, A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: Need for sensitive, fast, and standardized technologies. Blood 2015, 125, 3996–4009. [Google Scholar] [CrossRef]
- Brüggemann, M.; Raff, T.; Flohr, T.; Gökbuget, N.; Nakao, M.; Droese, J.; Lüschen, S.; Pott, C.; Ritgen, M.; Scheuring, U.; et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood 2006, 107, 1116–1123. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef]
- Burkart, M.; Dinner, S. Advances in the treatment of Philadelphia chromosome negative acute lymphoblastic leukemia. Blood Rev. 2024, 66, 101208. [Google Scholar] [CrossRef]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef]
- Bhansali, R.S.; Pratz, K.W.; Lai, C. Recent advances in targeted therapies in acute myeloid leukemia. J. Hematol. Oncol. 2023, 16, 29. [Google Scholar] [CrossRef]
- Di Francia, R.; Crisci, S.; De Monaco, A.; Cafiero, C.; Re, A.; Iaccarino, G.; De Filippi, R.; Frigeri, F.; Corazzelli, G.; Micera, A.; et al. Response and Toxicity to Cytarabine Therapy in Leukemia and Lymphoma: From Dose Puzzle to Pharmacogenomic Biomarkers. Cancers 2021, 13, 966. [Google Scholar] [CrossRef]
- Blair, H.A. Daunorubicin/Cytarabine Liposome: A Review in Acute Myeloid Leukaemia. Drugs 2018, 78, 1903–1910. [Google Scholar] [CrossRef]
- Wang, Y.; Ronckers, C.M.; van Leeuwen, F.E.; Moskowitz, C.S.; Leisenring, W.; Armstrong, G.T.; de Vathaire, F.; Hudson, M.M.; Kuehni, C.E.; Arnold, M.A.; et al. Subsequent female breast cancer risk associated with anthracycline chemotherapy for childhood cancer. Nat. Med. 2023, 29, 2268–2277. [Google Scholar] [CrossRef]
- Ohtake, S.; Miyawaki, S.; Fujita, H.; Kiyoi, H.; Shinagawa, K.; Usui, N.; Okumura, H.; Miyamura, K.; Nakaseko, C.; Miyazaki, Y.; et al. Randomized study of induction therapy comparing standard-dose idarubicin with high-dose daunorubicin in adult patients with previously untreated acute myeloid leukemia: The JALSG AML201 Study. Blood 2011, 117, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Stroet, A.; Hemmelmann, C.; Starck, M.; Zettl, U.; Dörr, J.; Friedemann, P.; Flachenecker, P.; Fleischer, V.; Zipp, F.; Nückel, H.; et al. Incidence of therapy-related acute leukaemia in mitoxantrone-treated multiple sclerosis patients in Germany. Ther. Adv. Neurol. Disord. 2012, 5, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Yanishevski, Y.; Nemec, J.; Evans, W.E.; Boyett, J.M.; Behm, F.G.; Pui, C.H. Etoposide and antimetabolite pharmacology in patients who develop secondary acute myeloid leukemia. Leukemia 1998, 12, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Cotteret, C.; Rousseau, J.; Zribi, K.; Schlatter, J. Severe hypersensitivity reaction to etoposide phosphate: A case report. Clin. Case Rep. 2020, 8, 1821–1823. [Google Scholar] [CrossRef] [PubMed]
- Apiraksattayakul, N.; Jitprapaikulsan, J.; Sanpakit, K.; Kumutpongpanich, T. Potential neurotoxicity associated with methotrexate. Sci. Rep. 2024, 14, 18548. [Google Scholar] [CrossRef]
- Maiguma, T.; Hayashi, Y.; Ueshima, S.; Kaji, H.; Egawa, T.; Chayama, K.; Morishima, T.; Kitamura, Y.; Sendo, T.; Gomita, Y.; et al. Relationship between oral mucositis and high-dose methotrexate therapy in pediatric acute lymphoblastic leukemia. Int. J. Clin. Pharmacol. Ther. 2008, 46, 584–590. [Google Scholar] [CrossRef]
- Gomber, S.; Dewan, P.; Chhonker, D. Vincristine induced neurotoxicity in cancer patients. Indian J. Pediatr. 2010, 77, 97–100. [Google Scholar] [CrossRef]
- Yasu, T.; Ohno, N.; Kawamata, T.; Kurokawa, Y. Vincristine-induced paralytic ileus during induction therapy of treatment protocols for acute lymphoblastic leukemia in adult patients. Int. J. Clin. Pharmacol. Ther. 2016, 54, 471–473. [Google Scholar] [CrossRef]
