
CRISPRing KRAS: A Winding Road with a Bright Future in Basic and Translational Cancer Research



Abstract
Simple Summary
Abstract
1. Introduction
2. Application of CRISPR/Cas Technology to Basic and Translational Cancer Research on KRAS
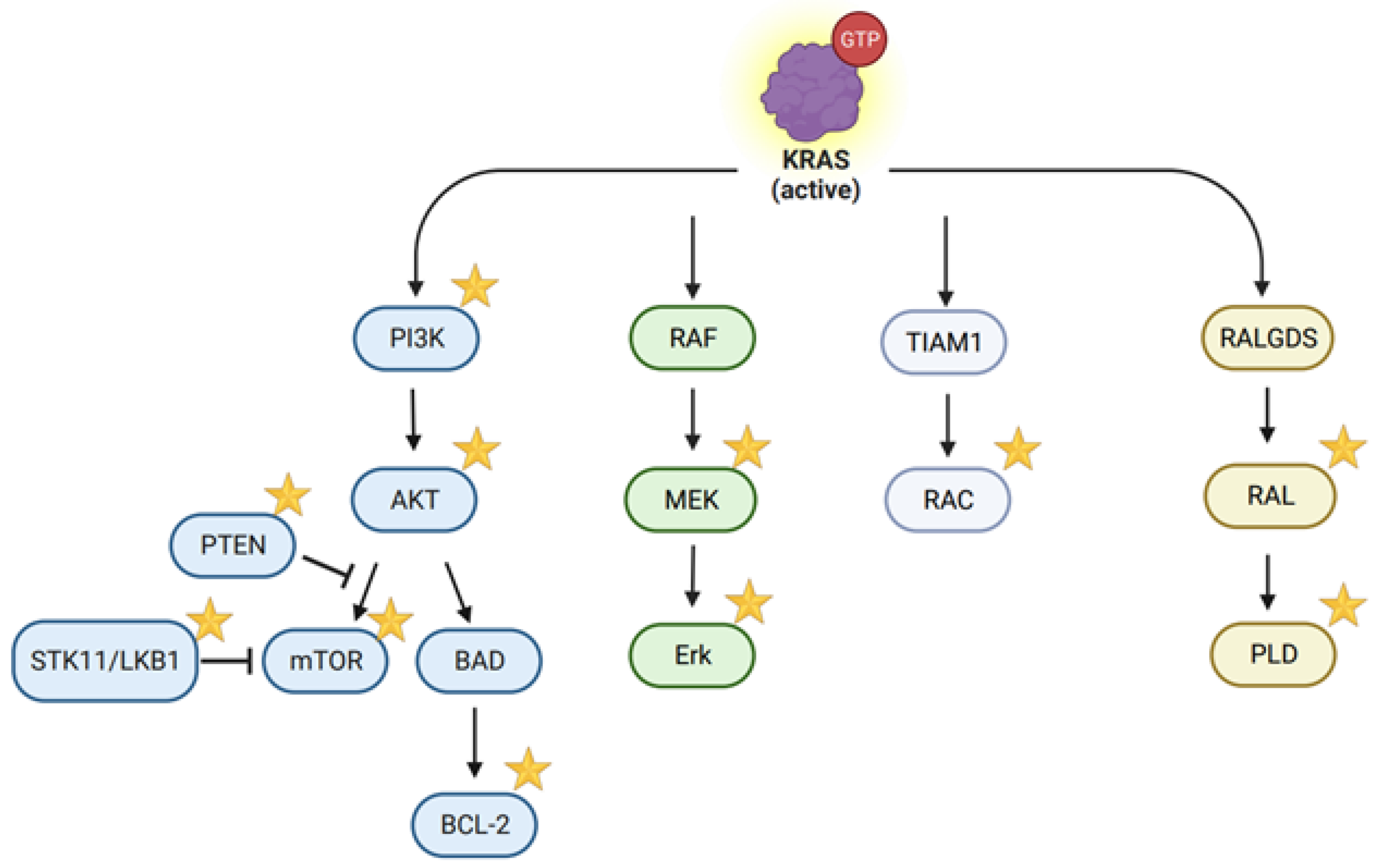
2.1. Mechanisms of KRAS-Dependent Oncogenesis
2.2. Mechanisms of Resistance to KRAS Pathway Inhibition
2.3. Epigenetic Regulatory Networks in the Development of KRAS-Mutant Tumors
2.4. Crosstalk between KRAS Signaling and Anti-Cancer Immunity
2.5. KRAS Synthetic Lethal Interactions
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Palomba, G.; Doneddu, V.; Cossu, A.; Paliogiannis, P.; Manca, A.; Casula, M.; Colombino, M.; Lanzillo, A.; Defraia, E.; Pazzola, A.; et al. Prognostic impact of KRAS, NRAS, BRAF, and PIK3CA mutations in primary colorectal carcinomas: A population-based study. J. Transl. Med. 2016, 14, 292. [Google Scholar] [CrossRef] [PubMed]
- Papke, B.; Der, C.J. Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Plowman, S.J.; Berry, R.L.; Bader, S.A.; Luo, F.; Arends, M.J.; Harrison, D.J.; Hooper, M.L.; Patek, C.E. K-ras 4A and 4B are co-expressed widely in human tissues, and their ratio is altered in sporadic colorectal cancer. J. Exp. Clin. Cancer Res. 2006, 25, 259–267, Erratum in J. Exp. Clin. Cancer Res. 2007, 26, 2. [Google Scholar]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef]
- Nimnual, A.; Bar-Sagi, D. The two hats of SOS. Sci. STKE 2002, 2002, pe36. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976, Erratum in Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Bannoura, S.F.; Uddin, H.; Nagasaka, M.; Fazili, F.; Al-Hallak, M.N.; Philip, P.A.; El-Rayes, B.; Azmi, A.S. Targeting KRAS in pancreatic cancer: New drugs on the horizon. Cancer Metastasis Rev. 2021, 40, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; de Vos, A.M.; Milburn, M.V.; Kim, S.-H. Crystal structures at 2.2 Å resolution of the catalytic domains of normal ras protein and an oncogenic mutant complexed with GDP. J. Mol. Biol. 1991, 217, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465, Erratum in Nat. Rev. Cancer 2003, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Crown, J. Drugging “undruggable” genes for cancer treatment: Are we making progress? Int. J. Cancer 2021, 148, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Bery, N.; Miller, A.; Rabbitts, T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Commun. 2020, 11, 3233. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mendiratta, S.; Ehrhardt, K.; Kashyap, N.; White, M.A.; Bleris, L. Exploiting the CRISPR/Cas9 PAM Constraint for Single-Nucleotide Resolution Interventions. PLoS ONE 2016, 11, e0144970. [Google Scholar] [CrossRef]
- Maresch, R.; Mueller, S.; Veltkamp, C.; Öllinger, R.; Friedrich, M.; Heid, I.; Steiger, K.; Weber, J.; Engleitner, T.; Barenboim, M.; et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat. Commun. 2016, 7, 10770. [Google Scholar] [CrossRef]
- Michels, B.E.; Mosa, M.H.; Streibl, B.I.; Zhan, T.; Menche, C.; Abou-El-Ardat, K.; Darvishi, T.; Członka, E.; Wagner, S.; Winter, J.; et al. Pooled In Vitro and In Vivo CRISPR-Cas9 Screening Identifies Tumor Suppressors in Human Colon Organoids. Cell Stem Cell 2020, 26, 782–792.e7. [Google Scholar] [CrossRef]
