
Tolerance of Oncogene-Induced Replication Stress: A Fuel for Genomic Instability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
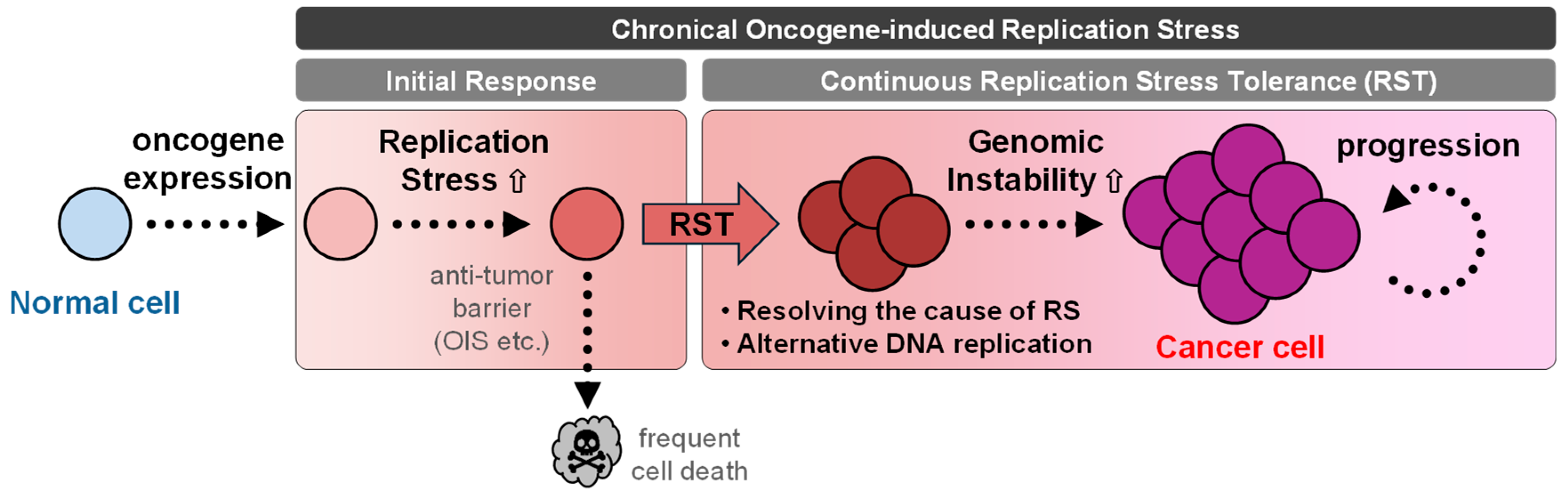
1. Introduction
2. The Impact of Oncogene Activation on DNA Replication
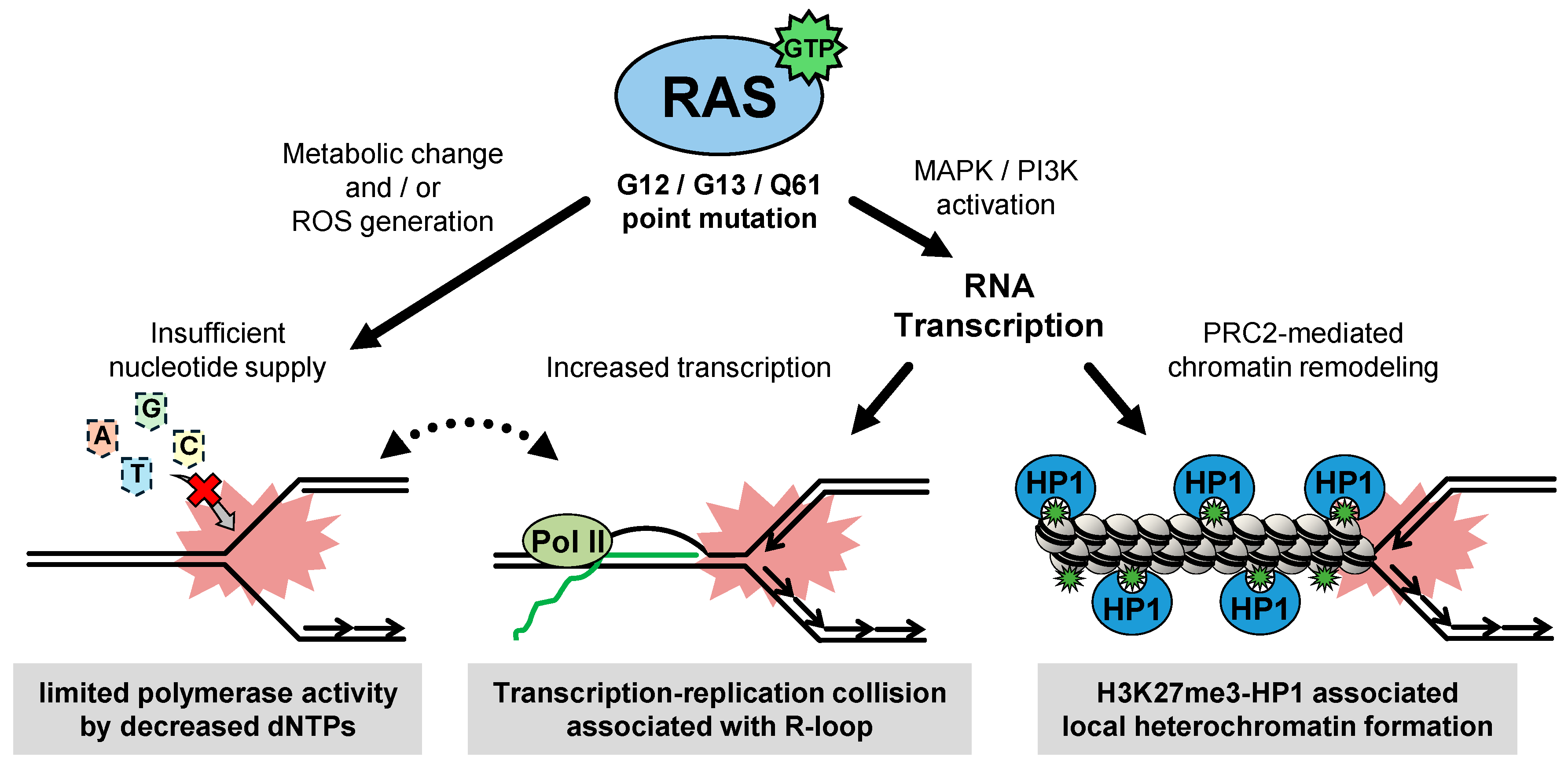
2.1. RAS-Induced RS
2.1.1. The Function of Oncogenic RAS
2.1.2. The Entity of Oncogenic-RAS Induced RS
2.1.3. The Consequence of RAS-Induced RS During Gaining of GIN
2.1.4. Exploring Causes of Oncogenic RAS-Induced RS
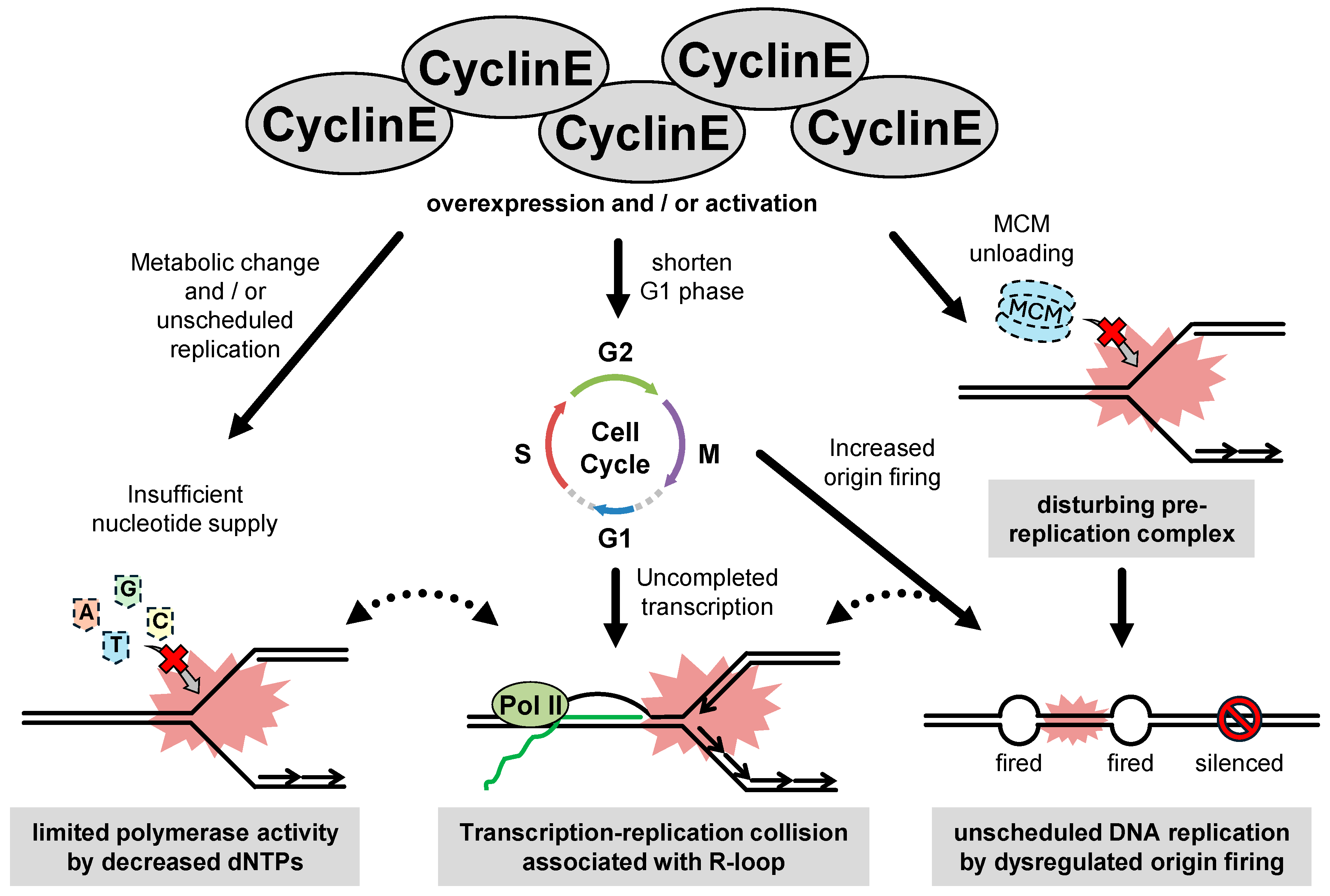
2.2. Cyclin E-Induced RS
2.2.1. The Function of Cyclin E
2.2.2. The Entity of Cyclin E-Induced RS
2.2.3. The Consequence of Cyclin E-Induced RS During Gaining of GIN
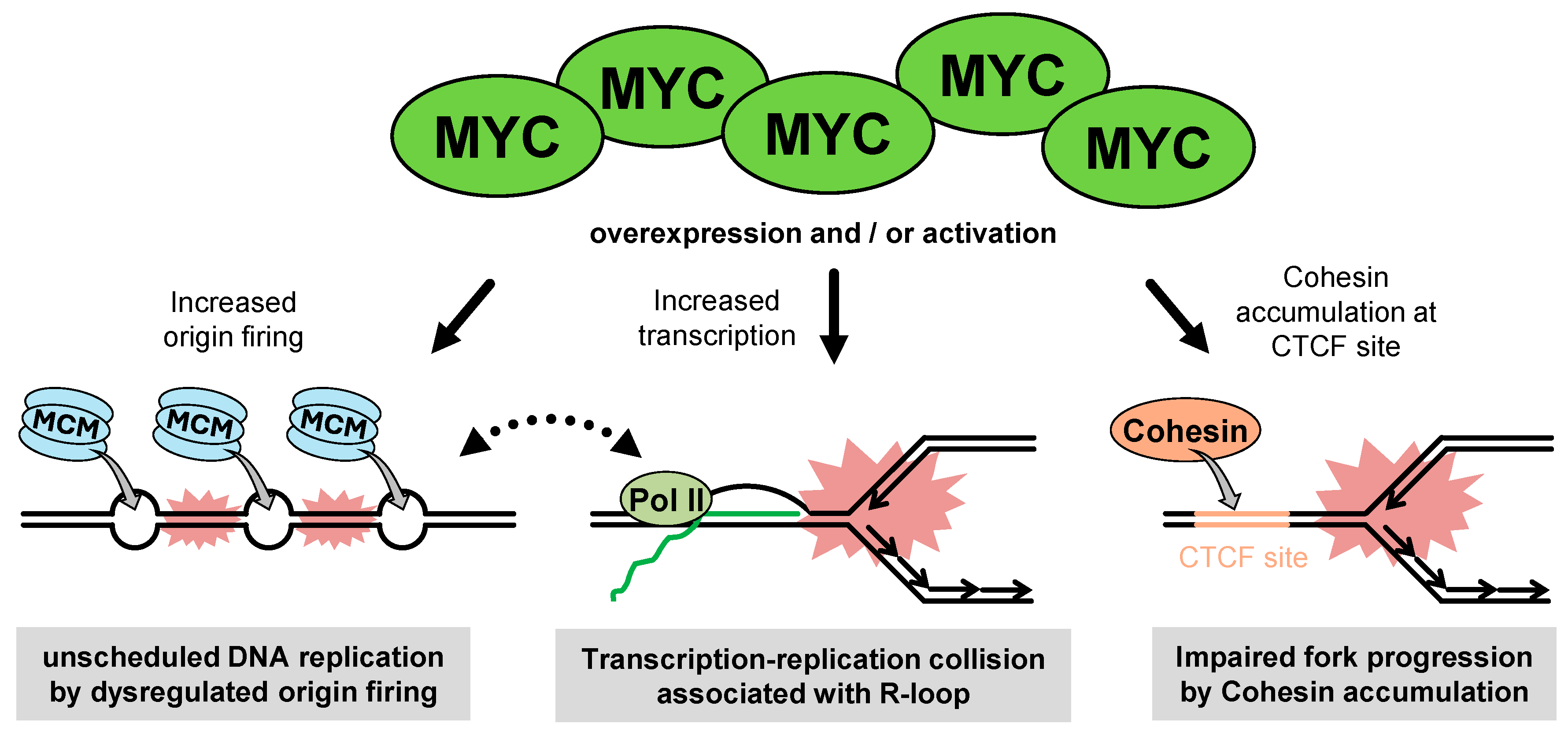
2.3. MYC-Induced RS
2.3.1. The Function of MYC
2.3.2. The Entity of MYC-Induced RS
2.3.3. Non-Transcriptional Role of MYC in RS
2.3.4. MYC-Mediated RST Mechanisms During Gaining of GIN
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roos, W.P.; Kaina, B. DNA Damage-Induced Cell Death: From Specific DNA Lesions to the DNA Damage Response and Apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Lee, J.K.; Choi, Y.-L.; Kwon, M.; Park, P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 283–312. [Google Scholar] [CrossRef]
- Loeb, L.A. Mutator Phenotype May Be Required for Multistage Carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar]
- Boland, C.R.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability—An Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased Global Transcription Activity as a Mechanism of Replication Stress in Cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Igarashi, T.; Mazevet, M.; Yasuhara, T.; Yano, K.; Mochizuki, A.; Nishino, M.; Yoshida, T.; Yoshida, Y.; Takamatsu, N.; Yoshimi, A.; et al. An ATR-PrimPol Pathway Confers Tolerance to Oncogenic KRAS-Induced and Heterochromatin-Associated Replication Stress. Nat. Commun. 2023, 14, 4991. [Google Scholar] [CrossRef]
- Kakiuchi, N.; Ogawa, S. Clonal Expansion in Non-Cancer Tissues. Nat. Rev. Cancer 2021, 21, 239–256. [Google Scholar] [CrossRef]
- Brickner, J.R.; Garzon, J.L.; Cimprich, K.A. Walking a Tightrope: The Complex Balancing Act of R-Loops in Genome Stability. Mol. Cell 2022, 82, 2267–2297. [Google Scholar] [CrossRef]
- Petermann, E.; Lan, L.; Zou, L. Sources, Resolution and Physiological Relevance of R-Loops and RNA–DNA Hybrids. Nat. Rev. Mol. Cell Biol. 2022, 23, 521–540. [Google Scholar] [CrossRef]
- Pai, C.C.; Kearsey, S.E. A Critical Balance: DNTPs and the Maintenance of Genome Stability. Genes 2017, 8, 57. [Google Scholar] [CrossRef]