- Rank, C.U.; Wolthers, B.O.; Grell, K.; Albertsen, B.K.; Frandsen, T.L.; Overgaard, U.M.; Toft, N.; Nielsen, O.J.; Wehner, P.S.; Harila-Saari, A.; et al. Asparaginase-Associated Pancreatitis in Acute Lymphoblastic Leukemia: Results From the NOPHO ALL2008 Treatment of Patients 1-45 Years of Age. J. Clin. Oncol. 2020, 38, 145–154. [Google Scholar] [CrossRef]
- Hernández-Espinosa, D.; Miñano, A.; Martínez, C.; Pérez-Ceballos, E.; Heras, I.; Fuster, J.L.; Vicente, V.; Corral, J. L-asparaginase-induced antithrombin type I deficiency: Implications for conformational diseases. Am. J. Pathol. 2006, 169, 142–153. [Google Scholar] [CrossRef]
- Hongo, T.; Okada, S.; Ohzeki, T.; Ohta, H.; Nishimura, S.; Hamamoto, K.; Yagi, K.; Misu, H.; Eguchi, N.; Suzuki, N.; et al. Low plasma levels of hemostatic proteins during the induction phase in children with acute lymphoblastic leukemia: A retrospective study by the JACLS. Japan Association of Childhood Leukemia Study. Pediatr. Int. 2002, 44, 293–299. [Google Scholar] [CrossRef]
- Mitchell, L.; Hoogendoorn, H.; Giles, A.R.; Vegh, P.; Andrew, M. Increased endogenous thrombin generation in children with acute lymphoblastic leukemia: Risk of thrombotic complications in L’Asparaginase-induced antithrombin III deficiency. Blood 1994, 83, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Zerra, P.; Bergsagel, J.; Keller, F.G.; Lew, G.; Pauly, M. Maintenance Treatment With Low-Dose Mercaptopurine in Combination With Allopurinol in Children With Acute Lymphoblastic Leukemia and Mercaptopurine-Induced Pancreatitis. Pediatr. Blood Cancer 2016, 63, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Varma, P.P.; Subba, D.B.; Madhoosudanan, P. Cyclophosphamide Induced Haemorrhagic Cystitis (A Case Report). Med. J. Armed Forces India 1998, 54, 59–60. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, H.; Zhou, S.; Yu, M.; Wang, X.; Fu, K.; Qian, Z.; Zhang, H.; Qiu, L.; Liu, X.; et al. Risk of second malignant neoplasms after cyclophosphamide-based chemotherapy with or without radiotherapy for non-Hodgkin lymphoma. Leuk. Lymphoma 2013, 54, 1396–1404. [Google Scholar] [CrossRef]
- Naka, R.; Kondo, T.; Nishikubo, M.; Muranushi, H.; Ueda, Y.; Oka, T.; Wada, F.; Kanda, J.; Yamamoto, S.; Watanabe, M.; et al. PB1862: Venetoclax and Azacitidine Therapy in Acute Myeloid Leukemia Patients with Severe Renal Impairment. HemaSphere 2023, 7, e8463360. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Michael, L.; Björn, H.R.; Rainer, C.; Claudia, S.; Mathias, S.; Ulrich, G.; Andrea, K.; Volker, R.; Arnold, G.; Uwe, P.; et al. A multicenter phase II trial of decitabine as first-line treatment for older patients with acute myeloid leukemia judged unfit for induction chemotherapy. Haematologica 2012, 97, 393–401. [Google Scholar] [CrossRef]
- Rowe, J.M.; Tallman, M.S. How I treat acute myeloid leukemia. Blood 2010, 116, 3147–3156. [Google Scholar] [CrossRef]
- Rowe, J.M.; Buck, G.; Burnett, A.K.; Chopra, R.; Wiernik, P.H.; Richards, S.M.; Lazarus, H.M.; Franklin, I.M.; Litzow, M.R.; Ciobanu, N.; et al. Induction therapy for adults with acute lymphoblastic leukemia: Results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood 2005, 106, 3760–3767. [Google Scholar] [CrossRef]
- Esparza, S.; Muluneh, B.; Galeotti, J.; Matson, M.; Richardson, D.R.; Montgomery, N.D.; Coombs, C.C.; Jamieson, K.; Foster, M.C.; Zeidner, J.F. Venetoclax-induced tumour lysis syndrome in acute myeloid leukaemia. Br. J. Haematol. 2020, 188, 173–177. [Google Scholar] [CrossRef]