- Junttila, M.R.; Karnezis, A.N.; Garcia, D.; Madriles, F.; Kortlever, R.M.; Rostker, F.; Brown Swigart, L.; Pham, D.M.; Seo, Y.; Evan, G.I.; et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010, 468, 567–571. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9–mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes 2020, 11, 704. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; Leboeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar] [PubMed]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H.; et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, P.E.; Eichner, L.J.; Brun, S.N.; Kamireddy, A.; Svensson, R.U.; Vera, L.I.; Ross, D.S.; Rymoff, T.J.; Hutchins, A.; Galvez, H.M.; et al. The AMPK-Related Kinases SIK1 and SIK3 Mediate Key Tumor-Suppressive Effects of LKB1 in NSCLC. Cancer Discov. 2019, 9, 1606–1627. [Google Scholar] [CrossRef]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301, Erratum in Nat. Rev. Cancer 2011, 11, 458. [Google Scholar] [CrossRef]
- Berthelsen, M.F.; Leknes, S.L.; Riedel, M.; Pedersen, M.A.; Joseph, J.V.; Hager, H.; Vendelbo, M.H.; Thomsen, M.K. Comparative Analysis of Stk11/Lkb1 versus Pten Deficiency in Lung Adenocarcinoma Induced by CRISPR/Cas9. Cancers 2021, 13, 974. [Google Scholar] [CrossRef]
- Cheng, R.; Li, F.; Zhang, M.; Xia, X.; Wu, J.; Gao, X.; Zhou, H.; Zhang, Z.; Huang, N.; Yang, X.; et al. A novel protein RASON encoded by a lncRNA controls oncogenic RAS signaling in KRAS mutant cancers. Cell Res. 2023, 33, 30–45. [Google Scholar] [CrossRef]
- Chen, H.; Garbutt, C.C.; Spentzos, D.; Choy, E.; Hornicek, F.J.; Duan, Z. Expression and Therapeutic Potential of SOX9 in Chordoma. Clin. Cancer Res. 2017, 23, 5176–5186. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Carrasco-Garcia, E.; Garcia-Puga, M.; Aldaz, P.; Montes, M.; Fernandez-Reyes, M.; de Oliveira, C.C.; Lawrie, C.H.; Araúzo-Bravo, M.J.; Ribeiro, M.L.; et al. SOX9 Elevation Acts with Canonical WNT Signaling to Drive Gastric Cancer Progression. Cancer Res 2016, 76, 6735–6746. [Google Scholar] [CrossRef] [PubMed]
- Aleman, A.; Adrien, L.; Lopez-Serra, L.; Cordon-Cardo, C.; Esteller, M.; Belbin, T.J.; Sanchez-Carbayo, M. Identification of DNA hypermethylation of SOX9 in association with bladder cancer progression using CpG microarrays. Br. J. Cancer 2007, 98, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Lian, P.; Zheng, P.-S. SOX9, a potential tumor suppressor in cervical cancer, transactivates p21WAF1/CIP1 and suppresses cervical tumor growth. Oncotarget 2015, 6, 20711–20722. [Google Scholar] [CrossRef]
- Zhong, H.; Lu, W.; Tang, Y.; Wiel, C.; Wei, Y.; Cao, J.; Riedlinger, G.; Papagiannakopoulos, T.; Guo, J.Y.; Bergo, M.O.; et al. SOX9 drives KRAS-induced lung adenocarcinoma progression and suppresses anti-tumor immunity. Oncogene 2023, 42, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; Swaroop, A.; Troche, C.; Licht, J.D. The Role of Nuclear Receptor–Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026708. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Zeng, L.; Li, Y.; Hausmann, S.; Ghosh, D.; Yuan, G.; Nguyen, T.N.; Lyu, R.; Caporicci, M.; Morales Benitez, A.; et al. NSD2 dimethylation at H3K36 promotes lung adenocarcinoma pathogenesis. Mol. Cell 2021, 81, 4481–4492.e9. [Google Scholar] [CrossRef]
- Yang, H.; Liang, S.-Q.; Schmid, R.A.; Peng, R.-W. New Horizons in KRAS-Mutant Lung Cancer: Dawn after Darkness. Front. Oncol. 2019, 9, 953. [Google Scholar] [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552, Erratum in Nat. Rev. Drug Discov. 2020, 19, 902. [Google Scholar] [CrossRef]