- Saxena, S.; Nabel, C.S.; Seay, T.W.; Patel, P.S.; Kawale, A.S.; Crosby, C.R.; Tigro, H.; Oh, E.; Vander Heiden, M.G.; Hata, A.N.; et al. Unprocessed Genomic Uracil as a Source of DNA Replication Stress in Cancer Cells. Mol. Cell 2024, 84, 2036–2052.e7. [Google Scholar] [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding Pancreatic Cancer Proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, H.N.; Jeong, M.S.; Jang, S.B. Oncogenic KRAS: Signaling and Drug Resistance. Cancers 2021, 13, 5599. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Gideon, P.; John, J.; Frech, M.; Lautwein, A.; Clark, R.; Scheffler, J.E.; Wittinghofer, A. Mutational and Kinetic Analyses of the GTPase-Activating Protein (GAP)-P21 Interaction: The C-Terminal Domain of GAP Is Not Sufficient for Full Activity. Mol. Cell. Biol. 1992, 12, 2050–2056. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from Biology to Therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Quinet, A.; Carvajal-Maldonado, D.; Lemacon, D.; Vindigni, A. DNA Fiber Analysis: Mind the Gap! Methods Enzymol. 2017, 591, 55–82. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-Induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Carlos, A.R.; Escandell, J.M.; Kotsantis, P.; Suwaki, N.; Bouwman, P.; Badie, S.; Folio, C.; Benitez, J.; Gomez-Lopez, G.; Pisano, D.G.; et al. ARF Triggers Senescence in Brca2-Deficient Cells by Altering the Spectrum of P53 Transcriptional Targets. Nat. Commun. 2013, 4, 2697. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-Induced Senescence Is Part of the Tumorigenesis Barrier Imposed by DNA Damage Checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras Oncogenes Engage Different Energy Metabolism Programs and Evoke Distinct Patterns of Oxidative and DNA Replication Stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef]
- Leikam, C.; Hufnagel, A.; Schartl, M.; Meierjohann, S. Oncogene Activation in Melanocytes Links Reactive Oxygen to Multinucleated Phenotype and Senescence. Oncogene 2008, 27, 7070–7082. [Google Scholar] [CrossRef]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; D’Adda Di Fagagna, F. Oncogene-Induced Reactive Oxygen Species Fuel Hyperproliferation and DNA Damage Response Activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef]
- Weyemi, U.; Lagente-Chevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.M.; et al. ROS-Generating NADPH Oxidase NOX4 Is a Critical Mediator in Oncogenic H-Ras-Induced DNA Damage and Subsequent Senescence. Oncogene 2012, 31, 1117–1129. [Google Scholar] [CrossRef]
- Andrs, M.; Stoy, H.; Boleslavska, B.; Chappidi, N.; Kanagaraj, R.; Nascakova, Z.; Menon, S.; Rao, S.; Oravetzova, A.; Dobrovolna, J.; et al. Excessive Reactive Oxygen Species Induce Transcription-Dependent Replication Stress. Nat. Commun. 2023, 14, 1791. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS Oncogenes: Weaving a Tumorigenic Web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-Loop-Mediated Genomic Instability Is Caused by Impairment of Replication Fork Progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Karakaidos, P.; Herlyn, M.; Liloglou, T.; Zacharatos, P.; Venere, M.; Kittas, C.; Vassiliou, L.-V.F.; Levy, B.; Kastrinakis, N.G.; DiTullio, R.A.; et al. Activation of the DNA Damage Checkpoint and Genomic Instability in Human Precancerous Lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR Suppression with Oncogenic Ras Synergistically Increases Genomic Instability, Causing Synthetic Lethality or Tumorigenesis in a Dosage-Dependent Manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic Stress Sensitizes Murine Cancers to Hypomorphic Suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef]