- McMahon, C.M.; Canaani, J.; Rea, B.; Sargent, R.L.; Qualtieri, J.N.; Watt, C.D.; Morrissette, J.J.D.; Carroll, M.; Perl, A.E. Gilteritinib induces differentiation in relapsed and refractory FLT3-mutated acute myeloid leukemia. Blood Adv. 2019, 3, 1581–1585. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; Botton, S.d.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.; Wang, E.S. Gemtuzumab ozogamicin for the treatment of acute myeloid leukemia. Expert Rev. Clin. Pharmacol. 2018, 11, 549–559. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; By, K.; Subramaniam, S.; Zhuang, L.; Del Valle, P.L.; Przepiorka, D.; Shen, Y.-L.; Sheth, C.M.; Liu, C.; Leong, R.; et al. FDA Approval Summary: Glasdegib for Newly Diagnosed Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6021–6025. [Google Scholar] [CrossRef]
- Ottmann, O.G.; Druker, B.J.; Sawyers, C.L.; Goldman, J.M.; Reiffers, J.; Silver, R.T.; Tura, S.; Fischer, T.; Deininger, M.W.; Schiffer, C.A.; et al. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome–positive acute lymphoid leukemias. Blood 2002, 100, 1965–1971. [Google Scholar] [CrossRef]
- Shen, S.; Chen, X.; Cai, J.; Yu, J.; Gao, J.; Hu, S.; Zhai, X.; Liang, C.; Ju, X.; Jiang, H.; et al. Effect of Dasatinib vs Imatinib in the Treatment of Pediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 358–366. [Google Scholar] [CrossRef]
- Özgür Yurttaş, N.; Eşkazan, A.E. Dasatinib-induced pulmonary arterial hypertension. Br. J. Clin. Pharmacol. 2018, 84, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Abu Rmilah, A.A.; Lin, G.; Begna, K.H.; Friedman, P.A.; Herrmann, J. Risk of QTc prolongation among cancer patients treated with tyrosine kinase inhibitors. Int. J. Cancer 2020, 147, 3160–3167. [Google Scholar] [CrossRef]
- Stein, A.S.; Schiller, G.; Benjamin, R.; Jia, C.; Zhang, A.; Zhu, M.; Zimmerman, Z.; Topp, M.S. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: Management and mitigating factors. Ann. Hematol. 2019, 98, 159–167. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Advani, A.S.; Stelljes, M.; Kebriaei, P.; Cassaday, R.D.; Merchant, A.A.; Fujishima, N.; Uchida, T.; Calbacho, M.; et al. Hepatic adverse event profile of inotuzumab ozogamicin in adult patients with relapsed or refractory acute lymphoblastic leukaemia: Results from the open-label, randomised, phase 3 INO-VATE study. Lancet Haematol. 2017, 4, e387–e398. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah (accessed on 17 November 2024).
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Dubey, M.; Randhawa, A.S.; Khanikar, D.; Hazarika, M.; Roy, P.S.; Dutta, C.; Barbhuiyan, S.; Deka, R. Improved Treatment Outcomes With Modified Induction Therapy in Acute Myeloid Leukemia (AML): A Retrospective Observational Study From a Regional Cancer Center. Cureus 2024, 16, e53303. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Saul, D.; Braun, M.; Tohumeken, S.; Völkl, S.; Kischel, R.; Lutteropp, M.; Dos Santos, C.; Mackensen, A.; Mougiakakos, D. CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J. Immunother. Cancer 2018, 6, 116. [Google Scholar] [CrossRef]
- Ebinger, S.; Zeller, C.; Carlet, M.; Senft, D.; Bagnoli, J.W.; Liu, W.H.; Rothenberg-Thurley, M.; Enard, W.; Metzeler, K.H.; Herold, T.; et al. Plasticity in growth behavior of patients’ acute myeloid leukemia stem cells growing in mice. Haematologica 2020, 105, 2855–2860. [Google Scholar] [CrossRef]
- Mukherjee, A.; Ahmed, N.; Rose, F.T.; Ahmad, A.N.; Javed, T.A.; Wen, L.; Bottino, R.; Xiao, X.; Kilberg, M.S.; Husain, S.Z. Asparagine Synthetase Is Highly Expressed at Baseline in the Pancreas Through Heightened PERK Signaling. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 1–13. [Google Scholar] [CrossRef]