- Bennouna, J.; Lang, I.; Valladares-Ayerbes, M.; Boer, K.; Adenis, A.; Escudero, P.; Kim, T.-Y.; Pover, G.M.; Morris, C.D.; Douillard, J.-Y. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Investig. New Drugs 2011, 29, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Bodoky, G.; Timcheva, C.; Spigel, D.R.; La Stella, P.J.; Ciuleanu, T.E.; Pover, G.; Tebbutt, N.C. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig. New Drugs 2012, 30, 1216–1223, Erratum in Investig. New Drugs. 2012, 30, 1272–1273. [Google Scholar] [CrossRef]
- Lake, D.; Corrêa, S.A.L.; Müller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [PubMed]
- Klomp, J.E.; Klomp, J.A.; Der, C.J. The ERK mitogen-activated protein kinase signaling network: The final frontier in RAS signal transduction. Biochem. Soc. Trans. 2021, 49, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Klomp, J.E.; Lee, Y.S.; Goodwin, C.M.; Papke, B.; Klomp, J.A.; Waters, A.M.; Stalnecker, C.A.; DeLiberty, J.M.; Drizyte-Miller, K.; Yang, R.; et al. CHK1 protects oncogenic KRAS-expressing cells from DNA damage and is a target for pancreatic cancer treatment. Cell Rep. 2021, 37, 110060. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Jänne, P.A. Circumventing Cancer Drug Resistance in the Era of Personalized Medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef]
- Lou, K.; Steri, V.; Ge, A.Y.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.M.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. KRAS G12C inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal. 2019, 12, eaaw9450. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Wu, Q.; Tian, Y.; Zhang, J.; Tong, X.; Huang, H.; Li, S.; Zhao, H.; Tang, Y.; Yuan, C.; Wang, K.; et al. In vivo CRISPR screening unveils histone demethylase UTX as an important epigenetic regulator in lung tumorigenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E3978–E3986. [Google Scholar] [CrossRef]
- Morgens, D.W.; Wainberg, M.; Boyle, E.A.; Ursu, O.; Araya, C.L.; Tsui, C.K.; Haney, M.S.; Hess, G.T.; Han, K.; Jeng, E.E.; et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 2017, 8, 15178. [Google Scholar] [CrossRef]
- Tycko, J.; Wainberg, M.; Marinov, G.K.; Ursu, O.; Hess, G.T.; Ego, B.K.; Aradhana; Li, A.; Truong, A.; Trevino, A.E.; et al. Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements. Nat. Commun. 2019, 10, 4063. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, M.; Cho, K.B.; Gao, X.; Guo, B. A CRISPR-Cas9 repressor for epigenetic silencing of KRAS. Pharmacol. Res. 2021, 164, 105304. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ng, W.-L.; Luster, T.A.; Hu, H.; Sviderskiy, V.O.; Dowling, C.M.; Hollinshead, K.E.; Zouitine, P.; Zhang, H.; Huang, Q.; et al. Epigenetic CRISPR Screens Identify Npm1 as a Therapeutic Vulnerability in Non–Small Cell Lung Cancer. Cancer Res 2020, 80, 3556–3567. [Google Scholar] [CrossRef] [PubMed]
- Pennock, G.K.; Chow, L.Q.M. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. Oncologist 2015, 20, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ishida, T.; Yoshikawa, K.; Ueda, R. Current status of immunotherapy. Jpn. J. Clin. Oncol. 2016, 46, 191–203. [Google Scholar] [CrossRef] [PubMed]
- D’Incecco, A.; Andreozzi, M.; Ludovini, V.; Rossi, E.; Capodanno, A.; Landi, L.; Tibaldi, C.; Minuti, G.; Salvini, J.; Coppi, E.; et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br. J. Cancer 2015, 112, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef]