- López-Contreras, A.J.; Gutierrez-Martinez, P.; Specks, J.; Rodrigo-Perez, S.; Fernandez-Capetillo, O. An Extra Allele of Chk1 Limits Oncogene-Induced Replicative Stress and Promotes Transformation. J. Exp. Med. 2012, 209, 455–461. [Google Scholar] [CrossRef]
- Bianco, J.N.; Bergoglio, V.; Lin, Y.-L.; Pillaire, M.-J.; Schmitz, A.-L.; Gilhodes, J.; Lusque, A.; Mazières, J.; Lacroix-Triki, M.; Roumeliotis, T.I.; et al. Overexpression of Claspin and Timeless Protects Cancer Cells from Replication Stress in a Checkpoint-Independent Manner. Nat. Commun. 2019, 10, 910. [Google Scholar] [CrossRef]
- Mehta, K.P.M.; Thada, V.; Zhao, R.; Krishnamoorthy, A.; Leser, M.; Lindsey Rose, K.; Cortez, D. CHK1 Phosphorylates PRIMPOL to Promote Replication Stress Tolerance. Sci. Adv. 2022, 8, eabm0314. [Google Scholar] [CrossRef]
- Saldanha, J.; Rageul, J.; Patel, J.A.; Phi, A.L.; Lo, N.; Park, J.J.; Kim, H. The TIMELESS and PARP1 Interaction Suppresses Replication-Associated DNA Gap Accumulation. Nucleic Acids Res. 2024, 52, 6424–6440. [Google Scholar] [CrossRef]
- Petropoulos, M.; Karamichali, A.; Rossetti, G.G.; Freudenmann, A.; Iacovino, L.G.; Dionellis, V.S.; Sotiriou, S.K.; Halazonetis, T.D. Transcription–Replication Conflicts Underlie Sensitivity to PARP Inhibitors. Nature 2024, 628, 433–441. [Google Scholar] [CrossRef]
- Buchynska, L.; Gordiienko, I.; Glushchenko, N.; Iurchenko, N. The KRAS, ATR and CHEK1 Expression Levels in Endometrial Cancer Are the Risk Factors Predicting Recurrence. PLoS ONE 2024, 19, e0302075. [Google Scholar] [CrossRef]
- Sarni, D.; Barroso, S.; Shtrikman, A.; Irony-Tur Sinai, M.; Oren, Y.S.; Aguilera, A.; Kerem, B. Topoisomerase 1-Dependent R-Loop Deficiency Drives Accelerated Replication and Genomic Instability. Cell Rep. 2022, 40, 111397. [Google Scholar] [CrossRef]
- Lambert, S.; Carr, A.M. Impediments to Replication Fork Movement: Stabilisation, Reactivation and Genome Instability. Chromosoma 2013, 122, 33–45. [Google Scholar] [CrossRef]
- Kurashima, K.; Kashiwagi, H.; Shimomura, I.; Suzuki, A.; Takeshita, F.; Mazevet, M.; Harata, M.; Yamashita, T.; Yamamoto, Y.; Kohno, T.; et al. SMARCA4 Deficiency-Associated Heterochromatin Induces Intrinsic DNA Replication Stress and Susceptibility to ATR Inhibition in Lung Adenocarcinoma. NAR Cancer 2020, 2, zcaa005. [Google Scholar] [CrossRef]
- Schvartzman, J.; Forsyth, G.; Walch, H.; Chatila, W.; Taglialatela, A.; Lee, B.J.; Zhu, X.; Gershik, S.; Cimino, F.V.; Santella, A.; et al. Oncogenic IDH Mutations Increase Heterochromatin-Related Replication Stress without Impacting Homologous Recombination. Mol. Cell 2023, 83, 2347–2356.e8. [Google Scholar] [CrossRef]