- Merryman, R.; Stevenson, K.E.; Gostic, W.J., 2nd; Neuberg, D.; O’Brien, J.; Sallan, S.E.; Silverman, L.B. Asparaginase-associated myelosuppression and effects on dosing of other chemotherapeutic agents in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2012, 59, 925–927. [Google Scholar] [CrossRef]
- Kieslich, M.; Porto, L.; Lanfermann, H.; Jacobi, G.; Schwabe, D.; Böhles, H. Cerebrovascular Complications of l-Asparaginase in the Therapy of Acute Lymphoblastic Leukemia. J. Pediatr. Hematol./Oncol. 2003, 25, 484–487. [Google Scholar] [CrossRef]
- Rathod, S.; Ramsey, M.; Relling, M.V.; Finkelman, F.D.; Fernandez, C.A. Hypersensitivity reactions to asparaginase in mice are mediated by anti-asparaginase IgE and IgG and the immunoglobulin receptors FcεRI and FcγRIII. Haematologica 2019, 104, 319–329. [Google Scholar] [CrossRef]
- Kamal, N.; Koh, C.; Samala, N.; Fontana, R.J.; Stolz, A.; Durazo, F.; Hayashi, P.H.; Phillips, E.; Wang, T.; Hoofnagle, J.H. Asparaginase-induced hepatotoxicity: Rapid development of cholestasis and hepatic steatosis. Hepatol. Int. 2019, 13, 641–648. [Google Scholar] [CrossRef]
- Lavine, R.L.; Brodsky, I.; Garofano, C.D.; Rose, L.I. The effect of E. coli L-asparaginase on oral glucose tolerance and insulin release in man. Diabetologia 1978, 15, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Cetin, M.; Yetgin, S.; Kara, A.; Tuncer, A.M.; Günay, M.; Gümrük, F.; Gürgey, A. Hyperglycemia, ketoacidosis and other complications of L-asparaginase in children with acute lymphoblastic leukemia. J. Med. 1994, 25, 219–229. [Google Scholar]
- Erdem, A.; Marin, S.; Pereira-Martins, D.A.; Cortés, R.; Cunningham, A.; Pruis, M.G.; de Boer, B.; van den Heuvel, F.A.J.; Geugien, M.; Wierenga, A.T.J.; et al. The Glycolytic Gatekeeper PDK1 defines different metabolic states between genetically distinct subtypes of human acute myeloid leukemia. Nat. Commun. 2022, 13, 1105. [Google Scholar] [CrossRef]
- Chen, W.-L.; Wang, J.-H.; Zhao, A.-H.; Xu, X.; Wang, Y.-H.; Chen, T.-L.; Li, J.-M.; Mi, J.-Q.; Zhu, Y.-M.; Liu, Y.-F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef]
- Hermanova, I.; Arruabarrena-Aristorena, A.; Valis, K.; Nuskova, H.; Alberich-Jorda, M.; Fiser, K.; Fernandez-Ruiz, S.; Kavan, D.; Pecinova, A.; Niso-Santano, M.; et al. Pharmacological inhibition of fatty-acid oxidation synergistically enhances the effect of l-asparaginase in childhood ALL cells. Leukemia 2016, 30, 209–218. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 234. [Google Scholar] [CrossRef]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef]
- de Boer, B.; Prick, J.; Pruis, M.G.; Keane, P.; Imperato, M.R.; Jaques, J.; Brouwers-Vos, A.Z.; Hogeling, S.M.; Woolthuis, C.M.; Nijk, M.T.; et al. Prospective Isolation and Characterization of Genetically and Functionally Distinct AML Subclones. Cancer Cell 2018, 34, 674–689.e8. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Lutz, C.; van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J.; et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011, 469, 356–361. [Google Scholar] [CrossRef]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef]
- Poort, V.M.; Hagelaar, R.; van Roosmalen, M.J.; Trabut, L.; Buijs-Gladdines, J.G.C.A.M.; van Wijk, B.; Meijerink, J.; van Boxtel, R. Transient Differentiation-State Plasticity Occurs during Acute Lymphoblastic Leukemia Initiation. Cancer Res. 2024, 84, 2720–2733. [Google Scholar] [CrossRef]