- Huang, Q.; Li, F.; Hu, H.; Fang, Z.; Gao, Z.; Xia, G.; Ng, W.-L.; Khodadadi-Jamayran, A.; Chen, T.; Deng, J.; et al. Loss of TSC1/TSC2 sensitizes immune checkpoint blockade in non–small cell lung cancer. Sci. Adv. 2022, 8, eabi9533. [Google Scholar] [CrossRef]
- Falcomatà, C.; Bärthel, S.; Widholz, S.A.; Schneeweis, C.; Montero, J.J.; Toska, A.; Mir, J.; Kaltenbacher, T.; Heetmeyer, J.; Swietlik, J.J.; et al. Selective multi-kinase inhibition sensitizes mesenchymal pancreatic cancer to immune checkpoint blockade by remodeling the tumor microenvironment. Nat. Cancer 2022, 3, 318–336. [Google Scholar] [CrossRef]
- Dervovic, D.; Malik, A.A.; Chen, E.L.Y.; Narimatsu, M.; Adler, N.; Afiuni-Zadeh, S.; Krenbek, D.; Martinez, S.; Tsai, R.; Boucher, J.; et al. In vivo CRISPR screens reveal Serpinb9 and Adam2 as regulators of immune therapy response in lung cancer. Nat. Commun. 2023, 14, 3150, Erratum in Nat. Commun. 2023, 14, 5404. [Google Scholar] [CrossRef]
- Adeegbe, D.O.; Liu, Y.; Lizotte, P.H.; Kamihara, Y.; Aref, A.R.; Almonte, C.; Dries, R.; Li, Y.; Liu, S.; Wang, X.; et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non–Small Cell Lung Cancer. Cancer Discov. 2017, 7, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huang, Q.; Luster, T.A.; Hu, H.; Zhang, H.; Ng, W.-L.; Khodadadi-Jamayran, A.; Wang, W.; Chen, T.; Deng, J.; et al. In Vivo Epigenetic CRISPR Screen Identifies Asf1a as an Immunotherapeutic Target in Kras-Mutant Lung Adenocarcinoma. Cancer Discov. 2020, 10, 270–287. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Hahn, W.C. Synthetic Lethal Vulnerabilities in KRAS-Mutant Cancers. Cold Spring Harb. Perspect. Med. 2018, 8, a031518. [Google Scholar] [CrossRef]
- Morgens, D.W.; Deans, R.M.; Li, A.; Bassik, M.C. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 2016, 34, 634–636. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Šuštić, T.; van Wageningen, S.; Bosdriesz, E.; Reid, R.J.D.; Dittmar, J.; Lieftink, C.; Beijersbergen, R.L.; Wessels, L.F.A.; Rothstein, R.; Bernards, R. A role for the unfolded protein response stress sensor ERN1 in regulating the response to MEK inhibitors in KRAS mutant colon cancers. Genome Med. 2018, 10, 90, Erratum in Genome Med. 2021, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.R.; Kostyrko, K.; Han, K.; Mooney, N.A.; Jeng, E.E.; Spees, K.; Dinh, P.T.; Abbott, K.L.; Gwinn, D.M.; Sweet-Cordero, E.A.; et al. Combined Proteomic and Genetic Interaction Mapping Reveals New RAS Effector Pathways and Susceptibilities. Cancer Discov. 2020, 10, 1950–1967. [Google Scholar] [CrossRef]
- Du, R.; Sullivan, D.K.; Azizian, N.G.; Liu, Y.; Li, Y. Inhibition of ERAD synergizes with FTS to eradicate pancreatic cancer cells. BMC Cancer 2021, 21, 237. [Google Scholar] [CrossRef]
- Dance, M.; Montagner, A.; Salles, J.-P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Li, T.; Kikuchi, O.; Zhou, J.; Wang, Y.; Pokharel, B.; Bastl, K.; Gokhale, P.; Knott, A.; Zhang, Y.; Doench, J.G.; et al. Developing SHP2-based combination therapy for KRAS-amplified cancer. JCI Insight 2023, 8, e152714. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.R.; Oh, Y.; Na, D.; Min, S.; Kang, J.; Jang, D.; Shin, S.; Kim, J.; Lee, S.E.; Jeong, E.M.; et al. CRISPR screens identify a novel combination treatment targeting BCL-XL and WNT signaling for KRAS/BRAF-mutated colorectal cancers. Oncogene 2021, 40, 3287–3302. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liang, S.