- Serresi, M.; Siteur, B.; Hulsman, D.; Company, C.; Schmitt, M.J.; Lieftink, C.; Morris, B.; Cesaroni, M.; Proost, N.; Beijersbergen, R.L.; et al. Ezh2 Inhibition in Kras-Driven Lung Cancer Amplifies Inflammation and Associated Vulnerabilities. J. Exp. Med. 2018, 215, 3115–3135. [Google Scholar] [CrossRef]
- Shih, C.; Weinberg, R.A. Isolation of a Transforming Sequence from a Human Bladder Carcinoma Cell Line. Cell 1982, 29, 161–169. [Google Scholar] [CrossRef]
- Santos, E.; Tronick, S.R.; Aaronson, S.A.; Pulciani, S.; Barbacid, M. T24 Human Bladder Carcinoma Oncogene Is an Activated Form of the Normal Human Homologue of BALB- and Harvey-MSV Transforming Genes. Nature 1982, 298, 343–347. [Google Scholar] [CrossRef]
- Der, C.J.; Krontiris, T.G.; Cooper, G.M. Transforming Genes of Human Bladder and Lung Carcinoma Cell Lines Are Homologous to the Ras Genes of Harvey and Kirsten Sarcoma Viruses. Proc. Natl. Acad. Sci. USA 1982, 79, 3637–3640. [Google Scholar] [CrossRef]
- Weinberg, R.A. It Took a Long, Long Time: Ras and the Race to Cure Cancer. Cell 2024, 187, 1574–1577. [Google Scholar] [CrossRef]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human Cyclin E, a Nuclear Protein Essential for the G1-to-S Phase Transition. Mol. Cell. Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef]
- Kelly, B.L.; Wolfe, K.G.; Roberts, J.M. Identification of a Substrate-Targeting Domain in Cyclin E Necessary for Phosphorylation of the Retinoblastoma Protein. Proc. Natl. Acad. Sci. USA 1998, 95, 2535–2540. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and P53 Pathways in Cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Hwang, H.C.; Clurman, B.E. Cyclin E in Normal and Neoplastic Cell Cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef]
- Chu, C.; Geng, Y.; Zhou, Y.; Sicinski, P. Cyclin E in Normal Physiology and Disease States. Trends Cell Biol. 2021, 31, 732–746. [Google Scholar] [CrossRef]
- Guerrero Llobet, S.; van der Vegt, B.; Jongeneel, E.; Bense, R.D.; Zwager, M.C.; Schröder, C.P.; Everts, M.; Fehrmann, R.S.N.; de Bock, G.H.; van Vugt, M.A.T.M. Cyclin E Expression Is Associated with High Levels of Replication Stress in Triple-Negative Breast Cancer. npj Breast Cancer 2020, 6, 40. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; Weir, B.A.; Au-Yeung, G.; Alsop, K.; Mitchell, G.; George, J.; Davis, S.; D’Andrea, A.D.; Simpson, K.; Hahn, W.C.; et al. Synthetic Lethality between CCNE1 Amplification and Loss of BRCA1. Proc. Natl. Acad. Sci. USA 2013, 110, 19489–19494. [Google Scholar] [CrossRef]
- Siu, K.T.; Rosner, M.R.; Minella, A.C. An Integrated View of Cyclin E Function and Regulation. Cell Cycle 2012, 11, 57–64. [Google Scholar] [CrossRef]
- Ekholm-Reed, S.; Méndez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of Cyclin E in Human Cells Interferes with Prereplication Complex Assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased Replication Initiation and Conflicts with Transcription Underlie Cyclin E-Induced Replication Stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. Intragenic Origins Due to Short G1 Phases Underlie Oncogene-Induced DNA Replication Stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef]