- Morris, V.; Marion, W.; Hughes, T.; Sousa, P.; Sensharma, P.; Pikman, Y.; Harris, M.; Shalek, A.K.; North, T.E.; Daley, G.Q.; et al. Single-cell analysis reveals mechanisms of plasticity of leukemia initiating cells. bioRxiv 2020. preprint. [Google Scholar] [CrossRef]
- Nguyen, H.A.; Su, Y.; Zhang, J.Y.; Antanasijevic, A.; Caffrey, M.; Schalk, A.M.; Liu, L.; Rondelli, D.; Oh, A.; Mahmud, D.L.; et al. A Novel l-Asparaginase with low l-Glutaminase Coactivity Is Highly Efficacious against Both T- and B-cell Acute Lymphoblastic Leukemias In Vivo. Cancer Res. 2018, 78, 1549–1560. [Google Scholar] [CrossRef]
- Sengupta, S.; Biswas, M.; Gandhi, K.A.; Gupta, S.K.; Gera, P.B.; Gota, V.; Sonawane, A. Preclinical evaluation of engineered L-asparaginase variants to improve the treatment of Acute Lymphoblastic Leukemia. Transl. Oncol. 2024, 43, 101909. [Google Scholar] [CrossRef]
- Schmidt, M.P.; Ivanov, A.V.; Coriu, D.; Miron, I.C. L-Asparaginase Toxicity in the Treatment of Children and Adolescents with Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 4419. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Kaspers, G.J.; Pieters, R.; Ramakers-Van Woerden, N.L.; den Boer, M.L.; Wünsche, R.; Rottier, M.M.; Hählen, K.; van Wering, E.R.; Janka-Schaub, G.E.; et al. Cellular drug resistance profiles in childhood acute myeloid leukemia: Differences between FAB types and comparison with acute lymphoblastic leukemia. Blood 2000, 96, 2879–2886. [Google Scholar]
- Michelozzi, I.M.; Granata, V.; De Ponti, G.; Alberti, G.; Tomasoni, C.; Antolini, L.; Gambacorti-Passerini, C.; Gentner, B.; Dazzi, F.; Biondi, A.; et al. Acute myeloid leukaemia niche regulates response to L-asparaginase. Br. J. Haematol. 2019, 186, 420–430. [Google Scholar] [CrossRef]
- Kaspers, G.J.L. Acute myeloid leukaemia niche regulates response to L-asparaginase. Br. J. Haematol. 2019, 186, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Capizzi, R.L.; Davis, R.; Powell, B.; Cuttner, J.; Ellison, R.R.; Cooper, M.R.; Dillman, R.; Major, W.B.; Dupre, E.; McIntyre, O.R. Synergy between high-dose cytarabine and asparaginase in the treatment of adults with refractory and relapsed acute myelogenous leukemia—A Cancer and Leukemia Group B Study. J. Clin. Oncol. 1988, 6, 499–508. [Google Scholar] [CrossRef]
- Buaboonnam, J.; Cao, X.; Pauley, J.L.; Pui, C.H.; Ribeiro, R.C.; Rubnitz, J.E.; Inaba, H. Sequential administration of methotrexate and asparaginase in relapsed or refractory pediatric acute myeloid leukemia. Pediatr. Blood Cancer 2013, 60, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, J.; Zeng, H.; Zhang, Y.; Zhang, Y.; Zhou, X.; Zhou, H. Antiproliferative effects of L-asparaginase in acute myeloid leukemia. Exp. Ther. Med. 2020, 20, 2070–2078. [Google Scholar] [CrossRef]
- Martínez-Cuadrón, D.; Boluda, B.; Martínez, P.; Bergua, J.; Rodríguez-Veiga, R.; Esteve, J.; Vives, S.; Serrano, J.; Vidriales, B.; Salamero, O.; et al. A phase I-II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann. Hematol. 2018, 97, 763–772. [Google Scholar] [CrossRef]
- Mishra, S.K.; Millman, S.E.; Zhang, L. Metabolism in acute myeloid leukemia: Mechanistic insights and therapeutic targets. Blood 2023, 141, 1119–1135. [Google Scholar] [CrossRef]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef]
- Chan, W.K.; Horvath, T.D.; Tan, L.; Link, T.; Harutyunyan, K.G.; Pontikos, M.A.; Anishkin, A.; Du, D.; Martin, L.A.; Yin, E.; et al. Glutaminase Activity of L-Asparaginase Contributes to Durable Preclinical Activity against Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 2019, 18, 1587–1592. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).