-Q.; Zhao, L.; Yang, H.; Marti, T.M.; Hegedüs, B.; Gao, Y.; Zheng, B.; Chen, C.; Wang, W.; et al. Metabolic synthetic lethality by targeting NOP56 and mTOR in KRAS-mutant lung cancer. J. Exp. Clin. Cancer Res. 2022, 41, 25. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.M.; Waters, A.M.; Klomp, J.E.; Javaid, S.; Bryant, K.L.; Stalnecker, C.A.; Drizyte-Miller, K.; Papke, B.; Yang, R.; Amparo, A.M.; et al. Combination Therapies with CDK4/6 Inhibitors to Treat KRAS-Mutant Pancreatic Cancer. Cancer Res. 2023, 83, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.R.; Winter, P.S.; Lin, K.H.; Nussbaum, D.P.; Cakir, M.; Stein, E.M.; Soderquist, R.S.; Crawford, L.; Leeds, J.C.; Newcomb, R.; et al. A Landscape of Therapeutic Cooperativity in KRAS Mutant Cancers Reveals Principles for Controlling Tumor Evolution. Cell Rep. 2017, 20, 999–1015. [Google Scholar] [CrossRef]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017, 168, 890–903.e15. [Google Scholar] [CrossRef]
- Yau, E.H.; Kummetha, I.R.; Lichinchi, G.; Tang, R.; Zhang, Y.; Rana, T.M. Genome-Wide CRISPR Screen for Essential Cell Growth Mediators in Mutant KRAS Colorectal Cancers. Cancer Res. 2017, 77, 6330–6339. [Google Scholar] [CrossRef]
- Dompe, N.; Klijn, C.; Watson, S.A.; Leng, K.; Port, J.; Cuellar, T.; Watanabe, C.; Haley, B.; Neve, R.; Evangelista, M.; et al. A CRISPR screen identifies MAPK7 as a target for combination with MEK inhibition in KRAS mutant NSCLC. PLoS ONE 2018, 13, e0199264. [Google Scholar] [CrossRef]
- Szlachta, K.; Kuscu, C.; Tufan, T.; Adair, S.J.; Shang, S.; Michaels, A.D.; Mullen, M.G.; Fischer, N.L.; Yang, J.; Liu, L.; et al. CRISPR knockout screening identifies combinatorial drug targets in pancreatic cancer and models cellular drug response. Nat. Commun. 2018, 9, 4275. [Google Scholar] [CrossRef]
- Sulahian, R.; Kwon, J.J.; Walsh, K.H.; Pailler, E.; Bosse, T.L.; Thaker, M.; Almanza, D.; Dempster, J.M.; Pan, J.; Piccioni, F.; et al. Synthetic Lethal Interaction of SHOC2 Depletion with MEK Inhibition in RAS-Driven Cancers. Cell Rep. 2019, 29, 118–134.e8. [Google Scholar] [CrossRef]
- Milton, C.K.; Self, A.J.; Clarke, P.A.; Banerji, U.; Piccioni, F.; Root, D.E.; Whittaker, S.R. A Genome-scale CRISPR Screen Identifies the ERBB and mTOR Signaling Networks as Key Determinants of Response to PI3K Inhibition in Pancreatic Cancer. Mol. Cancer Ther. 2020, 19, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Pierce, S.E.; Li, A.; Spees, K.; Anderson, G.R.; Seoane, J.A.; Lo, Y.-H.; Dubreuil, M.; Olivas, M.; Kamber, R.A.; et al. CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 2020, 580, 136–141. [Google Scholar] [CrossRef]
- Wei, X.; Yang, J.; Adair, S.J.; Ozturk, H.; Kuscu, C.; Lee, K.Y.; Kane, W.J.; O’Hara, P.E.; Liu, D.; Demirlenk, Y.M.; et al. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28068–28079. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Khatib, T.O.; Papke, B.; Goodwin, C.M.; Hobbs, G.A.; Diehl, J.N.; Yang, R.; Edwards, A.C.; Walsh, K.H.; Sulahian, R.; et al. Targeting p130Cas- and microtubule-dependent MYC regulation sensitizes pancreatic cancer to ERK MAPK inhibition. Cell Rep. 2021, 35, 109291. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Luo, D.; Yu, J.; Zhang, M.; Zheng, X.; Xu, G.; Wang, J.; Wang, H.; Xu, Y.; Jiang, K.; et al. Genome-wide CRISPR-cas9 knockout screening identifies GRB7 as a driver for MEK inhibitor resistance in KRAS mutant colon cancer. Oncogene 2022, 41, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.