- Lõoke, M.; Reimand, J.; Sedman, T.; Sedman, J.; Järvinen, L.; Värv, S.; Peil, K.; Kristjuhan, K.; Vilo, J.; Kristjuhan, A. Relicensing of Transcriptionally Inactivated Replication Origins in Budding Yeast. J. Biol. Chem. 2010, 285, 40004–40011. [Google Scholar] [CrossRef]
- Resnitzky, D.; Gossen, M.; Bujard, H.; Reed, S.I. Acceleration of the G1/S Phase Transition by Expression of Cyclins Dl and E with an Inducible System. Mol. Cell. Biol. 1994, 14, 1669–1679. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Neelsen, K.J.; Zanini, I.M.Y.; Herrador, R.; Lopes, M. Oncogenes Induce Genotoxic Stress by Mitotic Processing of Unusual Replication Intermediates. J. Cell Biol. 2013, 200, 699–708. [Google Scholar] [CrossRef]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA Damage Response as a Candidate Anti-Cancer Barrier in Early Human Tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Spruck, C.H.; Won, K.A.; Reed, S.I. Deregulated Cyclin E Induces Chromosome Instability. Nature 1999, 401, 297–300. [Google Scholar] [CrossRef]
- Brunner, A.; Li, Q.; Fisicaro, S.; Kourtesakis, A.; Viiliäinen, J.; Johansson, H.J.; Pandey, V.; Mayank, A.K.; Lehtiö, J.; Wohlschlegel, J.A.; et al. FBXL12 Degrades FANCD2 to Regulate Replication Recovery and Promote Cancer Cell Survival under Conditions of Replication Stress. Mol. Cell 2023, 83, 3720–3739.e8. [Google Scholar] [CrossRef]
- Audrey, A.; Kok, Y.P.; Yu, S.; de Haan, L.; van de Kooij, B.; van den Tempel, N.; Chen, M.; de Boer, H.R.; van der Vegt, B.; van Vugt, M.A.T.M. RAD52-Dependent Mitotic DNA Synthesis Is Required for Genome Stability in Cyclin E1-Overexpressing Cells. Cell Rep. 2024, 43, 114116. [Google Scholar] [CrossRef]
- Marques, J.F.; Kops, G.J.P.L. Permission to Pass: On the Role of P53 as a Gatekeeper for Aneuploidy. Chromosom. Res. 2023, 31, 31. [Google Scholar] [CrossRef]
- Zeng, J.; Hills, S.A.; Ozono, E.; Diffley, J.F.X. Cyclin E-Induced Replicative Stress Drives P53-Dependent Whole-Genome Duplication. Cell 2023, 186, 528–542.e14. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC Protein Interactors in Gene Transcription and Cancer. Nat. Rev. Cancer 2021, 21, 579–591. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-Cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional Amplification in Tumor Cells with Elevated C-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. C-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef]
- Sabò, A.; Kress, T.R.; Pelizzola, M.; De Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective Transcriptional Regulation by Myc in Cellular Growth Control and Lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; Von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and Repression by Oncogenic MYC Shape Tumour-Specific Gene Expression Profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R Loops: From Transcription Byproducts to Threats to Genome Stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-Replication Encounters, Consequences and Genomic Instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef]