-H.; Winters, I.P.; Wang, J.; Naranjo, S.; Dudgeon, C.; Tamburini, F.B.; Brady, J.J.; Yang, D.; Grüner, B.M.; Chuang, C.-H.; et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 2015, 29, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; Tammela, T.; Cetinbas, N.M.; Akkad, A.; Roghanian, A.; Rickelt, S.; Almeqdadi, M.; Wu, K.; Oberli, M.A.; Sánchez-Rivera, F.J.; et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 2017, 35, 569–576, Erratum in Nat. Biotechnol. 2017, 35, 1211. [Google Scholar] [CrossRef]
- Erlangga, Z.; Wolff, K.; Poth, T.; Peltzer, A.; Nahnsen, S.; Spielberg, S.; Timrott, K.; Woller, N.; Kühnel, F.; Manns, M.P.; et al. Potent Antitumor Activity of Liposomal Irinotecan in an Organoid- and CRISPR-Cas9-Based Murine Model of Gallbladder Cancer. Cancers 2019, 11, 1904. [Google Scholar] [CrossRef]
- Ideno, N.; Yamaguchi, H.; Okumura, T.; Huang, J.; Brun, M.J.; Ho, M.L.; Suh, J.; Gupta, S.; Maitra, A.; Ghosh, B. A pipeline for rapidly generating genetically engineered mouse models of pancreatic cancer using in vivo CRISPR-Cas9-mediated somatic recombination. Lab. Investig. 2019, 99, 1233–1244. [Google Scholar] [CrossRef]
- Zafra, M.P.; Parsons, M.J.; Kim, J.; Alonso-Curbelo, D.; Goswami, S.; Schatoff, E.M.; Han, T.; Katti, A.; Fernandez, M.T.C.; Wilkinson, J.E.; et al. An In Vivo Kras Allelic Series Reveals Distinct Phenotypes of Common Oncogenic Variants. Cancer Discov. 2020, 10, 1654–1671. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Fushimi, K.; Yabuki, Y.; Maru, Y.; Hasegawa, S.; Matsuura, T.; Kurotaki, D.; Suzuki, A.; Kobayashi, N.; Yoneda, M.; et al. Precision modeling of gall bladder cancer patients in mice based on orthotopic implantation of organoid-derived tumor buds. Oncogenesis 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, I.; D’Amico, S.; Rao, M.; Li, J.; Hayman, M.J.; Powers, S.; Petrenko, O.; Reich, N.C. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat. Commun. 2021, 12, 1482. [Google Scholar] [CrossRef]
- Hartmann, O.; Reissland, M.; Maier, C.R.; Fischer, T.; Prieto-Garcia, C.; Baluapuri, A.; Schwarz, J.; Schmitz, W.; Garrido-Rodriguez, M.; Pahor, N.; et al. Implementation of CRISPR/Cas9 Genome Editing to Generate Murine Lung Cancer Models That Depict the Mutational Landscape of Human Disease. Front. Cell Dev. Biol. 2021, 9, 641618. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Creighton, C.J.; Heavener, K.S.; Gutierrez, C.; Dou, Y.; Ku, A.T.; Zhang, Y.; Jiang, W.; Urrutia, J.; Jiang, W.; et al. Efficient cancer modeling through CRISPR-Cas9/HDR-based somatic precision gene editing in mice. Sci. Adv. 2023, 9, eade0059. [Google Scholar] [CrossRef]
- Ely, Z.A.; Mathey-Andrews, N.; Naranjo, S.; Gould, S.I.; Mercer, K.L.; Newby, G.A.; Cabana, C.M.; Rideout, W.M., 3rd; Jaramillo, G.C.; Khirallah, J.M.; et al. A prime editor mouse to model a broad spectrum of somatic mutations in vivo. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef]
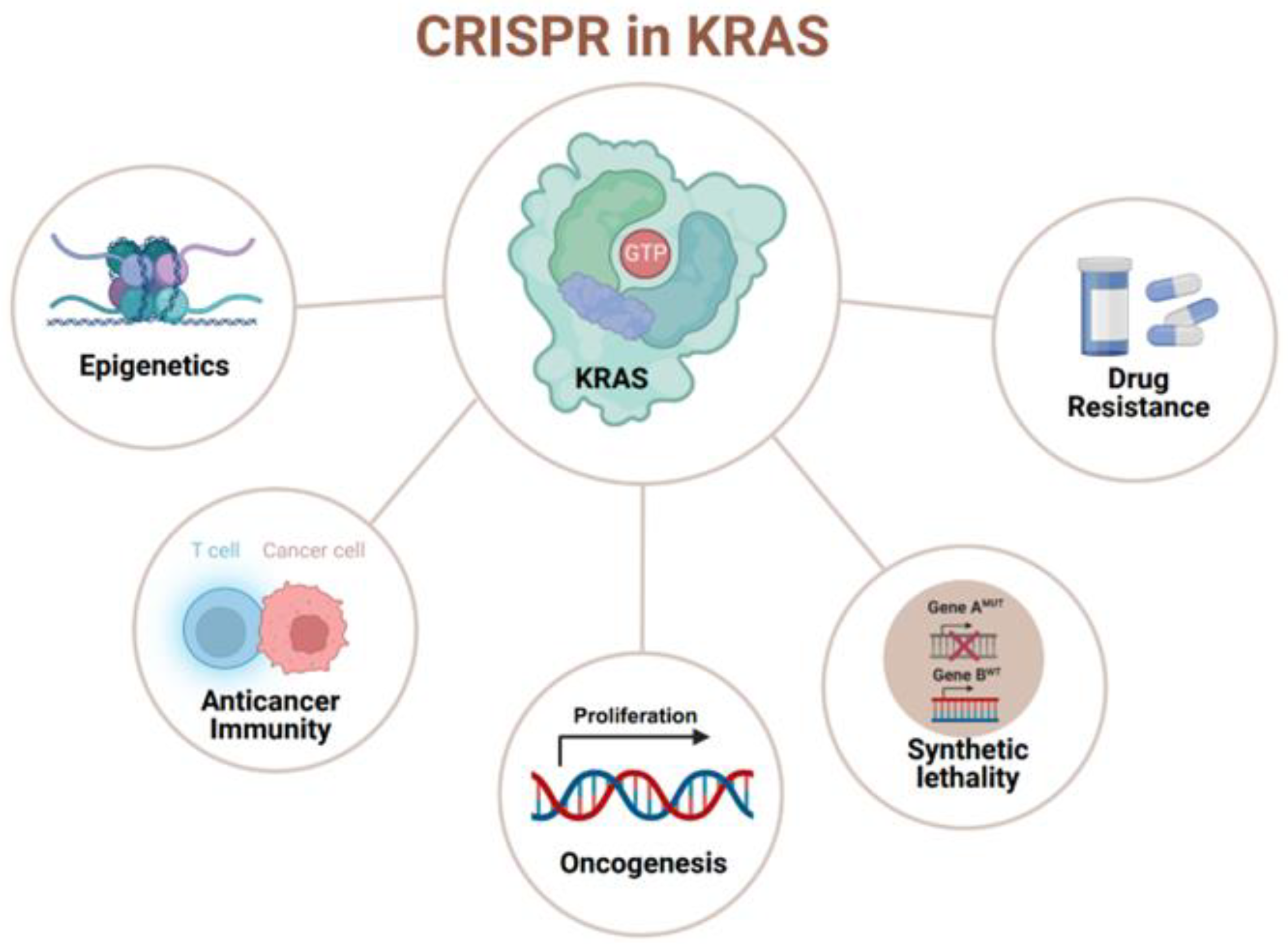
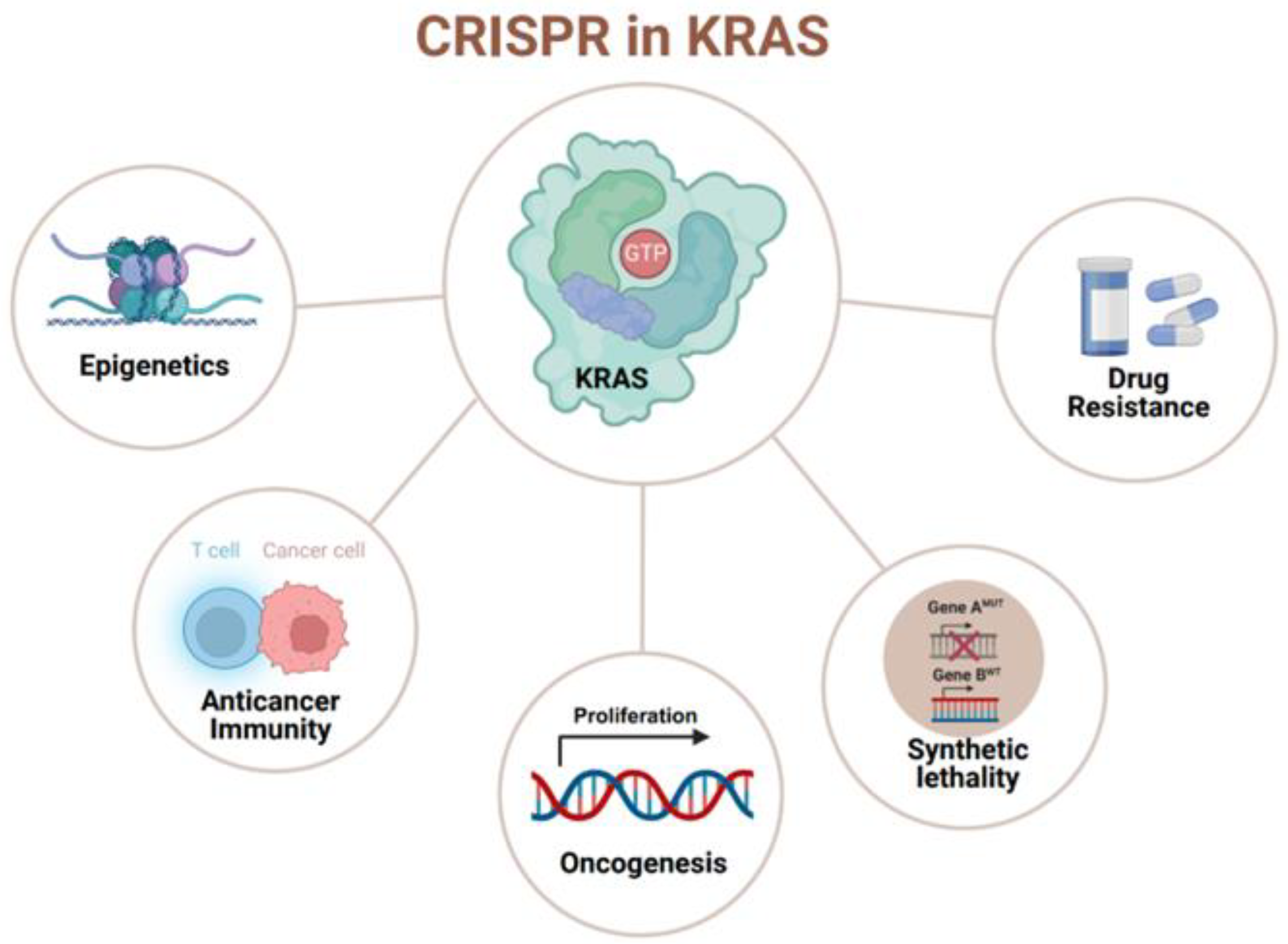
 : it has been targeted or studied by the CRISPR/Cas system. (created with BioRender.com, accessed on 29 December 2023).
: it has been targeted or studied by the CRISPR/Cas system. (created with BioRender.com, accessed on 29 December 2023).
: it has been targeted or studied by the CRISPR/Cas system. (created with BioRender.com, accessed on 29 December 2023).
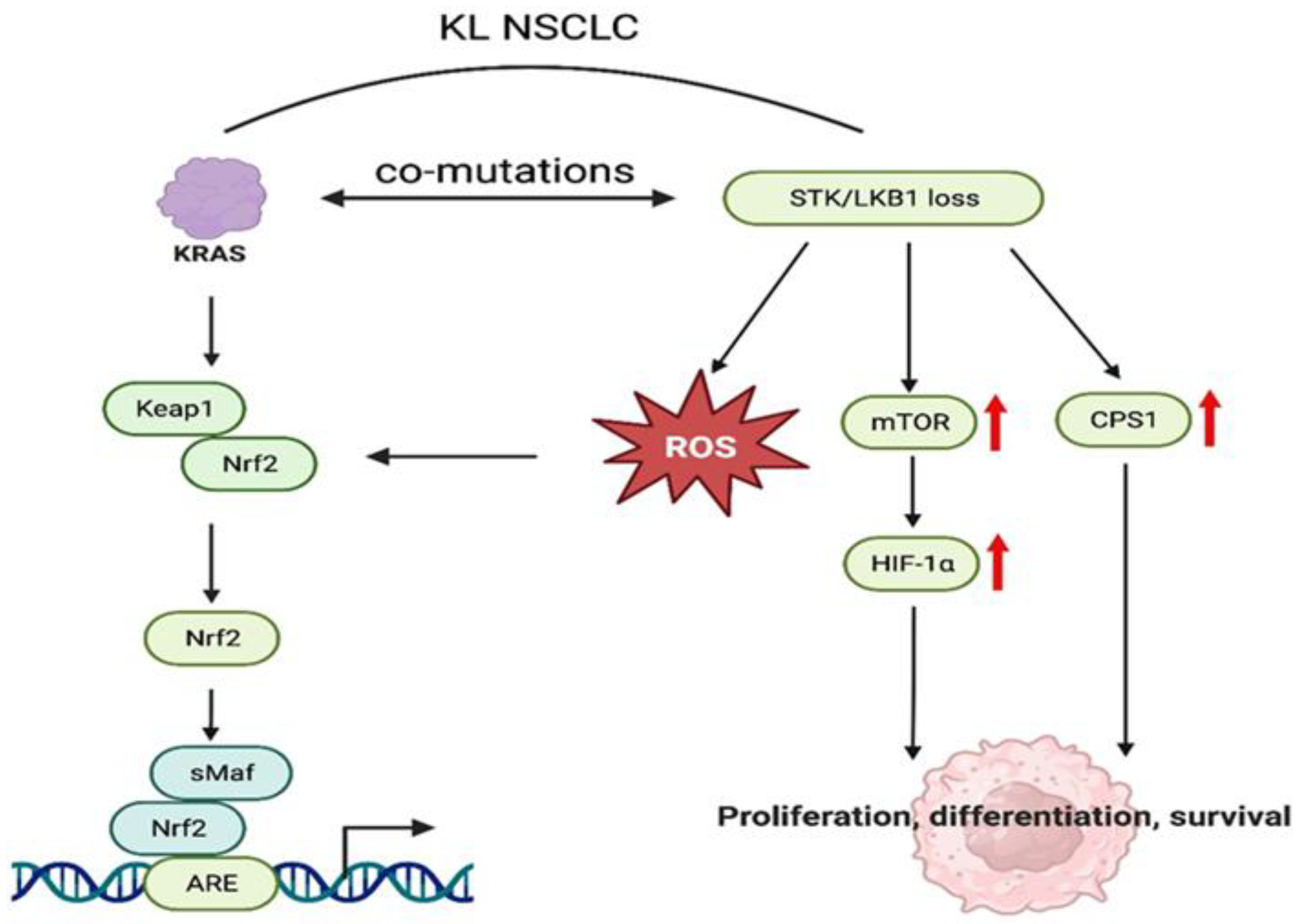
: it has been targeted or studied by the CRISPR/Cas system. (created with BioRender.com, accessed on 29 December 2023).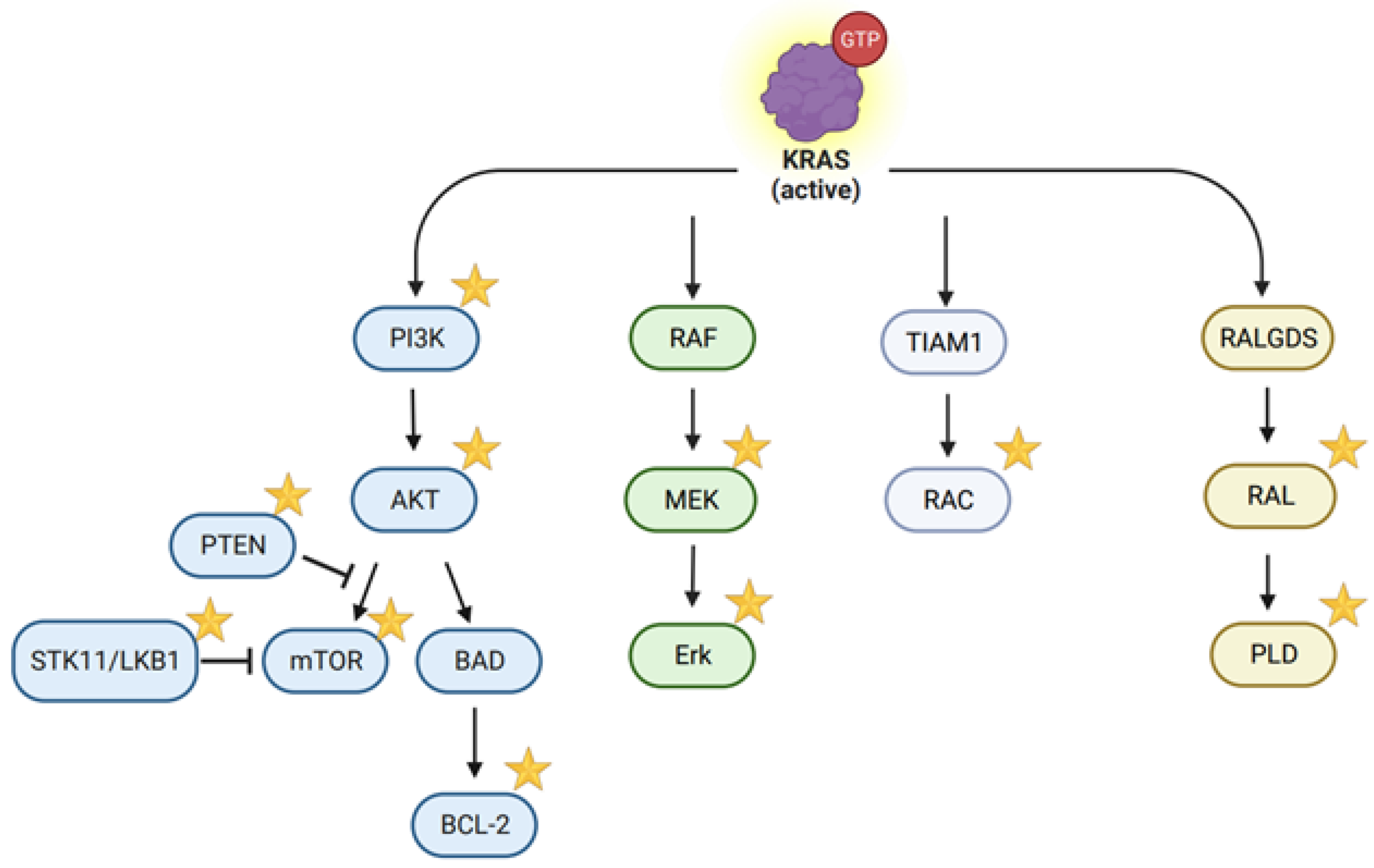

{kind=link}
{kind=link}
{kind=link}
| Cell Lines | CRISPR Library | Synthetic Lethal Genes or Pathways | Drug Inhibition | Types of Inhibition | Reference |
|---|---|---|---|---|---|
| HCT11, SW620 CRC119, CRC240 (CRC) | Subgenomic | MEK/ERK; SRC; BCL-XL | VX-11e Dasatinib WEHI-539 | KRAS effectors/tumor stress-response pathways | [85] |
| NB4; PL-21; SKM-1 (myeloid leukemia) | Genome-wide | RCE1; I CMT; RAF1; SHOC2; PREX1 | N/A | KRAS effectors | [86] |
| HCT116 (colorectal cancer) | Genome-scale | SUCLA2; NADK; KHK; SNRPC; POP5; SF3B2; NF2; RALGAPB; INO80C | N/A | Tumor stress response pathways | [87] |
| MOR (NSCLC) | “druggable genome” | MAPK7 | cobimetinib; XMD17-109 | KRAS effectors | [88] |
| PDX366 (PDAC) | “Nuclear” sgRNA library | CENPE; RRM1 | trametinib GSK923295 COH29 | KRAS effectors | [89] |
| ERN1 knockout (KO) LoVo, HCT-116, SW480, DLD1 (colon cancer) | The human GeCKO v. 2 | DUSP4; STK40, DET1; COP1; CBFB; RUNX2; JNK signaling | selumetinib (AZD6244),trametinib (GSK1120212); SR-3306; | KRAS effectors | [77] |
| Pancreatic and lung cancer cells | The Avana-4 barcoded sgRNA library | SHOC2/MEK | trametinib | Tumor stress-response pathways | [90] |
| MIA-PACA2 | The Avana4 lentiviral library | PI3K/ERBB-family RTK signaling/mTOR | BYL719; pelitinib; AZD2014 | KRAS effectors | [91] |
| H23 (NSCLC) | The genome-wide CRISPR library | IGF1R signaling pathways; CPD | N/A | KRAS effectors | [92] |
| PDX366 (PDAC) | 8031 sgRNA–containing library | PRMT5 | EPZ015666 and EPZ015938 | Tumor stress-response pathways | [93] |
| A549; H23; H2009 (lung cancer), | The large DrugTarget-CDKO library | RAP1GDS1/RHOA | N/A | KRAS effectors/Tumor stress-response pathways | [78] |