- Murayama, T.; Takeuchi, Y.; Yamawaki, K.; Natsume, T.; Li, M.; Marcela, R.C.N.; Nishimura, T.; Kogure, Y.; Nakata, A.; Tominaga, K.; et al. MCM10 Compensates for Myc-Induced DNA Replication Stress in Breast Cancer Stem-like Cells. Cancer Sci. 2021, 112, 1209–1224. [Google Scholar] [CrossRef]
- Lin, P.; Lourenco, C.; Cruickshank, J.; Palomero, L.; van Leeuwen, J.E.; Tong, A.H.Y.; Chan, K.; El Ghamrasni, S.; Pujana, M.A.; Cescon, D.W.; et al. Topoisomerase 1 Inhibition in MYC-Driven Cancer Promotes Aberrant R-Loop Accumulation to Induce Synthetic Lethality. Cancer Res. 2023, 183, 4015–4029. [Google Scholar] [CrossRef]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I Suppresses Genomic Instability by Preventing Interference between Replication and Transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef]
- Promonet, A.; Padioleau, I.; Liu, Y.; Sanz, L.; Biernacka, A.; Schmitz, A.L.; Skrzypczak, M.; Sarrazin, A.; Mettling, C.; Rowicka, M.; et al. Topoisomerase 1 Prevents Replication Stress at R-Loop-Enriched Transcription Termination Sites. Nat. Commun. 2020, 11, 3940. [Google Scholar] [CrossRef]
- Peripolli, S.; Meneguello, L.; Perrod, C.; Singh, T.; Patel, H.; Rahman, S.T.; Kiso, K.; Thorpe, P.; Calvanese, V.; Bertoli, C.; et al. Oncogenic C-Myc Induces Replication Stress by Increasing Cohesins Chromatin Occupancy in a CTCF-Dependent Manner. Nat. Commun. 2024, 15, 1579. [Google Scholar] [CrossRef]
- Das, S.K.; Kuzin, V.; Cameron, D.P.; Sanford, S.; Jha, R.K.; Nie, Z.; Rosello, M.T.; Holewinski, R.; Andresson, T.; Wisniewski, J.; et al. MYC Assembles and Stimulates Topoisomerases 1 and 2 in a “Topoisome”. Mol. Cell 2022, 82, 140–158.e12. [Google Scholar] [CrossRef]
- Solvie, D.; Baluapuri, A.; Uhl, L.; Fleischhauer, D.; Endres, T.; Papadopoulos, D.; Aziba, A.; Gaballa, A.; Mikicic, I.; Isaakova, E.; et al. MYC Multimers Shield Stalled Replication Forks from RNA Polymerase. Nature 2022, 612, 148–155. [Google Scholar] [CrossRef]
- Herold, S.; Kalb, J.; Büchel, G.; Ade, C.P.; Baluapuri, A.; Xu, J.; Koster, J.; Solvie, D.; Carstensen, A.; Klotz, C.; et al. Recruitment of BRCA1 Limits MYCN-Driven Accumulation of Stalled RNA Polymerase. Nature 2019, 567, 545–549. [Google Scholar] [CrossRef]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef]
- Roeschert, I.; Poon, E.; Henssen, A.G.; Dorado Garcia, H.; Gatti, M.; Giansanti, C.; Jamin, Y.; Ade, C.P.; Gallant, P.; Schülein-Völk, C.; et al. Combined Inhibition of Aurora-A and ATR Kinases Results in Regression of MYCN-Amplified Neuroblastoma. Nat. Cancer 2021, 2, 312–326. [Google Scholar] [CrossRef]
- Studzinski, G.P.; Brelvi, Z.S.; Feldman, S.C.; Watt, R.A. Participation of C-Myc Protein in DNA Synthesis of Human Cells. Science 1986, 234, 467–470. [Google Scholar] [CrossRef]
- Iguchi-Ariga, S.M.; Okazaki, T.; Itani, T.; Ogata, M.; Sato, Y.; Ariga, H. An Initiation Site of DNA Replication with Transcriptional Enhancer Activity Present Upstream of the C-Myc Gene. EMBO J. 1988, 7, 3135–3142. [Google Scholar] [CrossRef]
- Iguchi-Ariga, S.M.; Itani, T.; Kiji, Y.; Ariga, H. Possible Function of the C-Myc Product: Promotion of Cellular DNA Replication. EMBO J. 1987, 6, 2365–2371. [Google Scholar] [CrossRef]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-Transcriptional Control of DNA Replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef]
- Nepon-Sixt, B.S.; Bryant, V.L.; Alexandrow, M.G. Myc-Driven Chromatin Accessibility Regulates Cdc45 Assembly into CMG Helicases. Commun. Biol. 2019, 2, 110. [Google Scholar] [CrossRef]
- Srinivasan, S.V.; Dominguez-Sola, D.; Wang, L.C.; Hyrien, O.; Gautier, J. Cdc45 Is a Critical Effector of Myc-Dependent DNA Replication Stress. Cell Rep. 2013, 3, 1629–1639. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. C-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate P53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Luoto, K.R.; Meng, A.X.; Wasylishen, A.R.; Zhao, H.; Coackley, C.L.; Penn, L.Z.; Bristow, R.G. Tumor Cell Kill by C-MYC Depletion: Role of MYC-Regulated Genes That Control DNA Double-Strand Break Repair. Cancer Res. 2010, 70, 8748–8759. [Google Scholar] [CrossRef]
- Chiang, Y.C.; Teng, S.C.; Su, Y.N.; Hsieh, F.J.; Wu, K.J. C-Myc Directly Regulates the Transcription of the NBS1 Gene Involved in DNA Double-Strand Break Repair. J. Biol. Chem. 2003, 278, 19286–19291. [Google Scholar] [CrossRef]
- Petroni, M.; Sardina, F.; Heil, C.; Sahún-Roncero, M.; Colicchia, V.; Veschi, V.; Albini, S.; Fruci, D.; Ricci, B.; Soriani, A.; et al. The MRN Complex Is Transcriptionally Regulated by MYCN during Neural Cell Proliferation to Control Replication Stress. Cell Death Differ. 2016, 23, 197–206. [Google Scholar] [CrossRef]
- Petroni, M.; Sardina, F.; Infante, P.; Bartolazzi, A.; Locatelli, E.; Fabretti, F.; Di Giulio, S.; Capalbo, C.; Cardinali, B.; Coppa, A.; et al. MRE11 Inhibition Highlights a Replication Stress-Dependent Vulnerability of MYCN-Driven Tumors. Cell Death Dis. 2018, 9, 895. [Google Scholar] [CrossRef]
- Grandori, C.; Wu, K.J.; Fernandez, P.; Ngouenet, C.; Grim, J.; Clurman, B.E.; Moser, M.J.; Oshima, J.; Russell, D.W.; Swisshelm, K.; et al. Werner Syndrome Protein Limits MYC-Induced Cellular Senescence. Genes Dev. 2003, 17, 1569–1574. [Google Scholar] [CrossRef]
- Robinson, K.; Asawachaicharn, N.; Galloway, D.A.; Grandori, C. C-Myc Accelerates S-Phase and Requires WRN to Avoid Replication Stress. PLoS ONE 2009, 4, e5951. [Google Scholar] [CrossRef]
- Moser, R.; Toyoshima, M.; Robinson, K.; Gurley, K.E.; Howie, H.L.; Davison, J.; Morgan, M.; Kemp, C.J.; Grandori, C. MYC-Driven Tumorigenesis Is Inhibited by WRN Syndrome Gene Deficiency. Mol. Cancer Res. 2012, 10, 535–545. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; D’Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting Oncogene-Induced Replicative Stress for the Selective Killing of Myc-Driven Tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Ferrao, P.T.; Bukczynska, E.P.; Johnstone, R.W.; McArthur, G.A. Efficacy of CHK Inhibitors as Single Agents in MYC-Driven Lymphoma Cells. Oncogene 2012, 31, 1661–1672. [Google Scholar] [CrossRef]