| 4292; PANC-1; MIA PaCa-2 (PDAC) | Pooled mouse CRISPR lentiviral library | ERAD pathway | eeyarestatin I (EerI) | Tumor stress-response pathways | [79] |
| SW620; HCT116 (colorectal cancer) | Genome-wide CRISPR/Cas9 Knockout (MAGeCK) algorithm | WNT signaling/BCL-2 family genes | NCB-0846; Bcl-2i: ABT-263 | KRAS effectors | [82] |
| Pa16C (pancreatic cancer) | Not mentioned | SRC; ERK | KX2-391 (tirbanibulin) | Tumor stress-response pathways | [94] |
| HCT116, SW480, LS174T (colon cancer) | Genome-wide | GRB7 | N/A | KRAS effectors | [95] |
| H358, H460, A549, PF563, PF139 (lung cancer), MIA-PaCa, HPAF-II (pancreatic cancer) HCT-116, DLD-1 (colon cancer) | NOP56 Knockout | NOP56 and mTOR | rapamycin; shNOP56 | KRAS effectors/tumor stress-response pathways | [83] |
| Pa01C; Pa02C; Pa03C; Pa04C; Pa14C; Pa16C (PDAC) | “Druggable genome” | ERK; CDK2/4/6 | PF-06873600; SCH772984 | KRAS effectors | [84] |
| KE-39, HUG1-N (gastric cancer) | Genome-wide | SHP2/downstream MAPK pathways | SHP099; Ribociclib | KRAS effectors | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, X.; Du, J.; Peng, R.-W.; Chen, C.; Yang, Z. CRISPRing KRAS: A Winding Road with a Bright Future in Basic and Translational Cancer Research. Cancers 2024, 16, 460. https://doi.org/10.3390/cancers16020460
Gong X, Du J, Peng R-W, Chen C, Yang Z. CRISPRing KRAS: A Winding Road with a Bright Future in Basic and Translational Cancer Research. Cancers. 2024; 16(2):460. https://doi.org/10.3390/cancers16020460
Chicago/Turabian StyleGong, Xian, Jianting Du, Ren-Wang Peng, Chun Chen, and Zhang Yang. 2024. "CRISPRing KRAS: A Winding Road with a Bright Future in Basic and Translational Cancer Research" Cancers 16, no. 2: 460. https://doi.org/10.3390/cancers16020460
APA StyleGong, X., Du, J., Peng, R.-W., Chen, C., & Yang, Z. (2024). CRISPRing KRAS: A Winding Road with a Bright Future in Basic and Translational Cancer Research. Cancers, 16(2), 460. https://doi.org/10.3390/cancers16020460