- Höglund, A.; Nilsson, L.M.; Muralidharan, S.V.; Hasvold, L.A.; Merta, P.; Rudelius, M.; Nikolova, V.; Keller, U.; Nilsson, J.A. Therapeutic Implications for the Induced Levels of Chk1 in Myc-Expressing Cancer Cells. Clin. Cancer Res. 2011, 17, 7067–7079. [Google Scholar] [CrossRef]
- Puccetti, M.V.; Adams, C.M.; Kushinsky, S.; Eischen, C.M. SMARCAL1 and ZrAnB3 Protect Replication Forks from MYC-Induced DNA Replication Stress. Cancer Res. 2019, 79, 1612–1623. [Google Scholar] [CrossRef]
- Kurashima, K.; Sekimoto, T.; Oda, T.; Kawabata, T.; Hanaoka, F.; Yamashita, T. Polη, a Y-Family Translesion Synthesis Polymerase, Promotes Cellular Tolerance of Myc-Induced Replication Stress. J. Cell Sci. 2018, 131, jcs212183. [Google Scholar] [CrossRef]
- Leung, C.W.B.; Wall, J.; Esashi, F. From Rest to Repair: Safeguarding Genomic Integrity in Quiescent Cells. DNA Repair 2024, 142, 103752. [Google Scholar] [CrossRef]
- Brambati, A.; Sacco, O.; Porcella, S.; Heyza, J.; Kareh, M.; Schmidt, J.C.; Sfeir, A. RHINO Directs MMEJ to Repair DNA Breaks in Mitosis. Science 2023, 381, 653–660. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic P53-Independent P21 Expression Causes Genomic Instability by Deregulating Replication Licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef]
- Zampetidis, C.P.; Galanos, P.; Angelopoulou, A.; Zhu, Y.; Polyzou, A.; Karamitros, T.; Kotsinas, A.; Lagopati, N.; Mourkioti, I.; Mirzazadeh, R.; et al. A Recurrent Chromosomal Inversion Suffices for Driving Escape from Oncogene-Induced Senescence via SubTAD Reorganization. Mol. Cell 2021, 81, 4907–4923.e8. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Singh, V.K.; Rastogi, A.; Hu, X.; Wang, Y.; De, S. Mutational Signature SBS8 Predominantly Arises Due to Late Replication Errors in Cancer. Commun. Biol. 2020, 3, 421. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Variation in Cancer Risk among Tissues Can Be Explained by the Number of Stem Cell Divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-Related Remodelling of Oesophageal Epithelia by Mutated Cancer Drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Igarashi, T.; Yano, K.; Endo, S.; Shiotani, B. Tolerance of Oncogene-Induced Replication Stress: A Fuel for Genomic Instability. Cancers 2024, 16, 3507. https://doi.org/10.3390/cancers16203507
Igarashi T, Yano K, Endo S, Shiotani B. Tolerance of Oncogene-Induced Replication Stress: A Fuel for Genomic Instability. Cancers. 2024; 16(20):3507. https://doi.org/10.3390/cancers16203507
Chicago/Turabian StyleIgarashi, Taichi, Kimiyoshi Yano, Syoju Endo, and Bunsyo Shiotani. 2024. "Tolerance of Oncogene-Induced Replication Stress: A Fuel for Genomic Instability" Cancers 16, no. 20: 3507. https://doi.org/10.3390/cancers16203507
APA StyleIgarashi, T., Yano, K., Endo, S., & Shiotani, B. (2024). Tolerance of Oncogene-Induced Replication Stress: A Fuel for Genomic Instability. Cancers, 16(20), 3507. https://doi.org/10.3390/cancers16203507